
Abstract
In actinomycetes, many secondary metabolite biosynthetic genes are not expressed under typical laboratory culture conditions and various efforts have been made to activate these dormant genes. In this study, we focused on high-temperature culture. First, we examined the thermotolerance of 3160 actinomycete strains from our laboratory culture collection and selected 57 thermotolerant actinomycetes that grew well at 45 °C. These 57 thermotolerant actinomycetes were cultured for 5 days in liquid medium at both 30 °C and 45 °C. Culture broths were extracted with 1-butanol, and each extract was subjected to LC/MS analysis. The metabolic profiles of each strain were compared between the 30 °C and 45 °C cultures. We found that almost half of these thermotolerant actinomycetes produced secondary metabolites that were detected only in the 45 °C culture. This result suggests that high-temperature culture induces the production of dormant secondary metabolites. These compounds were named “heat shock metabolites (HSMs).” To examine HSM production in more detail, 18 strains were selected at random from the initial 57 strains and cultivated in six different media at 30 °C and 45 °C; as before, metabolic profiles of each strain in each medium were compared between the 30 °C and 45 °C cultures. From this analysis, we found a total of 131 HSMs. We identified several angucycline-related compounds as HSMs from two thermotolerant Streptomyces species. Furthermore, we discovered a new compound, murecholamide, as an HSM from thermotolerant Streptomyces sp. AY2. We propose that high-temperature culture of actinomycetes is a convenient method for activating dormant secondary metabolite biosynthetic genes.
Similar content being viewed by others
Introduction
Natural products have provided many small molecules in chemical biology research for drug discovery and elucidation of various biological phenomena. As a result of over 50 years of screening, 30,000 natural products derived from microbial sources have been reported [1]. In particular, secondary metabolites produced by actinomycetes have been widely used as sources of drug development. More than half of all naturally derived antibiotics were isolated from actinomycetes and 70% of these were found in Streptomyces [1]. Recent genomic analyses have predicted that one Streptomyces strain contains over 30 secondary metabolite biosynthetic gene clusters [2,3,4]. However, the predicted number of gene clusters in these strains exceeds the number of compounds isolated, which suggests that a considerable number of secondary metabolite biosynthetic genes are in a dormant state under laboratory culture conditions [4, 5]. To activate these dormant genes in actinomycetes, several strategies have been developed including ribosome engineering and combined-culture [6]. Ribosome engineering is based on the principle that the mutation of the ribosomal S12 protein, which is associated with the acquisition of antibiotic resistance, activates secondary metabolism [7]. Mutation of ribosomal S12 protein results in increased expression of translation factors, which leads to enhanced protein synthesis in secondary metabolism [7]. This method led to the production of blue-pigmented antibiotic, actinorhodin, in Streptomyces lividans [7]. Secondary metabolism is also activated by combined-culture, which is defined as the co-culture of actinomycetes with some mycolic acid-containing bacteria [8]. For example, combined-culture of Streptomyces endus with Tsukamurella pulmonis induced the production of a novel antibiotic, alchivemycin A, by S. endus [9]. Direct cell–cell contact is necessary for activation of dormant genes [10]. Both methods have made it possible to activate dormant genes in actinomycetes. Furthermore, the development of these activation methods is useful, not only for the discovery of novel secondary metabolites, but also for understanding the regulatory mechanisms that produce secondary metabolites.
Generally, actinomycetes have been cultured at 30–37 °C for the production or analysis of secondary metabolites; however, “thermotolerant actinomycetes” can grow even at 45–50 °C [11]. In some cases, production of secondary metabolites is enhanced by temporal heat shock [12, 13]. In the present study, we found that high-temperature culture under heat shock condition of thermotolerant actinomycetes often induced the production of secondary metabolites not produced by 30 °C culture. We named these metabolites “heat shock metabolites (HSMs).” Production of secondary metabolites by high-temperature culture of thrmophilic actinomycetes has been previously reported [14, 15]. However, since thermophilic actinomycetes can grow only at a high temperature, secondary metabolites obtained from high-temperature culture of thermophilic actinomycetes are not included in the HSM whose production was induced by heat shock. Using our heat shock culture, we isolated a new HSM compound from Streptomyces sp. AY2. These results indicate that high-temperature culture causing heat shock is a useful method for the activation of dormant biosynthetic genes for secondary metabolites.
