
Abstract
Duchenne muscular dystrophy (DMD) is a muscle wasting disorder caused by mutations in the gene encoding dystrophin. Gene therapy using micro-dystrophin (MD) transgenes and recombinant adeno-associated virus (rAAV) vectors hold great promise. To overcome the limited packaging capacity of rAAV vectors, most MD do not include dystrophin carboxy-terminal (CT) domain. Yet, the CT domain is known to recruit α1- and β1-syntrophins and α-dystrobrevin, a part of the dystrophin-associated protein complex (DAPC), which is a signaling and structural mediator of muscle cells. In this study, we explored the impact of inclusion of the dystrophin CT domain on ΔR4-23/ΔCT MD (MD1), in DMDmdx rats, which allows for relevant evaluations at muscular and cardiac levels. We showed by LC-MS/MS that MD1 expression is sufficient to restore the interactions at a physiological level of most DAPC partners in skeletal and cardiac muscles, and that inclusion of the CT domain increases the recruitment of some DAPC partners at supra-physiological levels. In parallel, we demonstrated that inclusion of the CT domain does not improve MD1 therapeutic efficacy on DMD muscle and cardiac pathologies. Our work highlights new evidences of the therapeutic potential of MD1 and strengthens the relevance of this candidate for gene therapy of DMD.
Similar content being viewed by others

Introduction
Duchenne Muscular Dystrophy (DMD) is a fatal X-linked degenerative neuromuscular disease affecting ≈1:5,000 male births [1]. DMD is caused by mutations in the DMD gene coding for dystrophin, a large sub-sarcolemmal protein (427 kDa) essential for the integrity of muscular fibers in skeletal and cardiac muscles [2]. Dystrophin comprises four structural regions: an actin-binding N-terminal (NT) domain, a central rod domain with 24 spectrin-like repeats and four hinges, a cysteine rich (CR) domain, and a carboxy-terminal (CT) domain. In myofibers, dystrophin plays a central role in the dystrophin-associated protein complex (DAPC) by maintaining the structural link between the actin cytoskeleton and the extracellular matrix, and by recruiting regulatory and signaling proteins (Fig. 1A) [3]. Its loss leads to DAPC disruption [4], susceptibility to contraction-induced injuries and replacement of muscle cells by connective (fibrosis) and adipose tissue, resulting in progressive loss of muscle function and cardiac involvement [5].

A Schematic representation of the Dystrophin Associated Protein Complex (DAPC). Dystrophin plays a central role in the maintenance of muscle membrane integrity and the recruitment of regulatory and signaling proteins. B Schematic representation of the dystrophin protein structure (with focus on the CT domain) and of the different micro-dystrophin (MD) constructs evaluated in this study. MD1 is deleted of CT domain (exons 71-78) except that it contains the last three amino acids of exon 79 of dystrophin. MD2 contains helix 1 (H1) of the coiled-coil motif of CT domain, with the interaction domains for α1- and β1- SNT and α-DTN, MD3 contains the entire coiled-coil motif (H1 + H2) of CT domain, and MD4 contains the entire dystrophin CT domain. C Schematic representation of the structure and size of the expression cassettes generated to produce rAAV2/9-MD vectors. Each MD cDNA sequence was subcloned into a pAAV plasmid that contained the 323 bp muscle-synthetic Spc5.12 promoter, a synthetic polyadenylation signal of 49 bp, and two flanking inverted terminal repeat (ITR) sequences of 130 pb from AAV serotype 2. The size of the resulting MD1, MD2, MD3 and MD4 expression cassettes were 4538 bp, 4721 bp, 4833 bp and 5114 bp, respectively.
Recombinant adeno-associated virus vectors (rAAVs) hold great promise for DMD gene therapy, as they allow efficient transduction of skeletal and cardiac muscles [6] and a long-term in vivo transgene expression [7]. One caveat is that rAAVs have limited packaging capacity of <5 kb [8] while full-length dystrophin cDNA is >14 kb in length. To overcome this DNA packaging limitation of rAAVs, several micro-dystrophin (MD) transgenes containing essential dystrophin functional domains have been developed [9, 10]. The rationale for the use of MDs to treat DMD patients is that Becker Muscular Dystrophy (BMD) patients who carry in-frame deletions that naturally produce MDs, exhibit a milder dystrophinopathy [11]. Several studies have shown body-wide expression and therapeutic efficacy of MDs in DMD animal models (mdx mice, DMDmdx rats and DMD dogs) following a single systemic administration of rAAV-MD vectors [12,13,14,15,16,17,18,19,20,21,22,23,24]. Based on these data, three clinical trials of systemic rAAV-MD gene therapy were initiated in DMD patients in the USA in 2017/2018. Three different MD transgenes are used in these clinical trials: ΔR4-23/ΔCT, ΔR3/ΔR4-21/ΔCT and ΔR2-15/ΔR18-22/ΔCT [18]. First results of the clinical trials using the ΔR4-23/ΔCT MD and the ΔR3/ΔR4-21/ΔCT MD showed safe and efficient expression of the transgene, associated with clinical improvements at one year post-injection [25, 26].
Notably, none of these constructs contain the dystrophin CT domain, which constitutes a large portion of the native protein (325 aa long and ≈36 kDa), and which is highly conserved between species [27]. It is known to interact directly with α1- and β1-syntrophins [28] (SNT) (binding site encoded by exon 74 of the DMD gene) and α-dystrobrevins [29] (α-DTN) (binding site on the helix 1 of the coiled-coil domain encoded by exons 74 and 75 of the DMD gene). These proteins play a major role in DAPC as α-DTN helps to strengthen the link between dystrophin and the sarcolemma through the sarcoglycan complex [30], while the SNT adaptor proteins help localize intracellular signaling proteins [31] such as sodium voltage-gated (NaV) [32] channels, transient receptor potential (TRP) [33,34,35] channels and neuronal nitric oxide synthase (nNOS) [36] (Fig. 1A). However, it was shown that the expression of a mini-dystrophin without CT domain in a transgenic mdx mouse allows restoration of expression of SNT and α-DTN at the sarcolemma of skeletal muscles, even in the absence of a direct link to dystrophin [37]. As a consequence, and due to the DNA packaging limitation of rAAV, inclusion of the 975 bp CT coding sequence in MD transgenes was dropped early in the development of many variants. However, regions in the CT domain encoded by exons 71–74 seem required to restore a normal expression level of α1- SNT [37]. In addition, one study showed that inclusion of helix 1 of the CT coiled-coil domain in MD1 facilitates increased expression of α1-SNT and α-DTN at the sarcolemma of skeletal muscle and increased muscle resistance to lesions induced by contraction [38]. To date, investigations on the role of the dystrophin CT domain has focused on skeletal muscles as the DMD animal model for these studies was the common mdx mouse, which shows minimal and late cardiac pathology [39]. Nevertheless, a recent genotype/phenotype correlation study conducted in 274 patients raised the possibility that the dystrophin CT domain may have a cardioprotective role [40]. Proteomics analysis also showed differences in DAPC composition, especially for some SNT and DTN isoforms, between skeletal muscle and heart in mouse [41]. Therefore, the precise impact of the inclusion of the dystrophin CT domain in a MD construct, remains to be explored, especially in the heart. The efficacy of MD expression on the DAPC recovery is classically evaluated by immunolabeling of the known DAPC partners. However, this method only provides information on the sarcolemmal colocalization of known proteins of the DAPC. Mass spectrometry (MS) analysis of MD enriched fractions by co-immunoprecipitation (co-IP) allows the identification of the MD interactome with DAPC partners whether known or not [41].
Moreover, the availability of the newly developed DMDmdx rat model, which exhibits systematic and stepwise severe phenotypes in both skeletal and cardiac muscles, close to what occurs in DMD patients [42], enables the functional exploration of new MD variants in a relevant model of DMD.
In this study, we evaluated for the first time the impact of inclusion of the dystrophin CT domain to ΔR4-23/ΔCT MD (also called “MD1”) when using rAAV-based gene transfer. Specifically, we evaluated, in both skeletal and cardiac muscles, (i) the DAPC composition and (ii) the therapeutic efficacy after injection of rAAV-MD1 +/− CT in DMDmdx rats. We show by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) analysis that, despite the absence of the dystrophin CT domain, MD1 can restore a normal level of interaction with most DAPC partners (including SNT and α-DTN) in both skeletal and cardiac muscles of DMDmdx rats. Surprisingly, the partial or complete inclusion of the CT domain to MD1 results in a supra-physiological association with some of these partners. At the functional level, we were unable to demonstrate that incorporation of the coil-coiled motif of the dystrophin CT provides a therapeutic benefit to MD1 in DMDmdx rats. This work strengthens the highly therapeutic potential of MD1 for systemic rAAV-MD gene therapy for DMD.
Materials and methods
Vector design and production
Murine-specific cDNA sequences of optimized MD version 1 and 2 (MD1 and MD2) have been previously described [13, 38]. The versions 3 and 4 (MD3 and MD4) were generated from these previous constructs [27]. The MD1 cDNA is deleted of spectrin-like repeat domain 4 to 23 and CT domain (exons 71–78) and contains the last three amino acids of exon 79 of dystrophin followed by three stop codons [13]. Based on this MD1 construct, the MD2 cDNA contains helix 1 of the coiled-coil motif of the CT domain of dystrophin [38], the MD3 cDNA contains the entire the coiled-coil motif (helix 1 and 2) and the MD4 cDNA contains the entire CT domain of dystrophin. Each murine MD cDNA sequence was subcloned into a pAAV plasmid that contained the 323 bp muscle-synthetic Spc5.12 promoter [43], a synthetic polyadenylation signal of 49 bp obtained from the pCI-neo plasmid (Promega, Madison, WI), and two flanking inverted terminal repeat (ITR) sequences of 130 pb from AAV serotype 2. The sizes of the resulting MD1, MD2, MD3 and MD4 expression cassettes (including Spc5.12 promoter, pCI-Neo polyA, and ITRs) were 4538 bp, 4721 bp, 4833 bp and 5114 bp, respectively (Fig. 1C). Recombinant pseudotyped AAV2/9-MD vectors were produced by the Vector Core of the UMR 1089 (CPV, INSERM and University of Nantes) by transient transfection of HEK293 cells followed by purification on cesium chloride density gradients. Final vectors were concentrated and formulated in Dulbecco’s phosphate-buffered saline (DPBS, Fisher Scientific, Illkirsch, France), sterile filtered, aliquoted and frozen at ≤70 °C. Vector genome titers (vg/mL) were determined using a qPCR assay specific for ITR2 [44]. Infectious particles (ip) titer (ip/mL) were determined using an Infectious Center Assay (ICA) [45].
