
Abstract
The double helix of a polysaccharide, xanthan was unwound by heating (denaturation) and rewound by cooling (renaturation) in acidic (0.01 M HCl) and basic (0.01 M NaOH) solutions. Circular dichroism spectra of xanthan renatured in the acidic and basic solutions were similar to those of native xanthan, which suggests that the local helical structures of the renatured xanthan were recovered by renaturation. The changes in the molar mass and intrinsic viscosity of xanthan induced by renaturation in the acidic solution can be explained by the renaturation scheme in the neutral solution, as reported previously. (Matsuda et al. Polym. J. 41, 526-532, 2009). Both the molar mass and radius of gyration of xanthan in the basic solution were decreased by denaturation and renaturation, and the molar mass dependence of the radius of gyration was similar to that of native xanthan. This change can be explained by the formation of a hairpin structure during renaturation in the basic solutions.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Some natural polysaccharides form helical structures in solutions, which determine their global structures and solution properties such as viscosity because the helical structures cause high rigidity of the polymer chains. Because helical structures can be unwound at high temperature or in solvents disturbing the hydrogen bonds formed by the helices, the stability of helical polysaccharides critically influences their solution properties.
Xanthan is one of the most important polysaccharides used as a viscosity enhancer [1]. The high viscosity of xanthan solutions is caused by the rigid main chains of xanthan with a persistence length of 100–120 nm [2,3,4] in aqueous solution at neutral pH, and the double helical structure of xanthan contributes to the rigidity of the main chains. It is known that the double helices can be unwound in aqueous solutions with low ionic strengths at high temperature (denaturation) and rewound in solutions with high ionic strengths at low temperature (renaturation) [5,6,7,8,9,10,11,12,13]. The double helical structure of xanthan is formed by hydrogen bonds. Xanthan has carboxy groups in its side chains, and the electrostatic repulsive interaction between the ionized carboxy groups destabilizes the helical structure in solution with low ionic strength.
There have been many studies on the thermal denaturation and renaturation of xanthan in neutral solution. Liu et al. [5, 6] proposed a partially unwound double helix with single coils on both ends based on the results of molar mass, radius of gyration, and intrinsic viscosity of xanthan at 80 °C. Capron et al. [7] carried out low angle light scattering measurements and viscometry, and proposed a model that a double helix of xanthan dissociates into two single coils at 110 °C, and forms a hairpin loop structure at 25 °C. On the other hand, Oviatt and Brant [8] argued for the formation of aggregates of renatured xanthan from the results of viscoelastic measurements. Kawakami et al. [9] carried out sedimentation equilibrium measurements and found the formation of both single coils and aggregates in the denaturation and renaturation process. These experimental results suggest very complicated structures for the denatured and renatured xanthan.
We investigated the complicated structural changes in xanthan induced by the denaturation and renaturation mainly with circular dichroism (CD) spectroscopy and size exclusion chromatography online multi angle light scattering (SEC-MALS) measurements [10]. The CD spectrum of the denatured xanthan was significantly different from that of native xanthan, but the CD spectrum of the renatured xanthan was very similar to that of native xanthan. Both the molar mass and radius of gyration determined by SEC-MALS increased after denaturation and renaturation in solutions at high xanthan concentrations, but the increase in radius of gyration was small compared with the increase in molar mass. The molar mass of xanthan denatured and renatured at low xanthan concentrations was approximately half that of native xanthan.
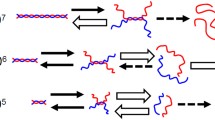
Based on the experimental results described above, a model as shown in Fig. 1 was proposed to describe the structural changes of xanthan. The coils in Fig. 1 are colored only to explain this figure, and all the coils are equivalent to each other. After denaturation at high xanthan concentrations, double helices are only partially unwound (Fig. 1b). Renaturation induces interdimer rewinding (between the blue and purple coils, and between the red and green coils), and the formation of branched aggregates, as shown in Fig. 1c. On the other hand, after denaturation at low xanthan concentrations, double helices are dissociated into two free coils (Fig. 1d). These coils produce two helices with a hairpin structure during renaturation (Fig. 1e). Atomic force microscopy of the renatured xanthan samples supported the model shown in Fig. 1 [11].