Results and discussion
Screening for thermotolerant actinomycete strains
We searched our laboratory actinomycete culture collection (3160 strains) for strains able to grow at high temperature (45 °C). The actinomycete strains were cultured initially on agar plates at 40 °C; against expectations, most of strains grew well at this temperature. We also observed that S. lividans TK21 and Streptomyces venezuelae NBRC13096, both of which were used as standard actinomycete strains, could grow at 40 °C (data not shown). Indeed, it has been reported that S. venezuelae can grow at 42 °C [12]. Therefore, we changed the culture temperature from 40 °C to 45 °C. From this screening, we obtained 57 actinomycete strains that can grow well at 45 °C (hit ratio: 1.8%). These “thermotolerant actinomycetes” can be divided into three groups by the pigmentation on an agar plate (Figs. 1a–c and S1). Thermotolerant actinomycetes in the first group show a similar pigmentation between the 30 °C and 45 °C cultures (51%). The second group produces more pigment(s) at 45 °C than at 30 °C (35%). In the third group, pigmentation is suppressed at 45 °C (14%). These results indicate that expression of secondary metabolite biosynthetic genes is affected by culture temperature in some thermotolerant actinomycetes.

Growth and pigmentation of nine thermotolerant Streptomyces strains. Thermotolerant actinomycete strains were cultured on K2 agar medium at 30 °C and 45 °C for 5 days. The 57 thermotolerant actinomycete strains tested in this study can be divided into three groups according to the pigmentation after cultivation. The three strains shown in a belong to the first group, which showed no change in pigmentation between the 30 °C and 45 °C cultures (including no pigmentation in both cultures). The three strains shown in b belong to the second group, which produced more pigment(s) at 45 °C than at 30 °C. The three strains shown in c belong to the third group, which produced more pigment(s) at 30 °C than at 45 °C
Analysis of metabolites produced by thermotolerant actinomycetes
We examined the effect of high-temperature culture (heat shock condition) on the production of secondary metabolites. For this purpose, the 57 thermotolerant actinomycetes were cultured for 5 days at both 30 °C and 45 °C in KG liquid medium as described in “Materials and methods.” Culture broths were extracted with 1-butanol, and each extract was analyzed by LC/MS. We compared the metabolic profiles of each strain between the 30 °C and 45 °C cultures. The results indicate that almost half of the 57 thermotolerant actinomycetes produced some compounds only in 45 °C culture. This result suggests that high-temperature culture induces the production of dormant secondary metabolites in certain actinomycetes; we named these compounds as HSMs.
In order to examine the HSM production in more detail, we randomly selected 18 strains from the 57 thermotolerant actinomycete strains and cultured them in six different media (KG medium, V medium, A3M medium, M medium, A11M medium, and T medium, see “Materials and methods” for the compositions of each medium) at 30 °C and 45 °C for 5 days, and screened for HSMs (Fig. 2a, b), (the peak highlighted in red). We summarized the number of metabolites of the 18 strains that were quantitatively changed by high-temperature culture in each of the six media in Table S1, and plotted the secondary metabolites based on their m/z and retention time in LC/MS in Fig. S2. As shown in Table S1, 16 of 18 strains produced an HSM in at least one of the six media. Furthermore, we found that these strains produced 0–17 HSMs per strain (average 7.3), while 0–34 metabolites per strain (average 16.4) disappeared in the 45 °C culture, when cultured in six different media. This indicates that high-temperature culture should cause the switching of some metabolic pathways. On the other hand, there was only one strain whose metabolic profile was not affected by the high-temperature culture in all tested media (strain JS33, Table S1(r)). In addition, Fig. S2 shows that the molecular weights of HSMs were relatively small (200–400), while the molecular weights of the compounds that disappeared in the 45 °C culture were relatively large (over 400). The causes of this tendency are unknown.