Animals and vector delivery protocols
Study to assess the impact of the dystrophin CT domain on the DAPC composition
A total of 21 DMDmdx rats and 4 Sprague Dawley WT rats (littermates) were used in this study. Animals were randomly assigned to the different experimental groups. They were obtained from the UTE IRS2 (University of Nantes, France). They were handled and housed in the UTE IRS2. The Institutional Animal Care and Use Committee of the Région des Pays de la Loire (University of Angers, France) as well as the French Ministry for National Education, Higher Education and Research approved the protocol (authorization #2018102616384887). Each vector (MD1, MD2, MD3 and MD4) was administrated without anesthesia to 4 newborn DMDmdx rats (between 4 and 6 days of age) by intraperitoneal (IP) injection of 1.2E14 vg/kg (for rAAV-MD1, MD2 and MD3 vectors) or 2.4E14 vg/kg (for rAAV-MD4 vector). Prior injection, the rAAV vectors were diluted in DPBS vehicle solution to obtain a fixed total volume corresponding to 20 mL of perfusate per kg of animal. These animals were euthanized 4.5 months post-injection by CO2 inhalation. Three 2-month-old DMDmdx rats were also injected intramuscularly in the tibialis anterior muscle with a total of 1E12 vg of the rAAV/MD4 vector, after anesthesia. These animals were euthanized 7.5 months post-injection by CO2 inhalation.
Study to assess the impact of the coil-coiled motif of the CT domain of dystrophin in DMDmdx rats
A total of 40 DMDmdx rats and 10 Sprague Dawley WT rats (littermates) were used in this study. Animals were randomly assigned to the different experimental groups. They were obtained from the Boisbonne Center for Gene Therapy (ONIRIS, Nantes, France). They were handled and housed in the Boisbonne Center for Gene Therapy. The Institutional Animal Care and Use Committee of the Région des Pays de la Loire (University of Angers, France) as well as the French Ministry for National Education, Higher Education and Research approved the protocol (authorization #2017040616371353). Prior injection, the rAAV vectors were diluted in DPBS vehicle solution to obtain a fixed total volume corresponding to 15 mL of perfusate per kg of animal. Injections were performed without anesthesia but under analgesic premedication. Vector or its vehicle was administered at the age of 1 month by the intravenous route in a tail vein. Animals were sacrificed after 3 months of follow-up. After anesthesia and incision of the abdomen, whole blood was obtained at the level of the vena cava and euthanasia was performed by intravenous injection of pentobarbital sodium (Dolethal, Vetoquinol, Paris, France). For animals dedicated to ex-vivo sketetal muscle contractility analysis and calcium measurements, euthanasia was performed by section of the heart, after anesthesia.
Co-immunoprecipitations
Antibody production from hybridomas (DSHB, University of Iowa), protein extractions and immunoprecipitations were performed as previously described [41]. Briefly, MANEX1011B (clone 1C7, developed by Glenn E. Morris, Center for Inherited Neuromuscular Disease, RJAH Orthopedic Hospital, Oswestry, UK) or control MW8 (developed by Paul H. Patterson, Division of Biology, California Institute of Technology, Pasadena, CA) antibodies were incubated with magnetic Dynabeads protein G (Invitrogen, Illkirsch-Graffenstaden, France; 1.2 mg antibody per 1 mL beads) in 100 mM sodium phosphate (pH 5.0) overnight at 4 °C. Antibodies were cross-linked to the beads by incubation in 0.2 M Triethanolamine (Fisher Scientific, Illkirsch, France) containing 20 mM dimethyl pimelimidate (Fisher Scientific, Illkirsch, France) for 30 min at 20 °C.
Right after sacrifice, pectoralis muscles (for rats injected via the IP route and non-injected rats) or tibialis anterior muscles (for rats injected via the IM route) and heart were dissected and 100 mg of tissues were homogenized for protein extraction (Fig. S2). This amount of tissue provided enough material for one experimental co-IP and one control co-IP from the same protein homogenate. Tissues were homogenized 1:10 w/v in ice cold extraction Buffer (1% digitonin (Fisher Scientific, Illkirsch, France), 0.05% NP-40 (IGEPAL CA-630, Sigma–Aldrich, Saint Quentin Fallavier, France), NaCl 150 mM (VWR International, Fontenay sous Bois, France), Tris 50 mM, pH 7.4 (VWR International, Fontenay sous Bois, France)) with Complete Protease Inhibitors and PhosSTOP (Roche Diagnostics, Meylan, France) using the PowerGen 125 homogenizer (Fisher Scientific, Illkirsch, France). Proteins were extracted on ice for 1 h, centrifuged at 80,000 g for 30 min and supernatant was pre-cleared with protein G agarose beads (Invitrogen, Illkirsch-Graffenstaden, France). Protein concentration was determined with the DC Protein Assay (Bio-Rad, Marnes-la-Coquette, France). Antibody conjugated beads were incubated with 2–2.5 mg protein at +4 °C for 3 h, washed in ice cold extraction buffer without digitonin, and proteins were eluted in 2% SDS (Sigma–Aldrich, Saint Quentin Fallavier, France), 100 mM DTT (Sigma–Aldrich, Saint Quentin Fallavier, France) for LC-MS/MS analysis.
LC-MS/MS
A total of 15 µL of the eluents were taken for trypsin digestion. First, 9 µL of 10% SDS (Affymetrix, Cleveland, OH) in 100 mM TEAB (Triethylamonium bicarbonate, Sigma, Saint Louis, MO) was added to the samples to make the final concentration of SDS at 5%. 5 µL of 5 µg/µL DTT (FisherScientific, Pittsburg, PA) in 50 mM ammonium bicarbonate (ABC, J.T Baker, purchased through FisherScientific, Pittsburg, PA) solution was added and the sample was incubated at 65 °C for 15 min followed by addition of 5 µL of 50 mM ABC containing 15 µg/µL iodoacetamide (Acros, purchased through FisherScientific, Pittsburg) followed by incubation at RT for 30 min in the dark. Samples were then acidified by adding 12% phosphoric acid (Sigma, St. Louis, MO) (1:10 v/v acid to sample). 165 µL of TEAB (Triethylammonium bicarbonate, 1 M)/MeOH (FisherScientific, Pittsburg, PA) (10:90 v/v) was added to the samples. Samples were then loaded to S-trap (Protifi, Farmingdale NY) and centrifuged at 4000 g for 3 min (at + 4 °C) to remove supernatant. 150 µL of TEAB (Triethylamonium bicarbonate, 1 M)/MeOH (10:90 v/v) was added to the trap as wash solution and the trap was washed 5 times. After the final wash, 30 µL of sequencing grade trypsin (33 ng/uL, Promega. Madison, WI) dissolved in 50 mM TEAB was added and the digestion was carried on O/N at 37 °C. The following day, peptides were eluted from the trap by adding 40 µL of 50 mM TEAB, 0.1% formic acid (FA, FisherScientific, Pittsburg, PA) in water and 0.1% FA in Acetonitrile (FisherScientific, Pittsburg, PA) (50:50), sequentially. The eluted peptides were pooled together and dried in a vacufuge and resuspended in 20 µL of 50 mM acetic acid solution (FisherScientific, Pittsburg, PA).
Nano-liquid chromatography-nanospray tandem mass spectrometry (Nano-LC/MS/MS) of protein identification was performed on a Thermo Scientific orbitrap Fusion mass spectrometer equipped with an nanospray FAIMS Pro™ Sources (Thermo Fisher Scientific, Waltham, MA) operated in positive ion mode. Samples (6 µL) were separated on an easy spray nano column (PepmapTM RSLC, C18 3 µ 100 A, 75 µm x 150 mm Thermo Fisher Scientific, Waltham, MA) using a 2D RSLC HPLC system (Thermo Fisher Scientific, Waltham, MA, USA). Each sample was injected into the µ-Precolumn Cartridge (Thermo Scientific) and desalted with 0.1% Formic Acid in water for 5 min. The injector port was then switched to inject and the peptides were eluted off of the trap onto the column. Mobile phase A was 0.1% Formic Acid in water and acetonitrile (with 0.1% formic acid) was used as mobile phase B. Flow rate was set at 300 nL/min. mobile phase B was increased from 2% to 16% in 105 min and then increased from 16% to 25% in 10 min and again from 25% to 85% in 1 min and then kept at 95% for another 4 min before being brought back quickly to 2% in 1 min. The column was equilibrated at 2% of mobile phase B (or 98% A) for 15 min before the next sample injection.
MS/MS data was acquired with a spray voltage of 1.95 KV and a capillary temperature of 305 °C is used. The scan sequence of the mass spectrometer was based on the preview mode data dependent TopSpeed™ method: the analysis was programmed for a full scan recorded between m/z 375–1500 and a MS/MS scan to generate product ion spectra to determine amino acid sequence in consecutive scans starting from the most abundant peaks in the spectrum in the next 3 s. To achieve high mass accuracy MS determination, the full scan was performed at FT mode and the resolution was set at 120,000 with internal mass calibration. Three compensation voltage (cv = −50, −65 and −80 v) were used for samples acquisition. The AGC Target ion number for FT full scan was set at 4 x 10E5 ions, maximum ion injection time was set at 50 ms and micro scan number was set at 1. MSn was performed using HCD in ion trap mode to ensure the highest signal intensity of MSn spectra. The HCD collision energy was set at 32%. The AGC Target ion number for ion trap MSn scan was set at 3.0E4 ions, maximum ion injection time was set at 35 ms and micro scan number was set at 1. Dynamic exclusion is enabled with a repeat count of 1 within 60 s and a low mass width and high mass width of 10 ppm.
Peptide sequence analysis
Data were searched using Mascot Daemon by Matrix Science version 2.7.0 (Matrix Science, Boston, MA) via ProteomeDiscoverer (version 2.4 Thermo Fisher Scientific, Waltham, MA) against the most recent Uniprot rat databases. Customized database was constructed by replacing dystrophin with the MD sequences to specifically identify and quantify the synthetic MD. The mass accuracy of the precursor ions was set to 10 ppm, accidental pick of 1 13C peaks was also included into the search. The fragment mass tolerance was set to 0.5 Da. Carbamidomethylation (Cys) was used as a fixed modification and considered variable modifications were oxidation (Met) and deamidation (N and Q). Four missed cleavages for the enzyme were permitted. A decoy database was also searched to determine the false discovery rate (FDR) and peptides were filtered according at 1% FDR. Proteins identified with at least two unique peptides were considered as reliable identification. Any modified peptides are manually checked for validation.
Label-free quantification
Label-free quantitation was performed using the spectral count approach. Scaffold (Proteomic Software, Inc., Portland, OR) was used for data analysis and calculation of emPAI (Exponentially Modified Protein Abundance Index) values to estimate abundance of each identified protein within the total set of identified proteins [46, 47]. This index is an exponential form of the number of observed peptides divided by the number of all possible tryptic peptides from a particular protein.
Two Scaffold files were generated for DAPC selection and to remove contaminating proteins from the MS analysis of co-IP samples from skeletal muscles and heart. Control co-IP samples using the MW8 antibody were grouped in the IgG_co-IP category (n = 20 for skeletal muscle and n = 19 for heart). Co-IP samples from non-injected WT rats using MANEX1011B antibody were grouped in “WT_DYS_co-IP” category (N = 4). Finally, co-IP samples from rAAV9_MD injected DMDmdx rats using MANEX1011B antibody were grouped in four categories: MD1_DYS_co-IP (N = 4), MD2_DYS_co-IP (N = 4), MD3_DYS_co-IP (N = 4) and MD4_DYS_co-IP (N = 4 for skeletal muscles and N = 3 for heart). In these two scaffold files, emPAIs (non-normalized) were calculated for each identified protein within the total set of identified proteins. Then, a quantitative profile was generated by a variance analysis (ANOVA). Proteins with no significant change of quantitative profile between the different categories and a significant enrichment in IgG_co-IP were considered as background proteins and removed from DAPC selection. Proteins with a significant enrichment in the WT_DYS_co-IP and/or MD_DYS_co-IP categories were selected as potential DAPC proteins. emPAI values of each selected protein were used to perform a 2-way ANOVA followed by a multiple comparison post-hoc “original FDR method of Benjamini and Hochberg” test between WT_DYS_co-IP and IgG_co-IP samples. Proteins with significant difference (Q-value or corrected P ≤ 0.05) between WT_DYS_co-IP and IgG_co-IP categories were identified as specific DAPC proteins. Statistics were performed using GraphPad Prism 8.