Schematic images of native, denatured, and renatured xanthan proposed in our previous paper [10]
We also investigated the molar mass dependence of the denaturation and renaturation behaviors of xanthan [12]. The ratios of molar mass, radius of gyration, and intrinsic viscosity for xanthan renatured in 10 g L−1 solutions divided by those for native xanthan were lower for xanthan samples with higher molar masses. This molar mass dependence can be explained by the lower mobility and lower number concentration of chain ends of the xanthan chain for the xanthan samples with higher molar masses. To form aggregates as shown in Fig. 1c, unwound coils belonging to different dimers such as the blue and purple coils or red and green coils in Fig. 1b need to be rewound at renaturation. Because xanthan dimers in samples with higher molar mass have a lower diffusion coefficient in solution and fewer chain ends, there is less chance for xanthan dimers to meet another dimer. Instead of rewinding coils belonging to different dimers, unwound coils were rewound between coils belonging to the same dimers, such as the blue and red coils or purple and green coils in Fig. 1b, and xanthan recovered the original double helical structure, as shown in Fig. 1a.
When xanthan was renatured in 1.0 g L−1 solution, the weight average molar mass of renatured sample Mw,re divided by that of native sample Mw,na was approximately 0.5 around Mw,na ~ 1 × 106 g mol−1, but Mw,re/Mw,na was higher than 0.5 for the samples with other Mw,na. During denaturation, the double helix is unwound from both ends, and divided into two coils. However, it may be difficult for high molar mass samples to dissociate into free coils completely because of its long double helix. Therefore, the partially wound double helix produced during denaturation is renatured into the original double helix, and Mw,re/Mw,na was ~ 1.0 for Mw,na > 106 g mol−1. On the other hand, low molar mass samples can be completely dissociated into two single coils during denaturation. In this case, the xanthan single chain can produce a hairpin structure (Fig. 1e) after renaturation at low xanthan concentrations. However, it may be unstable for samples at low molar masses to form the hairpin structure, because of the semi-rigid nature of the xanthan single chain. According to Liu et al. [5, 6], the single coil of xanthan can be expressed with the wormlike chain with a persistence length of 21 nm, which may inhibit the formation of the hairpin structure. This is the reason why xanthan coils did not form the hairpin structure but recovered the double helical structure for xanthan samples at Mw,na < 106 g mol−1 (Fig. 1d→a).
All the studies mentioned above are for xanthan in aqueous solution in a neutral state, but xanthan is also used in acidic or basic solutions. There have been a few papers on native xanthan in acidic or basic solutions. Zhang et al. [14] carried out static light scattering and intrinsic viscosity measurements for xanthan at 25 °C in aqueous solution containing 0.01 M HCl and 0.01 M NaCl. The authors fitted the molar mass dependence of the radius of gyration and intrinsic viscosity [η] with the wormlike chain model using 1900 nm−1 of molar mass per contour length ML, 68 ± 7 nm of persistence length q, and 2.0 ± 0.6 nm of bead diameter d. Causse et al. [15] dissolved xanthan in aqueous solution at pH 2.7 and increased the pH by adding aqueous solution of NaOH. The authors reported the CD spectrum of xanthan at pH 3 had a positive peak at approximately 210 nm with ellipticity less than that at neutral pH.
Because xanthan is used as a viscosity enhancer for foods that can be acidic or basic, changes in the structure and viscosity of xanthan in acidic and basic solutions are important for the industrial application of xanthan. In this study, the thermal denaturation, and renaturation behaviors of xanthan in aqueous solutions containing HCl or NaOH were investigated mainly by static light scattering, intrinsic viscosity, SEC-MALS measurements, and CD spectroscopy.
