
Abstract
The emergence of bacterial resistance poses a serious threat to public health. One of the most important resistance mechanisms against β-lactam antibiotics is the production of metallo-β-lactamases (MBLs). In this study, α-lipoic acid (LA) and methimazole (MMI), which have been used in clinical practice as non-antibacterial drugs and as a supplement, were chosen to explore their potential to be metallo-β-lactamases inhibitors (MBLIs). Enzyme inhibition assays showed that LA and MMI had moderate inhibitory activity against NDM-1 but no activity against VIM-2 and IMP-7. Antibacterial assays to determine synergy, demonstrated that the combination of LA or MMI with meropenem (MER) reduced the MIC value of MER against NDM-1 producing E. coli 16 times and 4 times, respectively, lower than that of MER alone. The fractional inhibitory concentration index (FICI) values were calculated to be less than 0.5, indicating that both LA and MMI had synergistic antibacterial effects with MER against all three MBLs expressing E. coli strains. The time-kill studies also suggested that LA and MMI were effective in restoring the antibacterial effect of MER. These findings revealed that LA and MMI are potential carbapenem enhancers, and provide a starting point for the development of potent MBLIs.
Similar content being viewed by others
Introduction
Ever since their discovery, β-lactam antibiotics have become the most commonly used antibiotics in the treatment of bacterial infections because of their high efficacy and low toxicity. However, the indiscriminate use of β-lactam antibiotics has accelerated the development and prevalence of resistant bacteria, which poses a serious threat to public health around the world [1]. The most important resistance mechanism to β-lactam antibiotics is the production of β-lactamases that hydrolyze the β-lactam ring [2].
β-Lactamases can be divided into two groups according to their catalytic mechanisms. The first is serine β-lactamases (SBLs) with a nucleophilic serine residue at the active site, and includes the Ambler class A β-lactamases, such as Klebsiella pneumoniae carbapenemase, Ambler class C β-lactamases (AmpC), and class D, such as oxacillinase. The second class consists of MBLs with one or two zinc ions at the active center, including class B β-lactamases. Different amino acid sequences and zinc coordination modes in active site further divide MBLs into three subclasses, B1, B2, and B3. The B1 subfamily is the most common MBLs in clinical isolates, especially the New Delhi metallo-β-lactamase-1 (NDM-1) [3]. The B1 subclass can inactivate nearly all β-lactam antibiotics including carbapenems, which are considered as the antibiotics of last resort. What’s worse, MBLs genes can not only spread quickly across bacterial species through plasmids which are prone to mutation due to rearrangement, but they also can be integrated into the chromosome of bacteria to make their inheritance and resistance more stable [4].
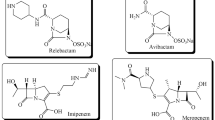
Inhibition of β-lactamase can effectively restore the sensitivity of β-lactam antibiotics to resistant bacteria. At present, clinically available β-lactamase inhibitors such as clavulanic acid, sulbactam, tazobactam, and avibactam, are all SBLIs, no inhibitor of MBLs to treat MBLs-positive bacterial isolates is approved for clinical use so far [5]. Therefore, there is a high unmet medical need to develop novel effective MBLIs to restore the activity of β-lactam antibiotics.
“The most fruitful basis for the discovery of a new drug is to start with an old drug” [6]. Drug repurposing refers to the strategy of using drugs that have been on the market, or are being developed or discontinued for lack of efficacy, for screening to identify lead for new therapeutic purposes. Among the reported MBLs inhibitors, sulfhydryl- or disulfide-containing compounds have a strong affinity to zinc ion which are considered to be a class of hit compounds with development potential [7,8,9,10]. Captopril, an angiotensin-converting enzyme inhibitor bearing a sulfhydryl group, is used as an antihypertensive agent for many decades. Several studies have showed that captopril has moderate inhibitory activity against NDM-1, and the mode of binding of captopril and NDM-1 has been demonstrated by mass spectrometry and X-ray crystallographic analysis [11,12,13].