Two new Scaffold files were generated for the comparison of selected DAPC proteins abundance between WT_DYS_co-IP, MD1_DYS_co-IP, MD2_DYS_co-IP, MD3_DYS_co-IP and MD4_DYS_co-IP experimental groups. In these files, IgG_co-IP samples were removed and normalized emPAI values were calculated for each previously selected DAPC proteins of WT_DYS_co-IP, MD1_DYS_co-IP, MD2_DYS_co-IP, MD3_DYS_co-IP and MD4_DYS_co-IP samples. Then, relative abundance of these DAPC partners were calculated using corresponding MD/dystrophin normalized emPAI. Relative abundances of each protein were compared between WT_DYS_co-IP, MD1_DYS_co-IP, MD2_DYS_co-IP, MD3_DYS_co-IP and MD4_DYS_co-IP experimental groups by a 2-way ANOVA followed by a multiple comparison post-hoc “original FDR method of Benjamini and Hochberg”. Statistics were performed using GraphPad Prism 8. Q-values (corrected P value) ≤0.05 were considered statistically significant.
Individual values of emPAI (non-normalized, normalized, and relative to MD/dystrophin normalized emPAI), as well as percentages of coverage, and numbers of unique peptides obtained for each analyzed sample, are available in Tables S4–S7.
Vector biodistribution analysis
Just before sacrifice, whole blood was collected from anesthetized rats in tubes containing EDTA as an anticoagulant, aliquoted in human DNA-free, RNAse/DNAse-free, and PCR inhibitor-free certified microtubes, and stored at ≤−70 °C before DNA extraction. Genomic DNA (gDNA) was extracted from 200 μL of whole blood using the NucleoSpin® Blood kit (Macherey Nagel, Hoerdt, France) according to the manufacturer’s instructions, which were adapted for a Microlab STAR Line liquid handling workstation (Hamilton, Villebon sur Yvette, France). gDNA was extracted from snap-frozen biceps femoris, pectoralis and EDL muscles, heart, diaphragm and liver as previously described [16]. Duplex Q-PCR analyses were conducted on a C1000 touch thermal cycler (Bio-Rad, Marnes-la-Coquette, France) using 50 ng of gDNA in duplicate and premix Ex taq (Ozyme, Saint-Cyr-L’Ecole, France). Vector copy numbers were determined using primers and probe specifically designed to amplify both MD1 and MD3 transgenes. Primers and probe designed to amplify the rat HPRT1 sequence were used to determine the endogenous gDNA copy numbers. For each sample, cycle threshold (Cq) values were compared with those obtained with different dilutions of linearized standard plasmids (containing either the mMD1 expression cassette or the rat HPRT1 gene).
Western blot analysis
Total proteins extraction from snap-frozen biceps femoris and heart samples, and MD1/MD3 expression analysis by western blot were performed as previously described [16]. Protein loading control analysis was assessed using a mouse anti-rat α-tubulin antibody (1:10,000, Sigma–Aldrich T5168, Saint Quentin Fallavier, France) and a secondary anti-mouse IgG HRP-conjugated antibody (1:5,000, Dako P0447, Les Ulis, France).
Histopathological analysis
Samples including biceps femoris, pectoralis and cardiac muscles were fixed in 10% neutral buffered formalin, embedded in paraffin wax. Sections were cut and stained with hematoxylin eosin saffron (HES) for histopathological evaluation. To avoid bias in the analysis, experiments were done in a blind fashion. Muscular lesions were then semi-quantitatively scored (as described in Fig. 4 legend), to identify any improvement of the histopathological pattern between the different experimental groups.
MD/dystrophin positive fibers and connective tissue quantification
Samples including biceps femoris, pectoralis and cardiac muscles were snap-frozen in isopentane cooled in liquid nitrogen and stored at ≤−70 °C until processing. Sections of 10 µm from frozen samples were permeabilized in PBS tween 0.2% (Sigma–Aldrich, Saint Quentin Fallavier, France) and saturated in 5% goat serum (Sigma–Aldrich, Saint Quentin Fallavier, France) in PBS tween 0.2%. Sections were then immunolabelled for detection of MD/dystrophin and connective tissue using the mouse monoclonal anti-dystrophin antibody NCL-DYSB (1:50, Novocastra Laboratories, Newcastle upon Tyne, UK) and Alexa Fluor 555 Wheat Germ Agglutinin (WGA) conjugate (1:500, Molecular Probes, Eugene, OR), diluted in 5% rat serum (Sigma–Aldrich, Saint Quentin Fallavier, France) in PBS tween 0.2%. Goat anti-mouse Alexa 488 IgG (H + L) (1:300, Invitrogen A28175, Illkirsch-Graffenstaden, France) was used as secondary antibody, diluted in 5% rat serum in in PBS tween 0.2%. Nuclei were stained with DAPI (1:1000, Invitrogen, Illkirsch-Graffenstaden, France). Fluorescent acquisitions of the entire sections were performed using the slide scanner Axio Scan.Z1 (Zeiss, Marly le Roi, France). For each section, 3 captures were randomly chosen to finally observe a minimum of 250 fibers. In these 3 captures, DYSB positive fibers and WGA positive areas (except for the heart) were quantified using ImageJ open-source image processing software (v 2.0.0-rc-49/1.51a). Additional sections from paraffin embedded samples of heart was stained with picrosirius red F3B (Sigma–Aldrich, Saint Quentin Fallavier, France) for collagen visualization. Picrosirius-positive areas in heart sections were quantified using Nikon Imaging software (Nikon, Champigny sur Marne, France).
Calcium measurements
Cell isolation
EDL muscles were harvested from animals just after euthanasia. Small bundles were dissected longwise tendon to tendon as described previously [48]. Cardiomyocytes were isolated by using the retrograde perfusion method through the aorta with enzyme-containing solutions [49]. Briefly, hearts were excised from heparinized (5000 U/kg body, Heparine choay, Sanofi, Gentilly, France) and anesthetized rats, mounted in a Langendorff perfusion apparatus, and perfused with a Ca2+-free NPS (Normal Physiological Solution) at 37 °C for 3 min. Perfusion was then switched to the same solution containing 300 U/ml Collagenase type 2 (Worthington Biochemical Corporation, Lakewood, NJ) for 20 min. The left ventricular tissue was excised, minced, and gently pipette dissociated. Collagenase activity was stopped by the addition of 4% bovine serum albumin (Sigma–Aldrich, Saint Quentin Fallavier, France). At this stage, cardiomyocytes were photographed with an inverted microscope, in bright field and with a X20 objective. Pictures were then used in ImageJ software to measure max and min Feret’s diameters to calculate length and width of the cells. Cell suspension was then sequentially washed and resuspended in NPS of increasing [Ca2+] values.
Fura-2 loading and fluorescence measurements
EDL muscle bundles, pinned in polydimethylsiloxane coated petri dish (Dow France S.A.S., Saint Denis, France), were incubated with NPS containing 5 µM Fura-2-AM (Molecular Probes, Illkirsch-Graffenstaden, France) at room temperature for 1 h under gentle agitation before washing. Isolated cardiomyocytes were resuspended in NPS containing 3 µM Fura-2-AM for 30 min before washing and Ca2+ measurements. Fura-2 fluorescence measurements were made using an integrated IonOptix device (Amsterdam, Netherlands) and excitation filters of 380 and 360 nm. Emitted fluorescence (510 nm) was background subtracted. For resting cytosolic [Ca2+] measurements, resting fluorescence ratios (360/380 nm) values were measured for each fiber composing EDL muscle bundles or isolated cardiomyocyte. [Ca2+] values were calculated from the Fura-2 fluorescence ratio using the Grynkiewics’s equation [50]. Main parameters of the equation were determined in situ [48].
Determination of sarcolemmal permeability to Ca2+ (SPCa)
The manganese quenching technique was used to measure the SPCa in EDL muscle fibers [48]. Muscle preparations were perfused by NPS containing 0.5 mM Mn2+ as a surrogate of Ca2+ (quenching solution). During the whole quenching protocol, the fluorescence of Fura-2 excited at 360 nm was acquired at 1 Hz. The quench rates were calculated in each fiber of the preparations using linear regression analysis of fluorescence signal and expressed as the decline per minute of the initial fluorescence intensity.
All experiments were done at room temperature. Solutions: NPS contained the following: 140 mM NaCl, 5 mM KCl, 1 mM MgC12, 10 mM HEPES, 10 mM glucose, and 1.8 mM CaCl2 (All chemicals form Sigma–Aldrich, Saint Quentin Fallavier, France) at pH 7.35.
Skeletal muscle force analysis
Ex-vivo sketetal muscle contractility
Isometric contractile properties of the EDL and soleus muscles were evaluated according to methods previously described [51, 52]. Briefly, muscles were removed from the hindlimb of anesthetized rats and mounted in an in vitro muscle test system (1205 A model; Aurora Scientific, Aurora, Canada). Muscles were placed between two platinum electrodes in a muscle bath containing 100 mL of bubbled mammalian Ringer solution (Na+ 140 mM, K+ 6 mM, Ca²+ 3 mM, Mg²+ 2 mM, Cl− 156 mM, pH 7.4, HEPES-buffered) at 25 °C. After a 5 min equilibration period, optimum muscle length was determined by gradual muscle length adjustments and eliciting isometric contractions (supramaximal square-wave pulses of 0.2 ms duration) until the maximum twitch tension was reached. After 5 min of rest, muscles were stimulated at 10, 20, 40, 60, 80, 100, 120 Hz for 500 ms at each frequency. Stimulus trains were separated by one-min interval. Maximum isometric tetanic force was determined from the plateau of this force frequency curve. Following force testing, muscles were removed from the bath, trimmed of tendons, and weighed. Muscle mass was then be used to calculate maximum tetanic specific force in g/g. To avoid bias in the analysis, experiments were done in a blind fashion.
In vivo grip test
For all animals included in the study, the grip test was performed, as previously described [42], at least 3 days before the cardiac evaluation at 3 months post-injection. To avoid bias in the analysis, experiments were done in a blind fashion.
Assessment of cardiac function
Cardiac function was assessed on anesthetized animals using 2D echocardiography and pulsed Doppler measurements. For all animals included in the study, these tests were performed at least 3 days after grip test at 3 months post-injection.
2D-echocardiography and pulsed Doppler were performed, as previously described [42], using a Vivid 7 ultrasound unit (GE Healthcare, Velizy Villacoublay, France). To avoid bias in the analysis, experiments were done in a blind fashion.