Experimental procedure
Sample preparation
The xanthan samples used in our previous study [12, 13] were also used in this study. The sample with the highest molar mass was purchased from Tokyo Chemical Industry Co., Ltd., Japan, and was used without decreasing the molar mass. The two other samples were kindly provided by Prof. Takahiro Sato. These samples were prepared by sonication, purification, fractionation, and neutralization. The detailed purification method for xanthan was described in their previous paper [2]. Before the preparation of solutions for the measurements, all the samples were dissolved in 0.01 M NaCl aqueous solution, neutralized with dilute aqueous HCl and NaOH, and precipitated in acetone. After the precipitate was washed with acetone containing 5 vol% water the residual salt was removed. After the sample was washed with pure acetone to remove residual water, it was dried under vacuum. The experimental data for the high molar mass sample purchased from Tokyo Chemical Industry are described in the following sections unless otherwise explicitly. Water was purified with a Millipore Direct-Q (Merck KGaA, Darmstadt, Germany). Other chemical reagents were purchased from FUJIFILM Wako Chemicals.
Because xanthan did not dissolve directly in 0.01 M HCl completely, acidic solutions of xanthan were prepared as follows. The dried xanthan sample was first dissolved in purified water, or 0.02 M NaCl aqueous solution. After the solutions were stirred overnight, an equal volume of 0.02 M HCl aqueous solution was added to obtain xanthan solution at pH 2. To prepare “native” xanthan solutions, dried samples were dissolved in 0.02 M NaCl aqueous solution and stirred overnight, then an equal volume of 0.02 M HCl aqueous solution was added to the xanthan solution. “Denatured” xanthan solution for CD spectroscopy was prepared by dissolving a dried xanthan sample in pure water, then the same volume of 0.02 M HCl aqueous solution was added to the xanthan solution. “Renatured” xanthan solutions were prepared as follows. First, the dried xanthan samples were dissolved in pure water to adjust the xanthan concentration to 2.0 or 10.0 g L−1, then an equal volume of 0.02 M HCl aqueous solution was added to the xanthan solution. The solutions were heated at 80 °C for 1 h by immersing the solutions in a water bath where the temperature was controlled at 80 °C. After heating, solid NaCl was added to the solutions to adjust its concentration to 0.01 M. After the solutions were stirred at room temperature for 24 h, they were diluted with 0.01 M HCl aqueous solution containing 0.01 M NaCl when the concentration was too high for the subsequent measurements. The NaCl concentration was fixed at 0.01 M to compare the data with those obtained by Zhang et al. [14].
Xanthan dissolved in 0.01 M NaOH aqueous solution, completely. To prepare “native” xanthan solutions, the dried samples were dissolved in 0.01 M NaOH aqueous solution containing 0.1 M NaCl and the solutions were stirred overnight. “Denatured” xanthan solution for CD spectroscopy was prepared by dissolving a dried xanthan sample directly in 0.01 M NaOH aqueous solution. “Renatured” xanthan solutions were prepared as follows. First, dried xanthan samples were dissolved in 0.01 M NaOH aqueous solution to adjust the xanthan concentrations to 1.0 or 10.0 g L−1. The solutions were heated at 80 °C for 1 h in a water bath. After heating, NaCl was added to the solutions to fix its concentration to 0.1 M. After the solutions were stirred at room temperature for 24 h, they were diluted with 0.01 M NaOH aqueous solution containing 0.1 M NaCl when the concentration was too high for the subsequent measurements.
Circular dichroism spectroscopy
CD spectroscopy at 25 °C, and 80 °C was carried out with a JASCO J-720WO spectropolarimeter [12] for the acidic solutions, and a JASCO J-820KS for the basic solutions. The spectra for native and renatured xanthan were measured at 25 °C while that of denatured xanthan was measured at 80 °C.