Analysis of the metabolites produced by two thermotolerant Streptomyces strains. Two thermotolerant actinomycetes were cultured in six different liquid media for 5 days at 30 °C and 45 °C from which 1-butanol extracts were analyzed by LC/MS. a PDA chromatograms of the crude extract of Streptomyces sp. HN66. b PDA chromatograms of the crude extract of Streptomyces sp. JA29. This figure shows a number of representative photodiode array (PDA) chromatograms. The peaks highlighted in red indicate heat shock metabolites (HSMs). It should be noted that many HSMs were produced only in a specific medium
Identification of heat shock metabolites (HSMs) produced by thermotolerant actinomycetes
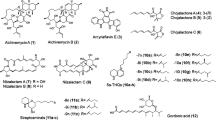
An attempt was made to identify HSMs produced by thermotolerant Streptomyces sp. CA40, as this strain produced seven HSMs (Fig. S3a). A time course experiment showed that an HSM production was the highest in A3M medium for 2 days. Therefore, strain CA40 was cultured in A3M for 2 days, and metabolites were extracted with acetone and ethyl acetate (EtOAc) from whole culture broth, followed by an analysis of the metabolic profile by LC/MS. A comparison with the LC/MS elution profile of metabolites obtained in the 30 °C culture, showed seven HSMs with similar UV spectra. Therefore, these HSMs were isolated by silica gel column chromatography and subsequent preparative HPLC monitoring UV spectra. Finally, we obtained CA40-HSM-A (12.7 mg), C (0.9 mg), E (2.0 mg), F (0.3 mg), and G (0.1 mg). By 1H NMR analyses (Tables S2–6), CA40-HSM-A, C, E, F, and G were identified as phenylacetic acid [16], PD 116,779 (2,5-dihydroxytetrangomycin) [17], tetrangomycin [18], dehydrorabelomycin [19], and tetrangulol [19], respectively; with the exception of phenylacetic acid, all are angucycline-related compounds (Fig. S3b). The yields of CA40-HSM-B and D were too low to determine their structures by NMR analysis. However, a database search based on their UV and MS spectra predicted that CA40-HSM-B and D were identical to gaudimycin B and gaudimycin A, respectively, both of which are also angucycline-related compounds [20]. Interestingly, gaudimycin B and gaudimycin A were originally isolated as artificial products from S. lividans TK24 harboring two different cryptic angucycline gene clusters [21].
HSMs produced by another thermotolerant Streptomyces strain HN66 were isolated by silica gel column chromatography and preparative HPLC by monitoring their UV spectra from which we obtained HN66-HSM-A (2.0 mg), B (2.7 mg), and C (2.7 mg). Using their 1H and 13C NMR results and a relevant database search, HN66-HSM-A, B, and C were identified as tetracenomycin D [22], resistoflavin [23], and resistomycin [24], respectively, all of which are angucycline-related compounds (Fig. S4b).
Jadomycin B is one of the most studied angucycline antibiotics, and the synthesis of jadomycin B was reported to be triggered by heat shock [12]. Jadomycin B binds directly to the pseudo gamma-butyrolactone receptor, ScbR2, and regulates various transcription cascades [25]. Thus, angucycline-type compounds produced as HSMs may also function as signaling molecules that regulate various transcription cascades to maintain homeostasis under heat stress. Further studies are necessary to reveal the function of these HSMs in thermotolerant actinomycetes.