Results
Assessment of the impact of the dystrophin CT domain on the DAPC composition: study design
In order to evaluate the impact of inclusion of the dystrophin CT domain on the efficacy of MD1 to recover the DAPC, we used 4 different murine-specific sequence–optimized MD constructs: MD1, MD2, MD3 and MD4. MD2 to 4 were derived from MD1 by progressively including the dystrophin CT domain sequence (Fig. 1B): (i) MD1 (ΔR4-23/ΔCT) is our reference, (ii) MD2 incorporates helix 1 (H1) of the coiled-coil motif, (iii) MD3 includes the entire coiled-coil motif (H1 + H2), and (iiii) MD4 (ΔR4-23) has the entire dystrophin CT domain.
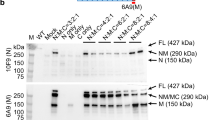
To obtain rat skeletal and cardiac muscles expressing these different MDs, we administered rAAV2/9 vectors encoding MD1, MD2, MD3 or MD4 cDNAs (Fig. 1C). Each vector was administered to 4 newborn DMDmdx rats by intraperitoneal (IP) injection to achieve efficient transduction of skeletal muscles and heart [53, 54]. As expected, the expression of the different MDs was detected by western blot in the pectoralis and cardiac muscles, except for MD4 that was weakly expressed in pectoralis muscle in 3 out of 4 animals (Fig. S1). The rAAV-MD4 vector carries an expression cassette that exceeds the DNA packaging capacity of rAAV (<5 kb, Fig. 1C) [8] thus limiting its infectivity. Supplemental skeletal muscles expressing MD4 were obtained after intramuscular (IM) injection in the tibialis anterior muscles of 3 DMDmdx rats at 2 months of age (Fig. S1).
Based on the methodology of Johnson et al. [41], we compared the composition of protein complexes assembled by full-length dystrophin (N = 4), MD1 (N = 4), MD2 (N = 4), MD3 (N = 4) and MD4 (n = 4 and N = 3 for skeletal muscle and for heart, respectively), in skeletal and cardiac muscles of wild-type (WT) and rAAV-MD treated DMDmdx rats. To identify proteins that selectively associate with MD/dystrophin, we performed a co-IP on proteins extracted from 50 mg of muscle using the MANEX1011B monoclonal antibody (DYS_co-IP) or an isotype-matched control antibody (MW8 antibody) (IgG_co-IP) (Fig. S2). LC-MS/MS analysis was performed on all co-IP samples and the abundance of each identified protein within the total set of identified proteins was estimated in Scaffold (Proteomic Software, Inc., Portland, OR) by calculation of the emPAI (Exponentially Modified Protein Abundance Index) [46, 47].
Identification of dystrophin protein partners in rat skeletal and cardiac muscles
We first confirmed that we can reliably identify proteins known to associate with dystrophin in rat skeletal and cardiac muscles co-IPs by MS analysis. Proteins that specifically co-purify with full-length dystrophin were identified based on significantly higher emPAI values in WT_DYS_co-IP compared to IgG_co-IP (Table S1). We noticed that utrophin, a homolog of dystrophin, was also detected in WT_DYS_co-IP, indicating that the MANEX1011B antibody slightly cross-reacts with utrophin. Since utrophin and dystrophin can both bind to DAPC proteins, we cannot exclude that we might have detected some proteins that bind to utrophin in our MD/dystrophin co-IPs. However, utrophin abundance in WT_DYS_co-IP remained minimal when compared to dystrophin, with levels much lower than dystrophin for skeletal muscles and heart (39- and 18-fold, respectively) (Table S1). This means proteins detected in WT_DYS_co-IP were mainly associated with dystrophin. Furthermore, utrophin contamination was low in all co-IP samples and did not significantly differ between the WT co-IP and MD co-IP samples (one-way ANOVA, data not shown). Therefore, utrophin did not interfere with our analysis of DAPC composition.
Proteins found to be significantly enriched in WT_DYS_co-IP compared to IgG_co-IP are shown in Table S1. Known DAPC components such as dystrophin, dystroglycan, α-, β-, δ-, and γ-sarcoglycans, α1- and β1-SNT, α-DTN and sarcospan were consistently and specifically detected in WT_DYS_co-IPs from both skeletal and cardiac muscles. As previously described in mice [41], nNOS was found binding to dystrophin in skeletal muscle but not in heart, confirming that this protein is also not part of the DAPC in cardiac tissue of rats. Cavin-1 and β2-SNT, that have been described as heart-specific DAPC partners [41], were detected here in association with both skeletal and cardiac muscle dystrophins. While we cannot rule out that these proteins may be present in skeletal muscle WT_DYS_co-IPs due to binding to utrophin, the fact that we are using LC-MS/MS instrumentation with higher sensitivity than in the previous study [41] could result in the detection of less abundant DAPC partners. Interestingly, additional proteins were specifically enriched in WT_DYS_co-IP compared to IgG_co-IP. Calmodulin-1 (known to associate with skeletal muscle dystrophin [55, 56]), pikachurin (a known extracellular ligand of dystroglycan [57]), β-DTN and perilipin-4 were all detected in skeletal muscle WT_DYS_co-IPs. The caveolar proteins caveolin-1 and cavin-2 were instead detected in cardiac muscle WT_DYS_co-IPs. Finally, cavin-3 was detected in association with both skeletal and cardiac muscle dystrophins.
Inclusion of the dystrophin CT domain in MD1 increases recruitment of some DAPC proteins
We next evaluated how the inclusion of progressively larger segments of the dystrophin CT domain impacted DAPC composition in DMDmdx rats. We reasoned that addition of the coil-coiled domain H1, known to bind to SNT and DTN, should lead to improved binding of MD2, MD3 and MD4 to these proteins, when compared to MD1. Scaffold was used to generate normalized emPAI values of each protein of the DAPC, within the total set of identified proteins for WT_DYS_co-IP, MD1_DYS_co-IP, MD2_DYS_co-IP, MD3_DYS_co-IP and MD4_DYS_co-IP experimental groups. We noticed that our western blot analyses indicated differences in abundance between MD/dystrophins (Fig. S1) that were also reflected in our mean emPAI values for MD/dystrophins co-purified from 50 mg of starting material (Fig. S3). Therefore, to be able to compare DAPC protein abundance between experimental groups, we calculated the ratio of the emPAI of each individual DAPC protein to the emPAI of MD or dystrophin in the same sample.
As shown in Table 1 and Fig. 2, we found several significant differences in the abundance of DAPC proteins that co-purify with different MDs. Some protein associations were significantly disrupted with all MD constructs in cardiac and/or skeletal muscles, suggesting that they require domains absent in our MDs or that these associations are decreased below our limit of detection. Protein associations falling in this category include perilipin-4, cavin-3, and caveolin-1 and nNOS, whose binding to dystrophin is known to be absent in our MDs (R16/17) [19]. Interestingly, in cardiac muscle where cavin-1 strongly associates with full-length dystrophin, we saw a significantly impaired association with MD1 and MD2 that was progressively rescued with MD3 and MD4 suggesting that it requires an intact coil–coil CT domain.

Relative abundance to MD/dystrophin were calculated by dividing the normalized emPAI of each DAPC partner by the normalized emPAI of the MD/dystrophin expressed in the same sample. Relative abundances of (A) α-Sarcoglycan, α-DTN, α1-, β1- and β2-SNT in co-IP from skeletal muscle, and of (B) α-Sarcoglycan, α1-SNT and Cavin-1 in co-IP from heart were compared between MD1_DYS_co-IP (N = 4), MD2_DYS_co-IP (N = 4), MD3_DYS_co-IP (N = 4), MD4_DYS_co-IP (N = 4 for skeletal muscles and N = 3 for heart) and WT_DYS_co-IP (N = 4) experimental groups. Data are presented as mean ± SEM. Statistics were done using a 2-way ANOVA followed by a multiple comparison post-hoc “original FDR method of Benjamini and Hochberg”. Q-values (corrected P value) ≤0.05 were considered statistically significant. Values sharing the same letter are not statistically different from each other.
Overall, in both skeletal and cardiac muscles, MD1 had a protein interaction profile that most closely matched that of full-length dystrophin. The only proteins that were significantly more abundant in MD1 co-IPs compared to full-length dystrophin were β- and γ-sarcoglycans in skeletal muscle (1.7- and 2.2-fold, respectively), and β-sarcoglycan (1.8-fold) in cardiac muscle. Interestingly, SNT and DTN that bind dystrophin’s CT, co-purified in similar amounts with MD1 and full-length dystrophin. By contrast, sequential addition of parts of the CT sequence in MD2, MD3 and MD4 constructs resulted in significant increases in their associations with several sarcoglycans, SNT and DTN compared to MD1 and to full-length dystrophin. In skeletal muscle, these involve α-, β-, δ- and γ-sarcoglycans, and α1- and β2-SNT. In cardiac muscles, proteins that are more abundant in MD2, MD3 and MD4 co-IPs are α- and β-sarcoglycans, and α1- and β1-SNT. Additionally, a few DAPC proteins were more abundant in co-IPs for specific MDs relative to all other MDs and to dystrophin. These are β1-SNT in MD4 skeletal muscle co-IPs; α-DTN in MD3 skeletal muscle co-IPs; and γ-sarcoglycan in MD3 cardiac muscle co-IPs. In general, the significant differences we found in protein associations depend not only on the MD sequence but also on the type of muscle, suggesting tissue-specific differences in the regulation of protein associations with MDs.
Assessment of the impact of the coil-coiled motif of the dystrophin CT domain on the therapeutic efficacy of a MD transgene: study design
To evaluate the impact of the dystrophin CT domain on the therapeutic efficacy of MDs in both skeletal and cardiac muscles, 1 month old male DMDmdx rats were treated systemically with the rAAV2/9-MD1, or the rAAV2/9-MD3 construct. MD3 was chosen because it contains the entire coil-coiled motif (H1 + H2) of the dystrophin CT domain, while not exceeding the <5 kb DNA packaging limitation of rAAV [8] (Fig. 1C). Both rAAV2/9-MD1 and -MD3 vector batches exhibited similar vg/mL and infectious titers (Table S2), implying that their efficacies can be confidently compared. The study design is summarized in Table S3. A group of 11 DMDmdx rats were injected with rAAV2/9-MD1 at a dose of 3E13 vg/kg, which we know as therapeutic (Bourdon et al., unpublished data). To compare the potential therapeutic benefit between MD3 and MD1, additional DMDmdx rats were injected with 1E13 vg/kg (N = 9 and N = 11 for rAAV2/9-MD1 and rAAV2/9-MD3, respectively). Groups of 10 WT and 10 DMDmdx control rats were also included and received buffer. All rats were followed for 3 months. Right before euthanasia, muscle function (evaluated by grip test) and cardiac function (evaluated by electrocardiography, echocardiography and pulsed Doppler) were evaluated. After sacrifice, 4 animals from each group were dedicated to assessments of ex-vivo muscle contractility analysis and calcium measurements. The other animals were dedicated to post-mortem sampling required for histological and molecular/biochemical analyses.