Intrinsic viscosity measurement
A four-bulb spiral capillary viscometer was used to determine the intrinsic viscosities [η] of the samples at 25 °C. Because the solution viscosity of xanthan samples with high molar masses depended on the shear rate, it was measured with a four-bulb spiral capillary viscometer. Detailed descriptions of our viscometer and the measurement conditions were reported in our previous paper [12].
Static light scattering measurement
Light scattering measurements at 25 °C were carried out with the same instrument used for neutral solutions in our previous study [12]. Debye plots were used for the sample with the highest molar mass because the radius of gyration of this sample was too large to determine the intercept and slope of the Berry plot. For the other samples, a Berry plot was adopted to calculate the weight average molar mass Mw and z-average radius of gyration <S2 > z1/2.
Size exclusion chromatography online multi angle light scattering
Size exclusion chromatography online multi angle light scattering (SEC-MALS) measurements were carried out to investigate changes in the molar mass and radius of gyration for xanthan samples renatured in the basic solutions. Because basic solvents at pH 12 were unavailable for our SEC-MALS instrument, the basic solutions were neutralized before injection into the SEC column, and analyzed using 0.1 M NaCl aqueous solution as the eluent at a flow rate of 0.5 ml/min at 40 °C. The xanthan solutions were neutralized with 0.01 M HCl aqueous solution, and then diluted to 0.1 M NaCl aqueous solution to decrease xanthan concentration to ~0.25 g/L for the SEC analyses. One guard column (TSKguardcolumn PWXL, Tosoh Co., Japan) and two analytical SEC columns (TSKgel GMPWXL, Tosoh Co., Japan) with an exclusion limit molecular weight of 107 for dextran were connected in series. The MALS detector and differential refractometer (RI) detector were a DAWN 8 manufactured by Wyatt, and a RI2031 manufactured by JASCO, respectively. A 60 mW laser with a wavelength of 658 nm was used as the light source.
Specific refractive index increment measurements
The specific refractive index increment for aqueous xanthan solutions in 0.01 M HCl was determined to be 0.137 cm3 g−1 at the wavelength used in this study (633 nm). Zhang et al. [14] reported the specific index increments at wavelengths of 436 and 546 nm, and the specific index increments at the wavelength used in this study were extrapolated from these data. The specific refractive index increment in the basic solution was determined with the same instruments and measurement conditions used in our previous study [16]. The solution was dialyzed in a dialyzer with two compartments containing the xanthan solution and solvent divided with a Visking cellulose membrane with a cutoff molecular weight of 1.2–1.4 × 104 [17]. The solution and solvent in the dialyzer were stirred, and the dialyzer was stored in an NRB-14A (Nihon Freezer) incubator at 25.0 °C until equilibrium. The refractive index increment of xanthan in the basic solution was determined with the above dialyzed solutions to be 0.139 cm3 g−1 at 633 nm and 25.0 °C.
Results and discussion
Denaturation and renaturation of xanthan in 0.01 M HCl
Figure 2 shows the CD spectra for the xanthan samples in 0.01 M HCl aqueous solution at 25 °C before denaturation (native xanthan), at 80 °C (denatured xanthan), and at 25 °C after renaturation (renatured xanthan). [θ] indicates specific ellipticity. The CD spectrum for denatured xanthan in 0.01 M HCl was different from that of native xanthan. The peak wavelength for the positive band at approximately 200 nm for native xanthan was slightly shifted to approximately 205 nm, and the peak strength of this band decreased after renaturation. Causse et al. [15] also reported similar changes in the CD spectrum obtained in the xanthan solution at pH 3. These results suggest that the double helix was unwound in 0.01 M HCl. The CD spectrum after renaturation was indistinguishable from that of native xanthan except for slight changes in the peak strength, as reported previously [10].