A new natural product, murecholamide, produced by thermotolerant Streptomyces sp. AY2
A database search based on UV and MS spectra of HSMs predicted that the HSMs produced by thermotolerant Streptomyces sp. AY2 contained an unknown compound, which was latter named murecholamide after its structure and the name of the town (Mure-cho) where the producing strain was collected (Fig. S5a). A time course experiment showed that an HSM production was highest in YMPD medium after a 3-day incubation using a shaking (Sakaguchi) flask. The flasks were shaken on a reciprocal shaker (120 rpm). Strain AY2 was cultured under these conditions, and then, the whole culture broth was extracted with acetone and 1-butanol. The extract was applied to silica gel and LH-20 column chromatography to produce murecholamide as a white powder. High-resolution ESITOF mass spectrometry suggested a molecular formula of C31H55N2O6 ([M + H]+ m/z = 551.4059). 1H and 13C NMR data in combination with the HMQC analysis revealed the presence of 31 carbons attributable to: two amides, two quaternary carbons, fourteen sp3 methylenes, nine sp3 methines (three of which are oxygenated), and four methyl groups.
The full planar structure of murecholamide was assigned through interpretation of 1D and 2D NMR spectroscopic data recorded in methanol-d4 (Table 1). A detailed analysis of the 1H–1H COSY spectrum revealed three partial proton–proton connectivities: (x) from H-1 to H-9, H-11 to H-12 and H-14, (y) from H-15 to H-17 and H-20 to H-23, (z) from H-25 to H-29.
Interpretation of the HMBC NMR data allowed the 1H–1H COSY-defined fragments of murecholamide to be connected as shown in Fig. 3a. The linkage of the partial structures between (x) and (y) was established by the HMBC correlations from H-18 to C-12, C-13, C-14, and C-17, and H-14 to C-16. On the basis of these HMBC correlations, C and D rings were formed. Furthermore, A and B rings were also formed by the HMBC correlations from H-19 to C-1, C5, C-9, and C-10. Thus, a 6-6-6-5-ring system was constructed. The partial structure (z) and the correlations from H-29 to C-30 and H-31 to C-30 formed a chain structure with an acetoamide terminus. This chain and the 6-6-6-5-ring system were connected through an amide linkage by HMBC correlations from H-23 to C-24 and H-25 to C-24. The molecular formula of murecholamide indicated three protons and one oxygen are left to be assigned, but these three protons could be assigned to the hydroxy protons at C-3 (δ 72.9), C-7 (δ 69.0), and C-12 (δ 74.0). Here, we noticed that the moiety from NH-H-25 to H-31 is the intermediate of a desferrioxamine analog [26] (Fig. 3c). In the desferrioxamine analog, the nitrogen atoms of the amides are hydroxylated. The methylene protons bonding to the hydroxylated amide (δ 3.60) were reported to show a slightly larger chemical shift value than those bonding to a usual amide group (δ 3.16) [27]. The chemical shift value of the methylene protons at C-29 (δ 3.56) is larger than that of the methylene protons at C-25 (δ 3.12), which indicates that the nitrogen atom of the terminal amide in murecholamide is hydroxylated.