The inclusion of the coil-coiled motif of the dystrophin CT domain in MD does not affect gene transfer efficacy and MD expression patterns
Using a quantitative PCR (qPCR) assay specific for both MD1 and MD3 sequences, vector genome copy numbers per diploid genome (vg/dg) were assessed in whole blood, skeletal muscles, heart, diaphragm and liver samples obtained at sacrifice (Fig. 3A). No qPCR signal was detected in whole blood, confirming that vector genome detection in rAAV-injected animals was restricted to the tissue itself. Both vectors had the same transduction pattern, with the highest vg/dg numbers detected in liver and heart, as expected after IV injection in rodents [58]. Lower vg/dg numbers levels (but similar between the different muscles) were detected in skeletal muscles. In correlation with the injected vector dose, significantly more vg/dg were found in tissues of rats injected with 3E13 vg/kg than with 1E13 vg/kg of rAAV2/9-MD1 (2.7- to 7.8-fold, respectively, depending on the tissue). Instead, the vg/dg values in tissues of rats injected with 1E13 vg/kg of rAAV2/9-MD1 or rAAV2/9-MD3 vector were similar, confirming that both vectors exhibited the same tropisms and infectivities. MD1/MD3 expression in skeletal muscles and heart was evaluated by both immunohistochemistry (IHC) staining (Fig. 3B, C) and western blot (Fig. 3D). Again, a dose effect between the two groups treated with 3E13 vg/kg or 1E13 vg/kg of rAAV2/9-MD1 was observed, with a mean of detected MD1-positive fibers of 85.7 ± 3.5% to 86.6 ± 2.7% vs. 33.6 ± 7.8% to 40.2 ± 43%, respectively, in skeletal muscles, and of 96 ± 0.8% vs. 86 ± 2.4% in heart. Similar levels of MD1 or MD3 positive fibers were detected in animals injected with 1E13 vg/kg of rAAV2/9-MD1 or rAAV2/9-MD3 (33.6 ± 7.8% to 40.2 ± 43% of MD1-positive fibers vs. 28.5 ± 4.8% to 43.5 ± 6.4% of MD3-positive fibers in skeletal muscles, and 86 ± 2.4% of MD1-positive fibers vs. 82 ± 0.8% of MD3-positive fibers in heart). Immunolabeling confirmed the proper localization of both MD1 and MD3 at the sarcolemma in biceps femoris muscle (Fig. 3C) and correlated with the detection of the 138 kDa MD1 and 160 kDa MD3 protein bands by western blot analysis (Fig. 3D). Moreover, the RTqPCR analysis showed that the MD1 and MD3 mRNA are equally expressed in skeletal muscle and heart samples when the two vectors are injected at the same dose. Only low levels of MD mRNA were detected in liver despite the presence of high vg/dg values, confirming the muscle restrictive activity of the Spc5.12 promoter (Fig. S4). Taken together, these results confirm that both MD1 and MD3 are similarly and properly expressed in skeletal muscles and heart after systemic injection, meaning that their therapeutic efficacies can be compared in a relevant manner.

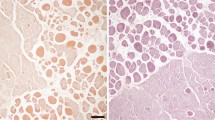
A Vector genome copy number per diploid genome (vg/dg) in tissues of the different experimental groups at euthanasia, determined by qPCR analysis. For each group, the analysis was performed on whole blood (N = 6 animals per group), liver (N = 4 animals for the DMDmdx + MD1 1E13 group; N = 6 animals for the other groups), diaphragm (N = 6 animals per group), heart (N = 5 animals for the DMDmdx + MD1 1E13 group; N = 6 animals for the other groups) and pectoral (N = 6 animals per group), biceps femoris (N = 6 animals per group) and EDL (N = 5 animals per group) muscles. The limit of quantification (LOQ) was 0.004 vg/dg. Data are presented as mean ± SEM. *P < 0.05 (ANOVA test, followed by post-hoc Tukey multiple comparison test). B Percentage of MD/Dystrophin positive fibers determined at euthanasia by immunolabelling in heart (N = 5 animals per group for the DMDmdx + MD1 1E13 group; N = 7 animals for the DMDmdx + MD1 3E13 group; N = 6 animals for the other groups), pectoralis muscle (N = 7 animals for the DMDmdx + MD1 3E13 group; N = 6 animals for the other groups), and biceps femoris muscle (N = 7 animals for the DMDmdx + MD1 3E13 group; N = 6 animals for the other groups). Data are presented as mean ± SEM. *p < 0.05 (ANOVA test, followed by post-hoc Tukey multiple comparison test). C Immunolabelling of transverse sections of biceps femoris muscle with DYSB antibody for MD1/MD3/dystrophin staining (green) and with WGA conjugate for connective tissue (red). Representative images corresponding to samples collected at euthanasia for each experimental group. Scale bar, 100 µm. D Western-blot analysis of total proteins (50 µg) extracted from biceps femoris muscle and heart samples collected at euthanasia for each rAAV injected group (N = 4 animals per group). The blots were stained with MANEX-1011C to reveal the presence of the 138 kDa MD1 and the 160 kDa MD3 protein. α-tubulin antibody as a loading control.
The inclusion of the coil-coiled motif of the dystrophin CT domain in MD1 does not improve its effectiveness to restore histopathological and calcium homeostasis alterations in DMDmdx rats
Similar to muscle damage in DMD patients, the histopathological pattern of the DMDmdx rats is characterized by increased variation in fiber size due to the simultaneous presence of hypertrophic fibers and small centronucleated regenerating fibers in skeletal muscles, and individual fiber necrosis associated with inflammatory cell infiltration and fibrosis in both skeletal muscle and heart [42]. To investigate the effect of MD1 and MD3 expression on skeletal and cardiac muscle histopathology, we performed a quantification of fibrotic connective tissue and a semi-quantification of muscle lesions in these tissues obtained at sacrifice. As shown in Fig. 4a, connective tissue represented 16.1 ± 0.7 to 19.4 ± 0.6% of skeletal muscle area in WT rats vs. 29.1 ± 1.8% to 30.5 ± 2.1% in untreated DMDmdx rats, showing an increase in connective tissue that corresponded to fibrosis. In all rAAV-treated groups, connective tissue amount was significantly reduced between 20.1 ± 0.4% and 23.7 ± 1.1%. In heart, connective tissue levels were 2.7 ± 0.5% in WT rats vs. 10.2 ± 2.2% in untreated DMDmdx rats and were also significantly reduced between 3.7 ± 0.8% and 4.2 ± 1% after rAAV2/9-MD administration. No significant difference was observed between the different rAAV-injected groups, whatever the injected dose or the nature of the transgene (MD1 or MD3). Semi-quantitative analysis of histological muscular lesions (scored according to Fig. S5) revealed typical myopathic lesions in both skeletal muscles and heart of untreated DMDmdx rats, resulting in a significantly higher score compared to WT rats (Fig. 4B). This score was improved after injection of rAAV2/9-MD, except with the MD3 vector in the pectoralis muscle. The improvement reached statistical significance in biceps femoris and heart of animals injected with 3E13 vg/kg of the MD1 vector. When comparing the impact of MD1 and MD3 vectors injected at 1E13 vg/kg, no significant difference was observed between these groups. However and as reported in a previous study [59], we observed a few isolated ring fibers with sarcomere disorientation (Fig. S6) in the skeletal muscles of 6/12 rats expressing MD1 (2/5 rats treated with 1E13 vg/kg and 4/7 rats treated with 3E13 vg/kg). None was observed in the muscles of the 6 rats expressing MD3.

A Connective tissue area (in %) was assessed using WGA for skeletal muscles and a specific picrosirius staining for the heart. Analysis were performed at sacrifice on heart (N = 5 animals for the DMDmdx + MD1 1E13 group; N = 7 animals for the DMDmdx + MD1 3E13 group; N = 6 animals for the other groups), pectoralis muscle (N = 7 for the DMDmdx + MD1 3E13 group; N = 6 animals for the other groups) and biceps femoris muscle (N = 7 animals for the DMDmdx + MD1 3E13 group; N = 6 animals for the other groups). Data are presented as mean ± SEM. *p < 0.05 (Parametric ANOVA test, followed by post-hoc Tukey multiple comparison test). B Muscle histopathological score in skeletal muscles and heart samples. Muscles lesions were scored during histopathological observation of the each muscle by a DVM, European-board certified anatomopathologist and using the following system: 0 = absence of lesions, 1 = presence of some regenerative activity as evidenced by centronucleated fibers, 2 = presence of degenerated/necrotic fibers, 3 = tissue remodeling and fiber replacement by fibrosis. In the heart, scoring was based on the presence of inflammatory and fibrotic areas (1 = 1 area, 2 = multiple isolated areas, 3 = extensive/coalescing multiple areas). Analysis were performed at sacrifice on heart (N = 5 animals for the DMDmdx + MD1 1E13 group; N = 7 animals for the DMDmdx + MD1 3E13 group; N = 6 animals for the other groups), pectoralis muscle (N = 7 animals for the DMDmdx + MD1 3E13 group; N = 6 animals for the other groups) and biceps femoris muscle (N = 7 animals for the DMDmdx + MD1 3E13 group; N = 6 animals for the other groups). Data are presented as mean ± SEM. *p < 0.05 (Nonparametric Kruskal–Wallis test, followed by a post-hoc Dunn’s multiple comparison test).
In correlation with muscle degeneration and necrosis and with what was described in mdx mice [60], alterations in Ca2+ homeostasis were observed in skeletal muscles and isolated cardiomyocytes of DMDmdx rats (Fig. 5; Fraysse et al., unpublished data). Lack of dystrophin expression in DMDmdx rats was associated with significant higher resting cytosolic Ca2+ levels as compared to WT rats in both extensor digitorum longus (EDL) muscle (Fig. 5A) and cardiomyocytes (Fig. 5B; increase of ≈49% in EDL muscle and of ≈31% in isolated cardiomyocytes). Treatment with the rAAV2/9-MD1 vector at 3E13 vg/kg showed a significant decrease in resting [Ca2+], although not completely rescued to WT levels. No statistically significant decrease in resting [Ca2+] was observed after injection of rAAV2/9-MD1 or –MD3 at 1E13 vg/kg. We also evaluated the sarcolemmal permeability to Ca2+ (SpCa) in EDL muscles using the manganese quenching technique [61], to assess if resting Ca2+ overload could be related to a greater Ca2+ influx. As shown in Fig. 5C, the mean quench rate of untreated DMDmdx rats was significantly higher than in WT rats, reflecting an increase in the permeability of the sarcolemma. This permeability was significantly decreased in the 3 rAAV2/9-MD treated groups, but without any difference between the different doses or transgenes. Overall, these data indicate that the inclusion of the coil-coiled motif of the dystrophin CT domain in MD1 doesn’t improve its effectiveness to restore the histopathology and the calcium homeostasis alterations of the DMDmdx rat.

Resting intracellular-free calcium concentration ([Ca2+]c) of EDL muscle fibers (A) and isolated cardiomyocytes (B) obtained from animals from the different experimental groups. [Ca2+]c was calculated in fura-2-loaded isolated muscle fibers using calibration parameters of Grynkiewicz’s equation determined in situ. A calibration was performed foreach muscle used. Data are presented as mean ± SEM from n muscle fibers and N animals (n/N). *p < 0.05 (ANOVA test, followed by post-hoc Bonferroni multiple comparison test). C Sarcolemma permeability to calcium ions (SPCa) in EDL muscle fibers obtained from animals of the different experimental groups. The sarcolemma permeability to Ca2+ was evaluated by measuring the quench rate of fura-2 fluorescence induced by Mn2+influx. Data are presented as mean ± SEM from n muscle fibers and N animals. *p < 0.05 (ANOVA test, followed by post-hoc Bonferroni multiple comparison test).