In this manuscript, we show that by screening two sulfhydryl- or disulfide-containing drugs, α-lipoic acid (LA) and methimazole (MMI) exhibit potent inhibitory activity toward NDM-1, both by cell-based and enzyme-based assays (Fig. 1). The combined use of LA or MMI with meropenem (MER) not only restores antibiotic activity, but also significantly reduces the NDM-1 evolution in NDM-1-positive Escherichia coli (E. coli).

Structures of sulfhydryl- or disulfide-containing drugs
Material and methods
General information
LA and MMI were purchased from Shanghai yuanye Bio-Technology Co., Ltd, Shanghai, China. MER was purchased from TCI (Shanghai) Development Co., Ltd, Shanghai, China. E. coli ATCC25922 and clinical isolates of E. coli BAA-2452 (blaNDM-1) were provided by the Department of Laboratory Medicine, General Hospital of Southern Theatre Command of PLA (Guangzhou, China).
Enzyme preparation
The amplified genes of NDM-1, VIM-2, and IMP-7 were cloned into pET-28a plasmid with an incorporated N-terminal His-Tag to build the vector pET-28a-NDM-1, pET-28a-VIM-2, and pET-28a-IMP-7, respectively. All the enzymes were overexpressed in E. coli BL21(DE3) transformed with the respective MBL using Luria-Bertani (LB) medium supplemented with 30 μg ml−1 kanamycin and 34 μg ml−1 chloramphenicol and incubated at 37 °C. Enzyme overexpression was induced using 0.5 mM IPTG when bacteria grew to OD600 of 0.6, The culture was incubated at 20 °C overnight. To purify the respective MBLs, the cultured bacteria were collected by centrifugation at 5000 rpm and resuspended in a lysis buffer (50 mM Tris, 300 mM NaCl, 0.2 mM PMSF, 0.1% Triton X-100, pH 8.0). The cells were ice-cooled and lysed by sonication and then centrifuged at 12,000 rpm for 20 min to remove cell debris. The supernatant was applied to a 5 ml Ni-NTA columns, followed by using washing buffer (50 mM Tris, 300 mM NaCl, 20/50 mM imidazole, pH 8.0). The protein was finally eluted out using elution buffer (50 mM Tris, 300 mM NaCl, 500 mM imidazole, pH 8.0). The N-terminal His-tag of the fusion protein was cleaved by adding TEV Protease. After dialysis with storage buffer (50 mM HEPES, 100 mM NaCl at pH 7.0), The enzymes were concentrated using PEG20000 and then filtered by 0.45 μm membrane filter. All the enzymes were separated into aliquots and stored at −80 °C.
Enzyme kinetics
The Michaelis–Menten constant (Km) is numerically equal to the concentration of the substrate at which the enzymatic reaction rate reaches half of the maximum reaction rate and can be obtained by the Michaelis–Menten equation: V = Vmax[S]/(Km + [S]). The assay was conducted in 96-well plates at 25 °C with MER as the hydrolyzed substrate ranging from 0 to 500 μM. NDM-1, VIM-2, and IMP-7 were added with the final concentrations of 1, 2, and 2.5 nM, respectively. The change of absorbance for MER was recorded every 17 s for 15 min. The Km was obtained by fitting the initial velocity data to the Michaelis−Menten equation using GraphPad Prism software (La Jolla, CA).
Enzyme inhibition
The experiment was performed at 25 °C in 96-well plates using MER as substrate. Freshly prepared NDM-1 (1 nM), VIM-2 (2 nM) and IMP-7 (2.5 nM) were first incubated with various concentrations of inhibitors (0.1 μM to 500 μM) for 15 min in the assay buffer (50 mM HEPES, 1 mM ZnSO4, 0.01% Triton X-100, 0.1 μg ml−1 BSA, pH 7.0), followed by adding 50 μM MER to initiate the reactions and monitoring the absorbance at 300 nm every second for 20 min. The IC50 values were obtained by plotting the initial reaction rate versus inhibitor concentration using GraphPad Prism software. Given that the concentration of the enzyme is much less than the inhibitor, The Ki values were calculated from the IC50 values, substrate concentration, and Km value using the Cheng−Prusoff equation as IC50/(1 + [S]/Km) [14]. The activity of each inhibitor was measured in triplicates.