Structure determination of murecholamide. a COSY (bold) and key HMBC (arrow) correlations for murecholamide. b Key NOESY (arrow) correlations for murecholamide. c The structure of desferrioxamine B
The A/B, B/C, and C/D ring junctions were deduced to be cis, trans, and trans, respectively, from the following NOESY correlations; CH3-19 (δ 0.88)–H-5 (δ 1.32) and H-6ax (δ 1.95) for A/B, H-9 (δ 2.22)–H-14 (δ 1.94) and H-6ax (δ 1.95)–H-8 (δ 1.51) for B/C, and H-8 (δ 1.51)–CH3-18 (δ 0.67) for C/D. The NOESY correlation between CH3-21 (δ 0.99) and CH3-18 (δ 0.67) assigned the pseudoequatorial orientation of the side chain at C-17. The axial orientation of the hydroxyl group at C-7 was substantiated by the NOESY correlation between H-7 (δ 3.76) and H-8 (δ 1.51) and by the inferred coupling constant of 2.9 Hz between H-7 and H-8. Also, the equatorial orientation of the hydroxyl group at C-3 was established by the NOESY correlation between H-2eq (δ 1.53) and H-3ax (δ 3.33). The axial orientation of the hydroxyl group at C-12 was confirmed by the NOESY correlation between H-12 (δ 3.91) and CH3-18 (δ 0.67). Finally, considering (i) the NOESY correlation of CH3-21 (δ 0.99) with CH3-18 (δ 0.67) and H-12 (δ 3.91), (ii) that of CH2-16 (δ 1.26) with CH2-22 (δ 1.74, 1.80), and (iii) the most stable conformer resulting from the steric effect caused by CH3-18, the configuration of C-20 was assigned as shown in Fig. 3b. As shown in Fig. 3b, the relative configurations of murecholamide were the same as common bile acids, such as glycocholic acid and taurocholic acid, produced from cholic acid. In particular, the optical rotation of murecholamide \((\left[\alpha\right]^{22}_{\mathrm{D}}\,=+17)\) was almost equal to that of glycocholic acid \((\left[\alpha\right]^{20}_{\mathrm{D}}\,=+26 \,\,{\mathrm{to}} + \!30)\) [28]. Therefore, it is estimated that the absolute stereochemistry of murecholamide is the same as glycocholic acid.
The bile acids described above are synthesized from cholic acid and an amide-containing compound such as glycine or taurine. In contrast, murecholamide is predicted to be synthesized from cholic acid and the intermediate of a desferrioxamine analog. Though the degradation of cholic acid in actinomycetes has been reported [29], little is known about the secondary metabolites derived from cholic acid.
Biological activities of murecholamide
A wound healing assay was used to assess the biological activity of murecholamide, and it was found to have inhibitory activity against cancer cell migration. As shown in Fig. 4, murecholamide weakly inhibited cell migration of EC17 cells at 30 μg ml−1, and completely inhibited at 100 μg ml−1, as judged from the residual area between the cells migrating from the edges of the scratch. However, murecholamide did not show cytotoxicity or growth inhibition after 72 h toward EC17 cells, up to 100 μg ml−1 (data not shown).

Effect of murecholamide on migration in EC17 cells. Wounding was introduced into a confluent culture of EC17 cells, as described in “Materials and methods.” The cells were incubated with various concentrations of murecholamide. MK571 (30 μM) which is an antagonist of cysteinyl leukotriene receptor 1 (CysLT1), that is a molecular target for antimetastatic activity, was used as a positive control
Conclusion
Natural products have great potential as they lead in drug development as well as being useful tools in chemical biology research; therefore, activation of secondary metabolism that is in a dormant state is expected to provide new metabolites for drug development or chemical biology. In this study, we found that high-temperature culture causing heat shock activates secondary metabolism of thermotolerant actinomycetes, and produced several HSMs. HSMs included angucycline-related compounds that were reported only as artificial products of Streptomyces and a new compound, murecholamide. Thus, high-temperature culture using thermotolerant actinomycetes is a useful and easy method for the activation of dormant secondary metabolism. We believe that high-temperature culture causing heat shock will provide potent secondary metabolites, and develop natural products chemistry.
Materials and methods
Bacterial strains
Actinomycete strains were isolated from soil samples collected in Japan and stored in 20% glycerol solution at −80 °C before use. Strain AY2 was isolated from a soil sample collected in Mure Town, Kagawa, Japan, and identified as a member of the genus Streptomyces on the basis of 100% similarity in the 16S rRNA gene sequence (1392 nucleotides; GenBank accession number LC511768) to Streptomyces sp. R8-11 (accession number AB841019) and Streptomyces coeruleorubidus strain KSRO2 (accession number JF682781).