The inclusion of the coil-coiled motif of the dystrophin CT domain in MD1 doesn’t improve its effectiveness to restore muscle and cardiac functions in DMDmdx rat
Ex-vivo force measurements were performed on isolated EDL and soleus muscles obtained at sacrifice. We evaluated the maximum contractile response amplitude following a series of electrical stimulations (tetanic) (Fig. 6A, B). When compared to WT rats, both EDL and soleus muscles of DMDmdx rats showed a decrease in maximum tetanic amplitude (decrease of ≈20% in EDL and significant decrease of ≈56% in soleus). In animals treated with rAAV2/9-MD1 at 3E13 vg/kg, a nearly complete normalization was observed in the EDL muscle and a partial one (≈48%) in the soleus. No effect was observed for the two other experimental groups.

Maximum tetanic amplitude force of isolated soleus (A) and EDL (B) muscles of the different experimental groups at sacrifice (N = 3 animals for the soleus of the DMDmdx + MD1 1E13 group; N = 4 animals for the other groups). Data are presented as mean ± SEM in grams (g) normalized to the muscle weight (g/g). *p < 0.05 (Nonparametric Kruskal–Wallis test, followed by a post-hoc Dunn’s multiple comparison test). C Maximum forelimb grip force of the different experimental groups at 3 months post-injection (N = 11 animals for the DMDmdx + MD1 3E13 group; N = 9 animals for the DMDmdx + MD1 1E13 group; N = 10 animals for the other groups). Data are presented as mean ± SEM in grams (g) normalized to the body weight (g/g BW). *p < 0.05 (Nonparametric Kruskal–Wallis test, followed by a post-hoc Dunn’s multiple comparison test). D Cross-trial evolution of forelimb grip force of the different experimental groups at 3 months post-injection (N = 11 animals for the DMDmdx + MD1 3E13 group; N = 9 animals for the DMDmdx + MD1 1E13 group; N = 10 animals for the other groups). Data are presented as mean ± SEM in grams (g) normalized to the body weight (g/g). *p < 0.05 vs. WT + Vehicle group; ¤p < 0.05 vs DMD mdx + Vehicle group; § p < 0.05 vs trial 1 of the same group (Friedman test followed by Dunn’s post hoc test).
We also evaluated in vivo forelimb force using the grip test. Five trials were performed and the highest grip value obtained during these trials was defined as maximum forelimb grip force. As illustrated in Fig. 6C, a reduction in maximum forelimb grip force was observed for untreated DMDmdx rats compared to WT rats. This value was fully normalized after injection of rAAV2/9-MD1 at 3E13 vg/kg, but only partially improved after injection of rAAV2/9-MD1 and rAAV2/9-MD3 at 1E13 vg/kg. The beneficial effect was slightly higher but not statistically significant with rAAV2/9-MD3 than with rAAV2/9-MD1.
Analysis of forelimb strength over the course of the test (Fig. 6D) showed progressive and marked decrease of forelimb strength in untreated DMDmdx rats, contrary to WT rats and to DMDmdx rats injected with rAAV2/9-MD1 at 3E13 vg/kg. This highlights the high efficiency of the MD1 vector when delivered at this dose. Animals treated with rAAV2/9-MD1 or rAAV2/9-MD3 vectors at 1E13 vg/kg show a delayed curve of force decrease. We can point out that, even if the final forelimb grip force at trial 5 is similar for these two groups, the significant difference of force compared to the WT rats appears later for the rats treated with the MD3 vector (trial 5) than for those treated with the MD1 vector (trial 4).
Since the majority of DMD patients die from heart failure [62], the cardiac dysfunction described in the DMDmdx rat model [42, 63] is a critical feature to evaluate the therapeutic potential of MD. Rapid concentric remodeling associated to an alteration of diastolic function, which progressed unfavorably with age towards systolic dysfunction are characteristics of this model (Toumaniantz, Le Guiner et al., unpublished data). In this study, cardiac function was assessed on anesthetized animals using electrocardiography (ECG), 2D echocardiography and pulsed Doppler measurements at 3 months post-injection. ECG analysis revealed no significant electrical conduction abnormality whatever the experimental group (data not shown). However, as shown in Fig. 7A, the left ventricular wall end-diastolic diameter measured by 2D-echocardiography tended to thicken in untreated DMDmdx rats compared to WT rats and is associated with a decrease in left ventricular diameter, reflecting the concentric hypertrophy of the heart in DMDmdx rats. This structural remodeling was prevented with injection of rAAV2/9-MD1 (whatever the dose), but not with rAAV2/9-MD3. The morphology of isolated cardiomyocytes was also assessed by measuring their length/width ratio. This ratio significantly decreased in untreated DMDmdx rats compared to WT rats, indicating a thickening of the isolated cardiomyocytes and correlating with the concentric hypertrophy pattern observed by 2D-echocardiography. This ratio was normalized in rats treated with rAAV2/9-MD1 at both doses, but not modified in DMDmdx rats treated with rAAV2/9-MD3 vector. As shown in Fig. 7B, ejection fraction, measured using pulsed Doppler, was significantly higher in untreated DMDmdx rats than in WT rats, indicating an adaptation of the systolic function explained by the hypertrophic process previously described. This parameter was almost normalized in the 3 groups of treated animals. Finally, diastolic dysfunction of the untreated DMDmdx rat was also shown in this study via the significant increase of deceleration time, the increase of IVRT and the decrease of E/A ratio, when compared to WT rats. Again, a clear trend of normalization of deceleration time and IVRT was observed in all the treated groups, whatever the injected dose and the transgene (Fig. 7C). This normalization was not observed with the E/A ratio parameter.

A Structural remodeling of the heart: free wall diastolic thickness and left ventricle diameter were assessed by Two-dimensional (2-D) echocardiography in the different experimental groups at 3 months post-injection (N = 11 animals for the DMDmdx + MD1 3E13 group; N = 9 animals for the DMDmdx + MD1 1E13 group; N = 10 animals for the other group). Cardiomyocytes morphology was assessed by the measuring length/width ratio of isolated cardiomyocytes obtained at sacrifice. Data are presented as mean ± SEM from n cardiomyocytes and N animals. *p < 0.05 (ANOVA test, followed by post-hoc Bonferroni multiple comparison test). Systolic (B) and diastolic (C) Functions of the heart: left ventricular ejection fraction, the early diastolic (E), the late diastolic (A), the E/A ratio, deceleration time and isovolumetric relaxation time was assessed by pulsed Doppler in the different experimental groups at 3 months post-injection (N = 11 animals for the DMDmdx + MD1 3E13 group; N = 9 animals for the DMDmdx + MD1 1E13 group; N = 10 animals for the other group). Left ventricular end-diastolic and end-systolic volumes (LVEDV and LVESV, respectively) were calculated from bi-dimensional long-axis parasternal views by means of the single-plane area-length method. These indexes were calculated in accordance with the standard, widely accepted formulas to study left ventricular dysfunction [78]. LV ejection fraction (EF) was calculated as [(LV end-diastolic volume − LV end-systolic volume)/LV end-diastolic volume] × 100. Systolic function was further assessed by calculation of LVEF using averaged measurements from 3 to 5 consecutive cardiac cycles in accordance with the guidelines of the American Society of Echocardiography. Data are presented as mean ± SEM. *p < 0.05 (Nonparametric Kruskal–Wallis test, followed by a post-hoc Dunn’s multiple comparison test).
These data showed no clear impact of the inclusion of the coil-coiled motif in MD1 on its efficacy to improve muscle and cardiac function in DMDmdx rats. Interestingly, MD3 expression in the heart do not seem to improve structural remodeling (in contrast to MD1) but still seems to prevent systolic and diastolic dysfunctions.
Discussion
Report of ≈46% shortened dystrophin in a 61-year-old ambulatory patient with Becker muscular dystrophy (BMD) [64] led to the design of MD transgenes that are compatible with additive gene therapy using rAAV vectors. Following preclinical success of systemic rAAV-MD gene therapy [12,13,14,15,16,17,18,19,20, 20,21,22,23], 3 independent clinical trials have been recently started [18]. However, due to the limitation of rAAV packaging capacity [8], current MD candidates cannot include all major domains of the full-length dystrophin. In this study, we evaluated if the inclusion of the dystrophin CT domain in MD1 could improve (i) its efficacy at recovering DAPC interactions, and (ii) its therapeutic potential, in both skeletal and cardiac muscles of DMDmdx rats.
Using dystrophin co-IP and LC-MS/MS analyses, we were first able to identify in the rat model most of the DAPC components that were previously identified in a study done in mice [41]. Only CRYAB, isoform 3 of cypher and ahnak1 were not identified as specific DAPC partners in our study since they were also detected in IgG_co-IP control group. However, we found that Cavin-1 and β2-SNT, previously described as heart specific DAPC partners, can be also detected at low levels in skeletal muscle DAPC, which is likely due to an increased sensitivity of our MS-based approach compared to this previous study. Calmodulin-1, was not identified as part of the DAPC in the Johnson et al. study [41], but was found associated with dystrophin in skeletal muscle of rabbits [55, 56]. We confirmed here this association in skeletal muscle in the rat. Finally, new DAPC partners were identified: β-DTN, Pikachurin and Perilipin-4 were specifically found in skeletal muscle DAPC; Caveolin-1 and Cavin-2 were specifically found in heart DAPC; and Cavin-3 was found in DAPC of both tissues.
We know from the literature that dystrophin CT domain is not critical for the expression of SNT and α-DTN partners at the membrane of skeletal muscle fibers [37]. In addition, always in skeletal muscle, MD1 (ΔR4-23/ΔCT) was shown to restore the expression of these proteins through the dystroglycan/sarcoglycan complex [37, 38, 65]. These observations agree with the fact that sarcoglycans interact with α-DTN [30] which itself interacts with SNT [29, 66], and that MD1 restores the dystroglycan/sarcoglycan complex through the ZZ domain present in the cysteine-rich domain [67]. Previous studies also shown that helix 1 of the CT coiled-coil domain is required to restore normal expression levels of SNT and α-DTN at the membrane of skeletal muscle fibers, suggesting a lower recruitment of these partners by MD1 compared to the full-length dystrophin [37, 38]. We hypothesized that addition of the dystrophin CT domain in MD1 should thus improve binding of these DAPC partners [29, 31]. In our study, we confirmed that MD1 is able to recruit SNT and α-DTN in skeletal muscle but also in heart. However, when compared to MD1, the results also showed that MD2, MD3 and MD4 increase the recruitment of proteins on the sarcoglycan and of the SNT complexes in heart and skeletal muscles, and of α-DTN in heart. Surprisingly, this is also the case when compared to full-length dystrophin. The abundances of these partners are equivalent in the complex associated to MD1 or to dystrophin, meaning that MD1 is sufficient to restore the physiological interactions of SNT and α-DTN in skeletal and cardiac muscles, and that supra-physiological levels of these partners are obtained when expressing MD2, MD3 or MD4. These results raise the question about the true path for the recruitment of SNT and α-DTN by the full-length dystrophin and whether it is mediated via the CT domain, via the sarcoglycan complex or both? Due to the different structures of MD, the affinity between the sarcoglycan complex and DTN could be modified. It is also possible that a recruitment via the sarcoglycan complex occurs only in MD associated protein complex, due to structure modification of MD compared to the native dystrophin. In that case, the inclusion of the CT domain would add a recruitment site for SNT and α-DTN, explaining the supra-physiological levels observed with MD2, MD3 and MD4 variants. We also showed that the recruitment of cavin-1 in heart reached equivalent levels than those observed with the full-length dystrophin only when the CT domain is present in MD, suggesting that this domain is required for an optimal recruitment of cavin-1 in heart. In skeletal muscle, it has been described that cavin-1 is rather a dystroglycan partner, independently to dystrophin, and that only a low amount of β-dystroglycan/cavin-1 complex is directly associated with dystrophin [68]. Such a mechanism probably occurs also in MD1-expressing heart, in which we observed partial recruitment of cavin-1 despite the absence of the dystrophin CT-domain. Knowing that the absence of cavin-1, which is required for caveolae formation [69], was linked to cardiomyopathic phenotypes [70, 71], further investigations are required to assess the functional consequence of a sub-physiological level of cavin-1 in MD1 associated protein complex in heart.