In vitro susceptibility testing
Minimum inhibitory concentration (MIC) was measured by the broth microdilution method according to the guidelines of American Clinical and Laboratory Standards Institute (CLSI) except that Mueller Hinton Broth was replaced by LB broth. MBL positive strains were inoculated on sterile LB agar medium at 37 °C overnight, a single colony was transferred to 3 ml LB liquid medium supplemented with 50 μg ml−1 kanamycin and incubated at 37 °C to the logarithmic growth phase. The bacterial suspension was diluted to 1 × 106 CFU ml−1 and the overexpression of MBL was induced by addition of 0.5 mM isopropyl β-d-thiogalactoside (IPTG). 100 μl of inoculum, 50 μl of MER (256 μg ml−1) and 50 μl of different concentrations of inhibitors (2-fold dilutions with concentrations ranging from 0.063 to 128 μg ml−1) were added to 96-well plates at 37 °C for 18–24 h. All MICs were determined in triplicates.
Fractional inhibitory concentration index (FICI) was calculated by the equation of FICI = FICA + FICB = CA/MICA + CB/MICB, where MICA and MICB were the MIC values of compound A and B used alone, and CA and CB were the inhibitor concentrations of compound A and B in an effective combination.
Time-kill assay
This experiment was conducted to further explore the synergy between MER and drugs. E. coli BL21(DE3)/pET26b-NDM-1 strain was cultured at 37 °C overnight and then inoculated into the LB broth containing 50 μg ml−1 kanamycin and 0.5 mM IPTG for 4 h to reach logarithmic phase. Bacteria suspension (final concentration of 5 × 105 CFU ml−1) were added into four sterile tubes containing MER alone (32 μg ml−1), drug alone (64 μg ml−1), combination (32 μg ml−1 MER + 64 μg ml−1 drug) and control groups (DMSO), respectively. Samples were taken from the tubes at 0, 2, 4, 8, 18, and 24 h, respectively, and then inoculated on LB agar medium after tenfold dilution at 37 °C for 24 h. Three independent experiments were performed, and the curve was plotted as log10 CFU ml−1 versus time.
Results and discussions
The crystal structure of l-captopril with NDM-1 shows that the sulfhydryl group could chelate to the zinc ions and the carboxyl group could form hydrogen bond with Asn220, suggesting that both sulfhydryl and carboxyl groups are essential for inhibiting NDM-1 activity. In addition, the active pocket of NDM-1 contains a large hydrophobic domain resembling a long corridor which might be compatible with linear alkyl or alkoxy links [15]. Reduced holomycin, is a natural product with dithiol moiety, showed potent NDM-1 inhibition [10]. We assume dithiol group or disulfide group may be a better alternative to a monothiol group to enhance the inhibitory activity of a compound. Based on the above consideration, we tried to find an approved drug bearing a disulfide group, a carboxyl group, and a linear hydrophobic link between these two groups. We were pleased to identify LA, a drug used to treat diabetic neuropathy in China, which met the above requirements perfectly. Furthermore, the linker between the disulfide bond and the carboxyl group of LA is also comparable to captopril in terms of length and hydrophobicity. To clarify the importance of hydrogen bonds formed by carboxyl groups on enzyme inhibitory activity, we chose MMI as a sulfhydryl-containing drug without a carboxyl group. MMI is used for the treatment of hyperthyroidism. MIC was measured to evaluate the ability of LA and MMI to restore antimicrobial activity of MER against a clinical isolate of NDM-1-positive E. coli using the microbroth dilution method. Captopril was used as the positive control. The NDM-1-expressing strain was confirmed to be resistant to MER as determined by its MIC of 16 μg ml−1, which is higher than the breakpoint value (≥4 μg ml−1) for MER against Enterobacterales defined by CLSI [16]. Notably, LA, MMI, and captopril alone did not exhibit any intrinsic antibacterial activity at 256 μg ml−1, however, when LA, MMI, or captopril were combined with MER, the MIC values of MER against NDM-1-positive isolate decreased to 4, 4 and 8 μg ml−1, respectively (Table 1). FICI were used to judge the interaction of two drugs when used in combination. It is calculated according to the following equation: FICI = FICA + FICB = CA/MICA + CB/MICB, where MICA and MICB are the MIC values of compound A and B used alone, and CA and CB are the inhibitor concentrations of compound A and B in an effective combination. FICI ≤ 0.5 indicates that the two compounds are considered to have synergistic effects [17]. The FICI of LA in combination with MER was calculated to be 0.38, which is lower than that of positive control captopril with MER (0.50), indicating LA could be a potential MBLI. The FICI of MMI combined with MER was calculated to be 0.63, indicative of the additive interaction only (0.5 < FICI ≤ 1). To further examine whether LA and MMI are able to resensitize MER against other clinically relevant B1 MBLs expressing bacteria, we genetically constructed three E. coli strains producing NDM-1, VIM-2, and IMP-7, which were confirmed by SDS-PAGE. As shown in Table 1, LA and MMI synergized with MER as determined by the FICI values ranging from 0.19 to 0.50, and the MIC of MER decreased by 4–16 fold, which showed better synergistic effects than with captopril. LA had better synergistic interaction against all tested MBLs producers, except the VIM-2 construct, than MMI, indicating that the large contribution of the carboxyl group to the inhibitory activity. Notably, compared with the clinical isolate expressing NDM-1, the constructed strain was more sensitive to the combination treatment, which might be attributed to the difference in NDM-1 expression and phenotype.
The synergistic effect of LA and MMI in combination with MER was confirmed by time-kill assays which provided dynamic measures of bactericidal activities of the combination over time. The experiment was divided into four groups, MER (1/2 MICA), drug (1/2 MICB), combination (1/2 MICA + 1/2 MICB), and blank control (no drug). Figure 2 shows that MER with LA can significantly reduce viable counts after 4 h incubation, and the bactericidal effect lasted at least for 24 h. while MEM or LA alone could not inhibit the growth of the NDM-1-producing strain. The time-kill curve of MER with MMI (Fig. 3) is similar to those of MER with LA and MER with captopril (Fig. 4), suggesting that MMI has a similar dynamic synergy profile to LA when combined with MER.

Time-kill curves of LA and MER alone or in combination against E. coli BL21(DE3)/pET26b-NDM-1 strain

Time-kill curves of MMI and MER alone or in combination against E. coli BL21(DE3)/pET26b-NDM-1 strain

Time-kill curves of captopril and MER alone or in combination against E. coli BL21(DE3)/pET26b-NDM-1 strain
To determine whether LA and MMI can inhibit enzymatic activity of MBLs, we carried out the enzyme kinetics assays using MER as the hydrolysis substrate (Fig. 5). Comparable half-maximum inhibitory concentration (IC50) values of LA, MMI, and captopril were observed for NDM-1, which were calculated to be 62.75, 98.84, and 33.44 μM, respectively (Table 2), whereas LA and MMI had no inhibitory effect on VIM-2 and IMP-7 (IC50 > 500 μM). Similar to the disulfide form of holomycin that showed no inhibitory effect on NDM-1, LA with disulfide bond is also unable to chelate zinc ions of MBLs to show any inhibitory activity. However, in cell-based studies, compounds with disulfide bond are internalized as prodrugs by bacteria and can be converted to dithiol form as a potential MBLIs. Like LA, MMI also showed inconsistent results in enzyme inhibition assays and synergistic antibacterial assays, we speculate that MMI is also a prodrug that needs to enter the bacteria to be converted into the active form as a MBLI.

Residual activities of NDM-1 enzyme with inhibitors
Conclusion
In this study, LA and MMI were selected and their inhibitory effect on MBLs and synergistic antibacterial activities against MBLs-positive strains combined with MER were evaluated. Both drugs exhibited synergy with MER against MBLs positive isolates, where LA was more effective than MMI. Notably, LA and MMI demonstrated moderate inhibition on NDM-1 and no inhibition on VIM-2 and IMP-7. The difference between the results of the enzyme inhibition assay and cell-based assay may be due to the fact that both drugs are prodrugs, which can only act as MBLIs when they were activated intracellularly. In the future, characterization of the active intracellular entity may be possible.