Media and culture conditions
All actinomycete strains were cultured on a K2 agar medium [soluble starch 0.4%, yeast extract 0.4%, malt extract 1%, and agar 2%] and subsequently inoculated into 500-ml Erlenmeyer flasks containing 100 ml of KG seed medium [glucose 2.5%, soybean meal 1.5%, yeast extract 2%, and CaCO3 0.4% (pH 7.2)]. The flasks were shaken on a rotary shaker (180 rpm) at 30 °C for 2 days. Seed cultures (3 ml) were transferred into 500-ml Erlenmeyer flasks each containing 100 ml of each production medium [1. KG: (see above), 2. V: glucose 2%, glycerol 2%, pharmamedia (Archer Daniels Midland Company, Ltd.) 1%, NaCl 0.5%, MnCl2・4H2O grain, Toast Sawyer 1.2%, CaCO3 0.3% (pH 7.4), 3. A3M: glucose 0.5%, glycerol 2%, soluble starch 2%, pharmamedia 1.5%, yeast extract 0.3%, HP-20 (Mitsubishi Chemical Co., Japan) 1% (pH 7.0), 4. M: glucose 2%, dextrin 2%, polypeptone 1%, yeast extract 0.3%, (NH4)2SO4 0.2%, CaCO3 0.2% (pH 7.4), 5. T: glucose 5%, polypeptone 0.4%, yeast extract 0.1%, meat extract 0.1%, NaCl 0.25%, soybean meal 1%, CaCO3 0.2% (pH 7.0), 6. A11M: glucose 0.2%, soluble starch 2.5%, yeast extract 0.5%, polypeptone 0.5%, NZ-amine 0.5%, CaCO3 0.3% (pH 7.0), 7. YMPD: yeast extract 0.2%, meat extract 0.2%, batch peptone 0.4%, NaCl 0.5%, MgSO4・7H2O 0.2%, glucose 1% (pH 7.2)]. The flasks were shaken on a rotary shaker (180 rpm) at 30 °C and 45 °C for 5 days.
Extraction
At the end of the fermentation period, 100 ml of acetone was added to each flask, and the mixtures were left to stand for 1 h. The mixture was transferred into a 15 ml tube, and centrifuged at 3500 rpm for 5 min. The supernatant (1 ml) was evaporated, and the aqueous layer (500 μl) extracted by 1-butanol or EtOAc (500 μl). Evaporation of the organic layer produced a crude extract which was solved by methanol (200 μl). This sample was then analyzed by liquid chromatography (LC)–photodiode array (PDA)–mass spectrometry (MS) system (Waters) with a photodiode array detector (2996) and mass analyzer (Micromass ZQ; Waters) using a reversed-phase C18 column (MGII, 250 × 20 mm ID) (SHISEIDO Co., Japan).
General experimental procedures
Optical rotation was measured using a JASCO P-1020 polarimeter (JASCO Co., Japan). UV spectrum was recorded on a Beckman DU530 UV/VIS spectrophotometer (Beckman coulter, Inc., California, USA). IR spectrum was recorded on a JASCO RT/IR-4200 instrument (JASCO Co., Japan). NMR spectra were obtained on a JEOL JNM-ECA500 spectrometer (JEOL Ltd., Japan). High-resolution (HR)-ESI-time of flight (TOF) MS was recorded on a Bruker micrOTOF mass spectrometer (Bruker Daltonics Inc., Massachusetts, USA). Silica gel column chromatography was performed with Silica gel 60 0.040–0.063 mm (Merck, Darmstadt, Germany). Gel filtration chromatography was performed with Sephadex LH-20 (GE Healthcare, Marlborough, USA).
Murecholamide
Evaporation of 1-butanol gave 1.9 g of crude extract from 5 l of culture, which was used for silica gel column chromatography with a step gradient of CHCl3–MeOH (100:1, 100:5, 100:10, 100:30, and 100:50 v/v). Fraction 4 (100:30) was concentrated to provide 0.17 g of sample, which was then fractionated by Sephadex LH-20 column chromatography with MeOH to yield murecholamide (6.4 mg).