Beyond its efficacy to restore the DAPC, we further investigated the impact of the dystrophin CT domain through the comparison of the therapeutic efficacy between MD1 and MD3 (containing the coil-coiled motif of the dystrophin CT domain) in the DMDmdx rat model. It is the first time that the therapeutic impact of the dystrophin CT domain has been assessed on both the muscular and cardiac pathology of a relevant DMD model and after the systemic administration of rAAV-MD. Crawford et al. showed that this domain was not critical for dystrophin functionality in the skeletal muscle of transgenic mice [37]. But this kind of approach did not allow evaluation of the therapeutic efficacy on an established pathology, representative to that which would be seen in DMD patients. In addition, until now, therapeutic efficacy of MD on the DMD related cardiomyopathy was only poorly described, as the most commonly used DMD animal models, shows minimal and late cardiac features [16, 22, 39, 72]. The recently developed DMDmdx rat model gives now the opportunity to evaluate new MD designs on both skeletal muscles and cardiac features of the DMD pathology [42]. Since MD1 construct already shows a significant therapeutic effect on animal models [13, 16, 38], we chose to compare MD1 and MD3 at a known sub-therapeutic dose for MD1 (1E13 vg/dg), to evaluate and potentially highlight a difference in efficacy between these two products. Whereas the 1E13 vg/kg dose was found to be sub-therapeutic in skeletal muscle, this was not fully the case in the heart, because of the preferential tropism of AAV9 serotype for this organ in rodents [58]. Similar efficacy of the MD1 product was observed at the 1E13 and 3E13 vg/kg vector doses for some of the criteria assessed, such as heart fibrosis, heart remodeling and heart function. Therefore, it would have been interesting to compare our two vectors at a lower dose, in order to detect in a more refined way the potential benefits of the CT domain specifically in the heart. However, the relevance of the DMDmdx rat model still allowed us to evaluate a wide range of parameters as the histological aspect, the calcium homeostasis in skeletal muscle and heart, the ex vivo and in vivo muscle strength and the cardiac function. Across all the assessed criteria, the inclusion of the coil-coiled motif of the dystrophin CT domain within MD1 did not show any clear therapeutic benefit. At the same dose, both MD constructs showed similar effects on fibrosis, histopathological scores and muscle force. One pointed difference is that cardiac structural parameters appeared to be less corrected with MD3 treatment than with MD1. Still, the DMDmdx rats treated with rAAV2/9-MD3 showed improved systolic and diastolic functions, suggesting that MD1 and MD3 could act on the DMD cardiac pathology via different mechanisms. The potential involvement of cavin-1 in this mechanism could be further investigated. Finally, as previously described [59, 73, 74], we observed some isolated ring fibers in some skeletal muscles expressing MD1, but not in the ones expressing MD3. This is quite surprising considering that formation of ring-shaped myofibers have been associated to the presence of the polyproline site in hinge 2 in this MD [59, 73, 74]. Formation of these ring fibers could also be prevented in MD3 expressing fibers by a change of conformation compared to MD1. Given the fact that these ring fibers were not observed in all MD1-expressing muscles, we cannot exclude that we could have detected some if we had injected more animals with rAAV-MD3. In any case, these ring fibers, in a very low number compared to other myopathies in which they are also described (e.g., myotonic myopathy and some mitochondrial myopathies [75]), do not seem to have any deleterious impact on muscle function.
Taken together, our results indicate that the inclusion of the CT domain does not provide a therapeutic benefit to MD1, at least on skeletal and cardiac muscles functions and after 3 months of treatment in DMDmdx rats. On the other hand, we cannot exclude that the dystrophin CT domain could be necessary to more effectively control disease progression at long-term, and/or useful in other organ like brain [76] and smooth muscles [77].
Importantly, this work also strengthened the evidence of the therapeutic efficacy of MD1. Our results indicate indeed that MD1 restores the DAPC at a physiological level, with the exception of Cavin-1 in the heart. We also confirmed in a relevant model of DMD (DMDmdx rats) that systemic administration of a rAAV-MD1 vector at 3E13 vg/kg significantly improved histological pattern, muscle force and DMD-related cardiac pathology. We also showed for the first time that MD1 significantly improves DMD-related calcium homeostasis alterations in skeletal muscles and heart. Very recently, a phase 1/2a nonrandomized controlled trial testing the safety and biological efficacy of a single systemic infusion of rAAV-MD1 in 4 DMD patients revealed encouraging results with marked reductions in creatine kinase levels and improvement of muscle histological and functional patterns such as NSAA (North Star Ambulatory Assessment) score [25]. These results will obviously have to be confirmed in a randomized clinical trial including more DMD patients, but our study shows new evidence that despite the absence of the dystrophin CT domain, rAAV-MD1 offers high therapeutic potential for a systemic gene therapy for DMD, both at the muscle and cardiac levels.
Data availability
The data that support the findings of this study are available from the corresponding authors on reasonable request.
References
Mendell JR, Lloyd-Puryear M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve. 2013;48:21–6.
Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–28.
Bhat HF, Mir SS, Dar KB, Bhat ZF, Shah RA, Ganai NA. ABC of multifaceted dystrophin glycoprotein complex (DGC). J Cell Physiol. 2018;233:5142–59.
Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–9.
Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93.
Wang D, Zhong L, Nahid MA, Gao G. The potential of adeno-associated viral vectors for gene delivery to muscle tissue. Expert Opin Drug Deliv. 2014;11:345–64.
Rivera VM, Gao G, Grant RL, Schnell MA, Zoltick PW, Rozamus LW, et al. Long-term pharmacologically regulated expression of erythropoietin in primates following AAV-mediated gene transfer. Blood. 2005;105:1424–30.
Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther Janv. 2010;18:80–6.
Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, et al. Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–61.
Athanasopoulos T, Graham I, Foster H, Dickson G. Recombinant adeno-associated viral (rAAV) vectors as therapeutic tools for Duchenne muscular dystrophy (DMD). Gene Ther. 2004;11:S109–21.
Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–40.
Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12:787–9.
Foster H, Sharp PS, Athanasopoulos T, Trollet C, Graham IR, Foster K, et al. Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 Gene Transfer. Mol Ther. 2008;16:1825–32.
Bostick B, Yue Y, Lai Y, Long C, Li D, Duan D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum Gene Ther. 2008;19:851–6.
Yue Y, Pan X, Hakim CH, Kodippili K, Zhang K, Shin J-H, et al. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum Mol Genet. 2015;24:5880–90.
Le Guiner C, Servais L, Montus M, Larcher T, Fraysse B, Moullec S, et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat Commun. 2017;8:16105.
Hakim CH, Wasala NB, Pan X, Kodippili K, Yue Y, Zhang K, et al. A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of duchenne muscular dystrophy. Mol Ther Methods Clin Dev. 2017;6:216–30.
Duan D, Systemic AAV. Micro-dystrophin gene therapy for duchenne muscular dystrophy. Mol Ther. 2018;26:2337–56.
Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–35.
Gregorevic P, Blankinship MJ, Allen JM, Chamberlain JS. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol Ther. 2008;16:657–64.
Shin J-H, Nitahara-Kasahara Y, Hayashita-Kinoh H, Ohshima-Hosoyama S, Kinoshita K, Chiyo T. et al. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther. 2011;18:910–9.
Bostick B, Shin J-H, Yue Y, Duan D. AAV-microdystrophin therapy improves cardiac performance in aged female mdx mice. Mol Ther. 2011;19:1826–32.
Wang B, Li J, Fu FH, Xiao X. Systemic human minidystrophin gene transfer improves functions and life span of dystrophin and dystrophin/utrophin-deficient mice. J Orthopaedic Res. 2009;27:421–6.
Guiner CL, McIntyre M, Larcher T, Adjali O, Lafoux A, Toumaniantz G, et al. Dose finding study in the DMDmdx rat model to determine the efficacious dose of a rAAV9 vector encoding a human mini-dystrophin after IV administration. Neuromuscular Disorders. 2017;27:S188.
Mendell JR, Sahenk Z, Lehman K, Nease C, Lowes LP, Miller NF, et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020;77:1122–31.
Moorehead T, Yong F, Neelakantan S, Beaverson K, Binks M. Safety and tolerability of PF-06939926 in ambulatory boys with duchenne muscular dystrophy: a phase 1b multicenter, open- label, dose ascending study. Mol Ther J Am Soc Gene Ther. 2020;28:272.
Koo T. Studies on gene transfer in skeletal muscle cells and tissues using recombinant adeno-associated virus (AAV) vectors. Thesis. 2010.
Suzuki A, Yoshida M, Ozawa E. Mammalian alpha 1- and beta 1-syntrophin bind to the alternative splice-prone region of the dystrophin COOH terminus. J Cell Biol. 1995;128:373–81.
Sadoulet-Puccio HM, Rajala M, Kunkel LM. Dystrobrevin and dystrophin: an interaction through coiled-coil motifs. Proc Natl Acad Sci USA. 1997;94:12413–8.
Yoshida M, Hama H, Ishikawa-Sakurai M, Imamura M, Mizuno Y, Araishi K, et al. Biochemical evidence for association of dystrobrevin with the sarcoglycan-sarcospan complex as a basis for understanding sarcoglycanopathy. Hum Mol Genet. 2000;9:1033–40.
Bhat HF, Adams ME, Khanday FA. Syntrophin proteins as Santa Claus: role(s) in cell signal transduction. Cell Mol Life Sci. 2013;70:2533–54.
Matamoros M, Pérez-Hernández M, Guerrero-Serna G, Amorós I, Barana A, Núñez M, et al. Nav1.5 N-terminal domain binding to α1-syntrophin increases membrane density of human Kir2.1, Kir2.2 and Nav1.5 channels. Cardiovasc Res. 2016;110:279–90.