In summary, the results yield a promising starting point for the development of potent MBLIs. Structure modification and detection of the active form of the prodrug are ongoing in our laboratories.
References
Lima LM, Silva BNMD, Barbosa G, et al. β-lactam antibiotics: an overview from a medicinal chemistry perspective. Eur J Med Chem. 2020;208:112829.
Bush K, Bradford PA. Epidemiology of beta-lactamase-producing pathogens. Clin Microbiol Rev. 2020;33:e00047–19.
Linciano P, Cendron L, Gianquinto E, et al. Ten years with New Delhi metallo-β-lactamase-1 (NDM-1): from structural insights to inhibitor design. ACS Infect Dis. 2019;5:9–34.
Yong D, Toleman MA, Giske CG, et al. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother. 2009;53:5046–54.
Egorov AM, Ulyashova MM, Rubtsova MY. Inhibitors of beta-lactamases. New life of beta-lactam antibiotics. Biochemistry (Mosc). 2020;85:1292–309.
Chen H, Wu J, Gao Y, et al. Scaffold repurposing of old drugs towards new cancer drug discovery. Curr Top Med Chem. 2016;16:2107–14.
Liu S, Jing L, Yu Z, et al. ((S)-3-Mercapto-2-methylpropanamido)acetic acid derivatives as metallo-β-lactamase inhibitors: synthesis, kinetic and crystallographic studies. Eur J Med Chem. 2018;145:649–60.
Wang YL, Liu S, Yu Z, et al. Structure-based development of (1-(3′-mercaptopropanamido)methyl)boronic acid derived broad-spectrum, dual-action inhibitors of metallo- and serine-β-lactamases. J Med Chem. 2019;62:7160–84.
Chang YN, Xiang Y, Zhang YJ, et al. Carbamylmethyl mercaptoacetate thioether: a novel scaffold for the development of L1 metallo-β-lactamase inhibitors. ACS Med Chem Lett. 2017;8:527–32.
Chan AN, Shiver AL, Wever WJ, et al. Role for dithiolopyrrolones in disrupting bacterial metal homeostasis. Proc Natl Acad Sci USA. 2017;114:2717–22.
Yusof Y, Tan DTC, Arjomandi OK, et al. Captopril analogues as metallo-β-lactamase inhibitors. Bioorg Med Chem Lett. 2016;26:1589–93.
Brem J, van Berkel SS, Zollman D, et al. Structural basis of metallo-β-lactamase inhibition by captopril stereoisomers. Antimicrob Agents Chemother. 2015;60:142–50.
Thomas CA, Cheng Z, Yang K, et al. Probing the mechanisms of inhibition for various inhibitors of metallo-β-lactamases VIM-2 and NDM-1. J Inorg Biochem. 2020;210:111123.
Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108.
Meng Z, Tang ML, Yu L, et al. Novel mercapto propionamide derivatives with potent New Delhi metallo-β-lactamase-1 inhibitory activity and low toxicity. ACS Infect Dis. 2019;5:903–16.
Performance Standards for Antimicrobial Susceptibility Testing. Document M100-S27. 27th Informational Supplement. Wayne, PA: Clinical and Laboratory Standards Institute; 2017.
Wachino JI, Jin W, Kimura K, et al. Sulfamoyl heteroarylcarboxylic acids as promising metallo-β-lactamase inhibitors for controlling bacterial carbapenem resistance. mBio. 2020;11:e03144–19.
Acknowledgements
This work was funded by Pearl River Nova Program of Guangzhou (201710010013) and Youth Program of Military Medical Science and Technology (19QNP041).
Author information
Authors and Affiliations
Contributions
ZJ and LS designed research and analyzed data. BZ, YY, JY, LC, TH, and HT conducted experiments. ZJ and LS supervised the whole study, wrote and revised the manuscript. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, B., Yang, Y., Yuan, J. et al. Methimazole and α-lipoic acid as metallo-β-lactamases inhibitors. J Antibiot 75, 282–286 (2022). https://doi.org/10.1038/s41429-022-00513-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-022-00513-x
- Springer Japan KK