CD spectra of native, renatured, and renatured xanthan in 0.01 M HCl aqueous solution. The temperature for native and renatured xanthan was controlled at 25 °C, and that for denatured xanthan was controlled at 80 °C
Figure 3 shows a typical result of viscosities at low shear rates for native xanthan in 0.01 M HCl aqueous solution, which reflects the dimension of xanthan in solutions; (a) shear rate dependence of ηsp/c; (b) concentration dependence of viscosity at a zero shear rate. The viscosity at zero shear rate estimated in Fig. 3a was extrapolated to determine the intrinsic viscosity in Fig. 3b, whereas ηsp, ηr and c represent the specific viscosity, relative viscosity, and xanthan concentration, respectively. The data points in Fig. 3a were vertically shifted with α, as described in the figure, to show the data points without overlapping.

A typical result of viscosity measurements to estimate intrinsic viscosity. (native xanthan in 0.01 M HCl). a Shear rate dependence, and (b) concentration dependence of viscosity at zero shear rate. The data points in (a) were shifted vertically with α shown in the figure
The molar mass of xanthan was obtained from the results of static light scattering measurements. Figure 4 is a typical result of static light scattering for native xanthan in 0.01 M HCl aqueous solution, whereas Rθ, and k represent excess Rayleigh ratio and the magnitude of scattering vector, respectively. The data shown in the open circles were extrapolated to zero concentration as shown in the filled circles. The Debye plots shown in the solid curves were used to estimate the molar masses for the samples with high molar masses.

A typical result of light scattering measurements to estimate the weight average molar mass and radius of gyration. (native xanthan in 0.01 M HCl). The data shown in the open circles were extrapolated to zero concentration shown in the filled circles
The molar mass dependence of the intrinsic viscosity in 0.01 M HCl aqueous solution is summarized in Fig. 5. The circles indicate the data for native xanthan. The triangles and squares are the data for xanthan renatured from the xanthan samples shown in circles with the same colors. The xanthan concentrations of the samples shown in triangles and squares are 1.0 g L−1 and 5.0 g L−1, respectively. The data shown in purple circles are the data for native xanthan obtained by Zhang et al. [14]. The solid curve was a theoretical curve calculated with the wormlike chain model to fit our data for native xanthan in this study and those for native xanthan reported by Zhang et al. [14]. The parameters used to calculate the theoretical curve were q = 61 nm, ML = 1900 nm−1, and d = 1.6 nm. All the data were distributed around the solid curve, which suggests that both native and renatured xanthan in this study formed the normal double helical structure reported by Zhang et al. [14]. The slight deviation of the data for the renatured samples from the solid curve was ascribed to partially branched aggregates, as shown in Fig. 1c or bending in the double helix. Because the rigidity of the xanthan chains is caused by the formation of the local helical structure, a few defects in the helix can effectively decrease the persistence length and intrinsic viscosity.

Molar mass dependence of the intrinsic viscosity of xanthan in 0.01 M HCl aqueous solution. The data shown in purple circles are obtained by Zhang et al. [14] for native samples. The triangles and squares are the data for xanthan renatured from the xanthan samples shown in the circles with the same colors. The solid curve was calculated with the wormlike chain model using q = 61 nm, ML = 1900 nm−1, and d = 1.6 nm
The weight average molar masses of the native xanthan (Mw,na) and renatured xanthan (Mw,re) were summarized as shown in Table 1. Here, the subscripts “na” and “re” denote the native and renatured samples, respectively. The xanthan concentrations at renaturation are also listed in cxan in Table 1. The molar mass ratios of renatured xanthan and native xanthan (Mw,re/Mw,na) are listed in Table 1. The Mw,re/Mw,na data at 1.0 g L−1 in 0.01 M aqueous HCl agree well with those for renatured xanthan in the neutral solution at 1.0 g L−1 (0.01 M NaCl), as reported previously (Mw,re/Mw,na = 0.839–0.955) [10]. These agreements strongly suggest that a similar structure was produced during renaturation in both solvents. On the other hand, the Mw,re/Mw,na values for renatured xanthan at 5.0 g L−1 ranged between 1.04 and 1.35. These values were higher than those for renatured xanthan both in 0.01 M HCl and in 0.01 M NaCl at 1.0 g L−1. These differences may be ascribed to the formation of the branched aggregates (Fig. 1c) after renaturation.