Leyva-Leyva M, Sandoval A, Felix R, González-Ramírez R. Biochemical and functional interplay between ion channels and the components of the dystrophin-associated glycoprotein complex. J Membr Biol. 2018;251:535–50.
Sabourin J, Lamiche C, Vandebrouck A, Magaud C, Rivet J, Cognard C, et al. Regulation of TRPC1 and TRPC4 cation channels requires an α1-syntrophin-dependent complex in skeletal mouse myotubes. J Biol Chem. 2009;284:36248–61.
Vandebrouck A, Sabourin J, Rivet J, Balghi H, Sebille S, Kitzis A, et al. Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J. 2007;21:608–17.
Dombernowsky NW, Ölmestig JNE, Witting N, Kruuse C. Role of neuronal nitric oxide synthase (nNOS) in Duchenne and Becker muscular dystrophies - Still a possible treatment modality? Neuromuscul Disord. 2018;28:914–26.
Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin cooh-terminal domain. J Cell Biol. 2000;150:1399–410.
Koo T, Malerba A, Athanasopoulos T, Trollet C, Boldrin L, Ferry A, et al. Delivery of AAV2/9-microdystrophin genes incorporating helix 1 of the coiled-coil motif in the C-Terminal domain of dystrophin improves muscle pathology and restores the level of α1-syntrophin and α-dystrobrevin in skeletal muscles of mdx mice. Hum Gene Ther. 2011;22:1379–88.
McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech. 2015;8:195–213.
Tandon A, Jefferies JL, Villa CR, Hor KN, Wong BL, Ware SM, et al. Dystrophin genotype-cardiac phenotype correlations in Duchenne and Becker muscular dystrophies using cardiac magnetic resonance imaging. Am J Cardiol. 2015;115:967–71.
Johnson EK, Zhang L, Adams ME, Phillips A, Freitas MA, Froehner SC, et al. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS ONE. 2012;7:e43515.
Larcher T, Lafoux A, Tesson L, Remy S, Thepenier V, François V, et al. Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PLoS ONE. 2014;9:e110371.
Li X, Eastman EM, Schwartz RJ, Draghia-Akli R. Synthetic muscle promoters: activities exceeding naturally occurring regulatory sequences. Nat Biotechnol. 1999;17:241–5.
D’Costa S, Blouin V, Broucque F, Penaud-Budloo M, François A, Perez IC, et al. Practical utilization of recombinant AAV vector reference standards: focus on vector genomes titration by free ITR qPCR. Mol Ther Methods Clin Dev. 2016;5:16019.
Salvetti A, Orève S, Chadeuf G, Favre D, Cherel Y, Champion-Arnaud P, et al. Factors influencing recombinant adeno-associated virus production. Hum Gene Ther. 1998;9:695–706.
Shinoda K, Tomita M, Ishihama Y. emPAI Calc—for the estimation of protein abundance from large-scale identification data by liquid chromatography-tandem mass spectrometry. Bioinformatics. 2010;26:576–7.
Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–72.
Fraysse B, Desaphy J-F, Rolland J-F, Pierno S, Liantonio A, Giannuzzi V, et al. Fiber type-related changes in rat skeletal muscle calcium homeostasis during aging and restoration by growth hormone. Neurobiol Dis. 2006;21:372–80.
Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol. 2011;51:288–98.
Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50.
Dyle MC, Ebert SM, Cook DP, Kunkel SD, Fox DK, Bongers KS, et al. Systems-based discovery of tomatidine as a natural small molecule inhibitor of skeletal muscle atrophy. J Biol Chem. 2014;289:14913–24.
Moorwood C, Liu M, Tian Z, Barton ER. Isometric and eccentric force generation assessment of skeletal muscles isolated from murine models of muscular dystrophies. J Vis Exp. 2013;31:e50036.
Chapdelaine P, Gérard C, Sanchez N, Cherif K, Rousseau J, Ouellet DL, et al. Development of an AAV9 coding for a 3XFLAG-TALEfrat#8-VP64 able to increase in vivo the human frataxin in YG8R mice. Gene Ther. 2016;23:606–14.
Mayra A, Tomimitsu H, Kubodera T, Kobayashi M, Piao W, Sunaga F, et al. Intraperitoneal AAV9-shRNA inhibits target expression in neonatal skeletal and cardiac muscles. Biochem Biophys Res Commun. 2011;405:204–9.
Madhavan R, Jarrett HW. Phosphorylation of dystrophin and alpha-syntrophin by Ca(2+)-calmodulin dependent protein kinase II. Biochim Biophys Acta. 1999;1434:260–74.
Madhavan R, Jarrett HW. Calmodulin-activated phosphorylation of dystrophin. Biochemistry. 1994;33:5797–804.
Sato S, Omori Y, Katoh K, Kondo M, Kanagawa M, Miyata K, et al. Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat Neurosci. 2008;11:923–31.
Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, et al. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006;99:e3–9.
Banks GB, Judge LM, Allen JM, Chamberlain JS. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010;6:e1000958.
Lorin C, Vögeli I, Niggli E. Dystrophic cardiomyopathy: role of TRPV2 channels in stretch-induced cell damage. Cardiovasc Res. 2015;106:153–62.
Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–30.
Hermans MCE, Pinto YM, Merkies ISJ, de Die-Smulders CEM, Crijns HJGM, Faber CG. Hereditary muscular dystrophies and the heart. Neuromuscul Disord. 2010;20:479–92.
Szabó PL, Ebner J, Koenig X, Hamza O, Watzinger S, Trojanek S, et al. Cardiovascular phenotype of the Dmdmdx rat - a suitable animal model for Duchenne muscular dystrophy. Dis Model Mech. 2021;22:14.
England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343:180–2.
Yue Y, Liu M, Duan D. C-Terminal-Truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double-knockout mice. Mol Ther. 2006;14:79–87.
Nakamori M, Takahashi MP. The role of α-dystrobrevin in striated muscle. Int J Mol Sci. 2011;12:1660–71.
Ishikawa-Sakurai M, Yoshida M, Imamura M, Davies KE, Ozawa EZZ. domain is essentially required for the physiological binding of dystrophin and utrophin to beta-dystroglycan. Hum Mol Genet. 2004;13:693–702.
Johnson EK, Li B, Yoon JH, Flanigan KM, Martin PT, Ervasti J, et al. Identification of new dystroglycan complexes in skeletal muscle. PLoS ONE. 2013;8:e73224.
Liu L. Lessons from cavin-1 deficiency. Biochem Soc Trans. 2020;48:147–54.
Taniguchi T, Maruyama N, Ogata T, Kasahara T, Nakanishi N, Miyagawa K, et al. PTRF/Cavin-1 deficiency causes cardiac dysfunction accompanied by cardiomyocyte hypertrophy and cardiac fibrosis. PLoS ONE. 2016;11:e0162513.
Kaakinen M, Reichelt ME, Ma Z, Ferguson C, Martel N, Porrello ER, et al. Cavin-1 deficiency modifies myocardial and coronary function, stretch responses and ischaemic tolerance: roles of NOS over-activity. Basic Res Cardiol. 2017;112:24.
Bostick B, Yue Y, Long C, Marschalk N, Fine DM, Chen J, et al. Cardiac expression of a mini-dystrophin that normalizes skeletal muscle force only partially restores heart function in aged mdx mice. Mol Ther. 2009;17:253–61.
Banks GB, Combs AC, Chamberlain JR, Chamberlain JS. Molecular and cellular adaptations to chronic myotendinous strain injury in mdx mice expressing a truncated dystrophin. Hum Mol Genet. 2008;17:3975–86.
Banks GB, Chamberlain JS, Froehner SC. Truncated dystrophins can influence neuromuscular synapse structure. Mol Cell Neurosci. 2009;40:433–41.
Sawicka E. Origin of the ring muscle fibers in neuromuscular diseases. Neuropatologia Polska. 1991;29:29–40.
Sekiguchi M. The role of dystrophin in the central nervous system: a mini review. Acta Myol: Myopathies and Cardiomyopathies: Official Journal of the Mediterranean Society of Myology. 2005;24:93–7.
Haenggi T, Fritschy J-M. Role of dystrophin and utrophin for assembly and function of the dystrophin glycoprotein complex in non-muscle tissue. Cell Mol Life Sci: CMLS. 2006;63:1614–31.
Chen L, Zhang J, Hu X, Philipson KD, Scharf SM. The Na+/Ca2+ exchanger-1 mediates left ventricular dysfunction in mice with chronic intermittent hypoxia. J Appl Physiol (1985). 2010;109:1675–85.
Acknowledgements
We thank all the personnel of the Boisbonne Center for Gene Therapy (ONIRIS, INSERM, Nantes, France) and of the UTE IRS2 (University of Nantes, France) for the handling and care of the rats included in this study. We also thank the vector core of UMR 1089 (CPV, INSERM and University of Nantes) for the cloning of the MDs pAAV plasmids and the production of the rAAV vectors used in this study. We thank the Mass Spectrometry and Proteomics Facility (Ohio State University, Columbus, Ohio, USA), which performed the proteomics experiments. We thank E. Marrosu (UCL Great Ormond Street Institute of Child Health, London, United Kingdom) for providing MANEX1011B and MW8 antibodies for co-immunoprecipitation experiments. We thank G. Potier for his valuable help during the analyses of label free quantitation. We also thank T. Cronin for editing the English language of this manuscript. Finally, we thank the MDA Monoclonal Antibody Resource for providing the MANEX 1011 C antibody.
Funding
This project was supported by the MDA (Muscular Dystrophy Association, Research Grant ID #513878), the AFM-Téléthon (Association Française contre les Myopathies), the “Fondation d’entreprise pour la thérapie génique en Pays de la Loire”, INSERM, INRA, the University of Nantes and the University Hospital of Nantes. The Fusion Orbitrap instrument used for proteomics study was supported by NIH Award Number Grant S10 OD018056.
Author information
Authors and Affiliations
Contributions
Study design and reviewing of the data: AB, VF, FM, CLG. Experimental investigation: AB, LZ, AL, BF, GT, TL, TG, ML, CL, AH, AC. Resources: VF, MA, BM, JG, VB, SR, IA, CH, AM, BK, ALH, LP and FM. Writing – original draft: AB, LZ, AL, BF and CLG. Writing – Review & Editing: all authors. Supervision: CLG and OA. Funding acquisition: CLG, PM, GD and OA.
Corresponding author
Ethics declarations
Competing interests
PM, GD and CLG are co-authors of a patent for systemic treatment of dystrophic pathologies (EP3044319A1, dated 27 June 2014). GD is an inventor on a PCT for production of large-sized micro-dystrophins in an AAV-based vector configuration (PCT/EP2016/060350, dated 17 May 2015). The remaining authors declare no competing financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bourdon, A., François, V., Zhang, L. et al. Evaluation of the dystrophin carboxy-terminal domain for micro-dystrophin gene therapy in cardiac and skeletal muscles in the DMDmdx rat model. Gene Ther 29, 520–535 (2022). https://doi.org/10.1038/s41434-022-00317-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41434-022-00317-6
- Springer Nature Limited
This article is cited by
-
Therapeutic approaches for Duchenne muscular dystrophy
Nature Reviews Drug Discovery (2023)
-
Micro-dystrophin gene constructs for repairing heart and muscle function in rats: the smaller is enough?
Gene Therapy (2022)