Murecholamide: white powder; \(\left[\alpha\right]^{22}_{\mathrm{D}} \,+ \,17\) (c 0.1, MeOH); UV (MeOH) end absorption only; IR (ATR) νmax 3418, 1651, 1019 cm−1; 1H and 13C NMR data, see Table 1; HR-ESITOFMS [M + H]+ 551.4059 (calcd for C31H55N2O6, 551.4060).
Cell culture
Human esophageal cancer EC17 cells were grown in RPMI 1640 medium supplemented with 5% fetal bovine serum (FBS), and maintained in a 37 °C incubator with 5% CO2.
Wound healing assay
EC17 cells were plated at 6 × 104 cells/well in 48-well plates and incubated in RPMI 1640 medium supplemented with 5% FBS for 24 h [30]. The confluent monolayer of cells was scratched with a micropipette tip to create a cell-free zone in each well, about 1 mm in width. The original medium was replaced by one containing 1% FBS in the presence of test compound. After 24 h, cells were observed under a microscope and photographs were taken.
References
Dictionary of natural products on DVD, version 23.1. London, UK: Chapman & Hall/CRC;2014.
Ohnishi Y, et al. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol. 2008;190:4050–60.
Bentley SD, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature. 2002;417:141–7.
Ikeda H, Kazuo SY, Omura S. Genome mining of the Streptomyces avermitilis genome and development of genome-minimized hosts for heterologous expression of biosynthetic gene clusters. J Ind Microbiol Biotechnol. 2014;41:233–50.
van Keulen G, Dyson PJ. Production of specialized metabolites by Streptomyces coelicolor A3(2). Adv Appl Microbiol. 2014;89:217–66.
Katz L, Baltz RH. Natural product discovery: past, present, and future. J Ind Microbiol Biotechnol. 2016;43:155–76.
Ochi K, et al. Ribosome engineering and secondary metabolite production. Adv Appl Microbiol. 2004;56:155–84.
Onaka H. Novel antibiotic screening methods to awaken silent or cryptic secondary metabolic pathways in actinomycetes. J Antibiot. 2017;70:865–70.
Onaka H, Mori Y, Igarashi Y, Furumai T. Mycolic acid-containing bacteria induce natural-product biosynthesis in Streptomyces species. Appl Environ Microbiol. 2011;77:400–6.
Asamizu S, Ozaki T, Teramoto K, Satoh K, Onaka H. Killing of mycolic acid-containing bacteria aborted induction of antibiotic production by Streptomyces in combined-culture. PLoS ONE. 2015;10:e0142372.
Keskar SS, Srinivasan MC, Deshpande VV. Chemical modification of a xylanase from a thermotolerant Streptomyces. Evidence for essential tryptophan and cysteine residues at the active site. Biochem J. 1989;261:49–55.
Doull JL, Ayer SW, Singh AK, Thibault P. Production of a novel polyketide antibiotic, jadomycin B, by Streptomyces venezuelae following heat shock. J Antibiot. 1993;46:869–71.
Wei ZH, Wu H, Bai L, Deng Z, Zhong JJ. Temperature shift-induced reactive oxygen species enhanced validamycin A production in fermentation of Streptomyces hygroscopicus 5008. Bioprocess Biosyst Eng. 2012;35:1309–16.
Akiyama H, Oku N, Harunari E, Panbangred W, Igarashi Y. Complete NMR assignment and absolute configuration of k4610422, a norditerpenoid inhibitor of testosterone-5α-reductase originally from Streptosporangium: rediscovery from a thermophilic Actinomadura. J Antibiot. 2019. https://doi.org/10.1038/s41429-019-0231-7.
Teta R, et al. Thermoactinoamide A, an antibiotic lipophilic cyclopeptide from the Icelandic thermophilic bacterium Thermoactinomyces vulgaris. J Nat Prod. 2017;80:2530–5.
Hwang BK, Lim SW, Kim BS, Lee JY, Moon SS. Isolation and in vivo and in vitro antifungal activity of phenylacetic acid and sodium phenylacetate from Streptomyces humidus. Appl Environ Chem. 2001;67:3739–45.