Denaturation and renaturation of xanthan in 0.01 M NaOH
Figure 6 shows the CD spectra of native, denatured, and renatured xanthan in 0.01 M NaOH aqueous solutions. The CD spectra significantly changed during the denaturation process. The peak wavelength for the negative CD band at 80 °C (denatured xanthan) was shifted from 225 nm for native xanthan to 215 nm, and the peak strength increased. These changes strongly suggest that the double helix of xanthan changed in denaturation at 80 °C in 0.01 M NaOH, differing from denaturation in 0.01 M HCl, as shown in Fig. 2. On the other hand, the CD spectrum at 25 °C after renaturation was similar to that for native xanthan. These results suggest that the xanthan chains recovered the double helical structure, after renaturation. However, the CD signal may be affected by the side chain conformation as noted by Tomofuji et al., [18] so that structured aggregates, such as hairpin-like double helices (Fig. 1e) and branched aggregates (Fig. 1c), cannot be identified only from CD spectra.

CD spectra of native, renatured, and renatured xanthan in 0.01 M NaOH aqueous solution. The temperature for native and renatured xanthan was controlled at 25 °C, and that for denatured xanthan was controlled at 80 °C
Figure 7 shows the elution curves and molar masses obtained by SEC-MALS measurements for xanthan renatured at 1 g L−1 and 10 g L−1 which were once dissolved in the basic solution, and then neutralized before injection into the SEC columns. The xanthan concentration was calculated from the signals of the RI detector and specific refractive index increment, and the molar mass was calculated from the signals of the MALS detectors and the xanthan concentration.

Elution curves and molar masses obtained from SEC-MALS measurements for xanthan renatured at (a) 1 g L−1 and (b) 10 g L−1. The SEC-MALS data were obtained at 40 °C in 0.1 M NaCl as an eluent after neutralization with 0.01 HCl
The weight average molar masses of the xanthan samples that were dissolved in 0.01 M NaOH aqueous solution, and then neutralized before the injection into the SEC columns are summarized in Table 1. It should be noted that the Mw of xanthan renatured in the basic solution was almost half of that of native xanthan, although xanthan with similar Mw (~5×106 g mol−1 in the native state) did not significantly decrease in Mw during the renaturation in acidic and neutral [13] solution because the long double helix prevented dissociation into two coils.
Figure 8 summarizes the molar mass dependence of the radius of gyration for the xanthan samples dissolved in the basic solutions and neutralized before the SEC measurements. The solid curve was calculated with the wormlike chain model using q = 100 nm, and ML = 1940 nm−1. The values of these parameters were essentially the same for those of native xanthan in the neutral solution reported by Sato et al. [4]. The solid curve fits the data points of all the samples measured in this study. Because the radii of gyration of branched polymers are smaller than those of the corresponding linear polymers, the results in Fig. 8 suggest the absence of branched structures of renatured xanthan as illustrated in Fig. 1c. The CD spectra of renatured xanthan suggest the absence of single coils without forming helices.

Molar mass dependence of the radius of gyration of xanthan, which was dissolved in 0.01 M NaOH solution, and subsequently neutralized before the injection to the SEC columns. The solid curve was calculated from the wormlike chain model using q = 100 nm, and ML = 1900 nm−1
As shown in Table 1, the Mw of the renatured xanthan samples was slightly lower than half of the Mw of the native sample. Because the dissociation of the double helices into two coils halves the Mw of native xanthan, the decrease in the Mw for renatured xanthan suggests the dissociation of the double helices into two coils. The experimental results showing that the CD spectra and molar mass dependence of the radius of gyration of renatured xanthan were very similar to those of native xanthan, suggest that the dissociated xanthan formed a hairpin structure as illustrated in Fig. 1e. The slightly smaller Mw than half of the native xanthan might be caused by the scission of the main chains at high temperature in the basic solution.
Denaturation and renaturation schemes for xanthan in aqueous solutions under acidic and basic conditions
Based on the experimental results described above, schematic images of renatured xanthan in acidic and basic solutions are proposed as illustrated in Fig. 9. In 0.01 M aqueous HCl solution, the double helical structure (a) of native xanthan was unwound during denaturation, and mainly recovered the original structure (b), but partially formed double helices with defects (c), branched aggregates (d) or hairpin structure (e). As shown in Fig. 5, some renatured samples showed no significant changes in molar mass, but their intrinsic viscosity was lower than that of native xanthan with the same molar mass. These changes in molar mass and intrinsic viscosity can be explained by the formation of double helices with defects as shown in Fig. 9c. The molar mass of some other renatured samples increased during the renaturation. The formation of the branched aggregates, as shown in Fig. 9d, increases the molar mass, but the increase in their intrinsic viscosity is insignificant due to their branching structure. The decrease in the molar mass of renatured xanthan induced by the renaturation can be explained by the formation of hairpin structure, as shown in Fig. 9e. The structures as illustrated in (b)-(e) contain helices and agree with the experimental result that CD spectra of renatured xanthan were virtually the same as that of native xanthan.

Schematic images of native, renatured, and heated xanthan in 0.01 M HCl and 0.01 M NaOH solutions
In 0.01 M NaOH aqueous solution, even the double helices of xanthan with a high molar mass dissociated into coils and formed a hairpin structure during renaturation, as shown in Fig. 9g. The dissociation of the double helix into two coils during the denaturation and the formation of the hairpin structure during the renaturation were enhanced in the basic solution. The molar mass after renaturation was slightly lower than half of the native sample because the main chains were partially decomposed by heating in alkali solution.
Conclusion
Structural changes induced by thermal denaturation and renaturation for xanthan in both acidic (0.01 M HCl) and basic (0.01 M NaOH) solutions were investigated. The CD spectra of renatured xanthan were very similar to those of native xanthan in both acidic and basic solutions, which suggests the recovery of the local helical structure for renatured xanthan. The molar masses of xanthan renatured in the acidic solutions increased or decreased depending on the molar masses of native xanthan and xanthan concentration but changes in molar mass were not significant compared with those in neutral solutions. The intrinsic viscosity of renatured xanthan was lower than that of native xanthan. These experimental results could be explained by the partial formation of defects in the double helices.
The molar mass of xanthan renatured in the basic solution was slightly lower than half that of the native sample. The molar mass dependence of the radius of gyration was similar to that in neutral solution. These results suggest the formation of a hairpin structure formed by dissociated coils partially decomposed by heating.
These experimental results suggested that the structural changes in xanthan induced by thermal denaturation and renaturation in aqueous solutions depend on the pH of the solution. The present results are summarized as follows. In the acidic solution, the xanthan double helix is dissociated by denaturation at 80 °C, and the subsequent renaturation recovered the double helical structure, which includes irregular components, such as the double helix with defects. At high xanthan concentrations, the double helices were linked to each other to produce branched aggregates during renaturation. At finite xanthan concentrations, these aggregates are produced competitively during renaturation. In the basic solution, the xanthan double helix was dissociated into single chains in denaturation at 80 °C, and the subsequent renaturation mainly produced the hairpin-like structure. At low xanthan concentrations and high molar masses, the hairpin structure becomes the major component during renaturation in the basic solution.
References
Norisuye T, Teramoto A. In Polymeric Materials Encyclopedia (ed. Salamone, JC) 8801-9 (RC Press, Boca Raton, USA, 1996).
Sato T, Norisuye T, Fujita H. Double-stranded helix of xanthan in dilute solution: evidence from light scattering. Polym J. 1984;16:341–50.
Sato T, Kojima S, Norisuye T, Fujita H. Double-stranded Helix of Xanthan in dilute solution: further evidence. Polym J. 1984;16:423–9.