Kern DL, Schaumberg JP, Hokanson GC, French JC. PD 116,779, a new antitumor antibiotic of the benz[a]anthraquinone class. J Antibiot. 1986;39:469–70.
Kesenheimer C, Kalogerakis A, Meissner A, Groth U. The cobalt way to angucyclinones: asymmetric total synthesis of the antibiotics (+)-rubiginone B2, (−)-tetrangomycin, and (−)-8-O-methyltetrangomycin. Chemistry. 2010;16:8805–21.
Yamashita N, Harada T, Shin-ya K, Seto H. 6-Hydroxytetrangulol, a new CPP32 protease inducer produced by Streptomyces sp. J Antibiot. 1998;51:79–81.
Kallio P, Liu Z, Mäntsälä P, Niemi J, Metsä-Ketelä M. Sequential action of two flavoenzymes, PgaE and PgaM, in angucycline biosynthesis: chemoenzymatic synthesis of gaudimycin C. Chem Biol. 2008;15:157–66.
Palmu K, Ishida K, Mäntsälä P, Hertweck C, Metsä-Ketelä M. Artificial reconstruction of two cryptic angucycline antibiotic biosynthetic pathways. Chembiochem. 2007;8:1577–84.
Rohr J, et al. Metabolic products of microorganisms. 249. Tetracenomycins B3 and D3, key intermediates of the elloramycin and tetracenomycin C biosynthesis. J Antibiot. 1988;41:1066–73.
Gorajana A, et al. Resistoflavine, cytotoxic compound from a marine actinomycete, Streptomyces chibaensis AUBN1/7. Microbiol Res. 2007;162:322–7.
Kock I, Maskey RP, Biabani MA, Helmke E, Laatsch H. 1-Hydroxy-1-norresistomycin and resistoflavin methyl ether: new antibiotics from marine-derived streptomycetes. J Antibiot. 2005;58:530–4.
Wang W, et al. Angucyclines as signals modulate the behaviors of Streptomyces coelicolor. Proc Natl Acad Sci USA. 2014;111:5688–93.
Barona-Gómez F, Wong U, Giannakopulos AE, Derrick PJ, Challis GL. Identification of a cluster of genes that directs desferrioxamine biosynthesis in Streptomyces coelicolor M145. J Am Chem Soc. 2004;126:16282–3.
Lee HS, et al. Cyclic peptides of the nocardamine class from a marine-derived bacterium of the genus Streptomyces. J Nat Prod. 2005;68:623–5.
Tokyo Chemical Industry Co., Ltd., Japan. https://www.tcichemicals.com/ja/jp/index.html (Accessed on Sep 29, 2019).
Drzyzga O, Fernández de las Heras L, Morales V, Navarro Llorens JM, Perera J. Cholesterol degradation by Gordonia cholesterolivorans. Appl Environ Microbiol. 2011;77:4802–10.
Nakae K, et al. Migrastatin, a new inhibitor of tumor cell migration from Streptomyces sp. MK929-43F1. Taxonomy, fermentation, isolation and biological activities. J Antibiot. 2000;53:1130–6.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research in Innovative Areas (Grant JP17H06401 to MI) from The Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Saito, S., Kato, W., Ikeda, H. et al. Discovery of “heat shock metabolites” produced by thermotolerant actinomycetes in high-temperature culture. J Antibiot 73, 203–210 (2020). https://doi.org/10.1038/s41429-020-0279-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-020-0279-4
- Springer Japan KK
This article is cited by
-
Winners of the 2021 JA Ōmura Awards for excellence
The Journal of Antibiotics (2023)
-
Heat stress enhanced perylenequinones biosynthesis of Shiraia sp. Slf14(w) through nitric oxide formation
Applied Microbiology and Biotechnology (2023)
-
Noaoxazole, a new heat shock metabolite produced by thermotolerant Streptomyces sp. HR41
The Journal of Antibiotics (2022)