Sato T, Norisuye T, Fujita H. Double-stranded Helix of Xanthan in dilute solution: dimensional and hydrodynamic properties in 0.1 M aqueous sodium chloride. Macromolecules. 1984;17:2696–2700.
Liu W, Sato T, Norisuye T, Fujita H. Thermally induced conformational change of Xanthan in 0.01 M aqueous sodium chloride. Carbohyd Res. 1987;160:267–81.
Liu W, Norisuye T. Order-disorder conformation change of Xanthan in 0.01 M aqueous sodium chloride: dimensional behavior. Biopolymers. 1988;27:1641–54.
Capron G, Brigand G, Muller G. About the native and renatured conformation of xanthan exopolysaccharide. Polymer. 1997;38:5289–95.
Oviatt HW Jr, Brant DA. Viscoelastic behavior of thermally treated aqueous Xanthan solutions in the semidilute concentration regime. Macromolecules. 1994;27:2402–8.
Kawakami K, Okabe Y, Norisuye T. Dissociation of dimerized xanthan in aqueous solution. Carbohydr Polym. 1990;14:189–203.
Matsuda Y, Bjiyajima Y, Sato T. Thermal denaturation, renaturation, and aggregation of a double-helical polysaccharide Xanthan in aqueous solutions. Polym J. 2009;41:526–32.
Matsuda Y, Sugiura F, Mays JW, Tasaka S. Atomic force microscopy of thermally renatured Xanthan with low molar mass. Polym J. 2015;47:282–5.
Matsuda Y, Sugiura F, Okumura KW, Tasaka S. Renaturation behavior of Xanthan with high molar mass and wide molar mass distribution. Polym J. 2016;48:653–8.
Matsuda Y, Okumura KW, Tasaka S. Molar mass dependence of structure of Xanthan thermally denatured and renatured in dilute solution. Polym J. 2018;50:1043–9.
Zhang L, Liu W, Norisuye T, Fujita H. Double-stranded helix of xanthan: Rigidity in 0.01M aqueous sodium chloride containing 0.01 N hydrochloric acid. Biopolymers. 1987;26:333–41.
Causse B, Spadini L, Sarret G, Faure A, Travelet C, Madern D, et al. Xanthan exopolysaccharide: Cu2+ complexes affected from the ph-dependent conformational state; implications for environmentally relevant biopolymers. Envir Sci Tech 2016;50:3477–85.
Matsuda Y, Ashizawa H, Fukui T, Akao R, Tasaka S. Fabrication of polymer structure among fibrous structure of poly(lactic acid) gel and improvement of physical properties. Macromol Chem Phys. 2018;219:1700317.
Sato T, Norisuye T, Fujita H. Triple helix of Schizophyllum commune polysaccharide in dilute solution. 5. Light scattering and refractometry in mixtures of water and dimethyl sulfoxide. Macromolecules. 1983;16:185–9.
Tomofuji F, Matsuo K, Terao K. Kinetics of denaturation and renaturation processes of double-stranded helical polysaccharide, xanthan in aqueous sodium chloride. Carbohydr Polym. 2022;275:118681.
Acknowledgements
This work was partially supported by MEXT Promotion of Distinctive Joint Research Center Program Grant Number# JPMXP 0621467946. The analysis of the samples was partly carried out with a JASCO J-720KS at the Instrumentation Center, The University of Kitakyushu. We thank Professor Takahiro Sato at the Open University of Japan who kindly provided the xanthan samples and the dialyzer and, allowed us to use the CD instrument. This work was supported by JSPS KAKENHI Grant Number 19H02773.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Matsuda, Y., Saiki, R., Sato, K. et al. Thermal denaturation and renaturation of a double-helical polysaccharide xanthan in acidic and basic solutions. Polym J 56, 629–637 (2024). https://doi.org/10.1038/s41428-024-00892-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-024-00892-y
- Springer Nature Limited