
Abstract
Several relaxation modes present in poly(propylene glycol) monobutyl ether [PPGMBE] were explored over a wide temperature range (120–310 K) and frequency band range (10 mHz–1.0 GHz) using wideband dielectric spectroscopy. Two resolvable secondary relaxations (β- and γ-processes) were observed below Tg, along with a structural relaxation mode (α-) above Tg. The β-relaxation process was well fitted with the Cole–Cole equation and was identified as true Johari Goldstein relaxation based on an aging experiment and a coupling model prediction. The spectra for the γ-process showed asymmetric behavior below Tg, hence it was well described by the Havriliak-Negami equation and did not show any of the peculiar behavior seen with many PPG families. The T-dependences of several dielectric properties, e.g., τα, τβ, τγ, τσ, and σdc, were thoroughly investigated. It was noticed that the T-variation of the relaxation time for the α-mode was not satisfactorily described with only one VFT equation, and the derivative plot indicated a crossover temperature of ~240 K. Our experimental findings demonstrated that a logarithmic plot of the σdc versus τα data showed nonlinear dependency and thus were expressed by the fractional Stokes-Einstein–Debye law, with a change in the exponent from ζ = −0.70 to −0.85 for high-T (>240 K) and low-T behavior (<240 K), respectively.

Broadband dielectric spectroscopy was employed to study the relaxation dynamics in poly(propylene glycol) monobutyl ether [PPGMBE]. Two relaxation processes (β- and γ-) were observed in the sub-Tg region, in addition to the α-process above Tg. The physical aging experiment performed below Tg suggested that the observed β-process is the genuine Johari Goldstein relaxation process and the γ-process is of intramolecular origin.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Relaxation dynamics in polymers have been studied extensively with various experimental tools, such as Brillouin light scattering [1,2,3], NMR techniques [4, 5], mechanical spectroscopy [6, 7], photon correlation spectroscopy (PCS) [8], calorimetry [9, 10] and dielectric spectroscopy (DS) [10,11,12,13,14,15,16,17,18,19,20]. Among various experimental tools, DS has proven to be the most valuable tool for investigating the relaxation behaviors of glass-forming materials and polymers because of its sensitivity and broad frequency range [20]. As with molecular glasses formed by supercooling monomeric liquids, the dielectric responses in polymers are generally dominated by the structural relaxation process commonly known as the α-process, for which the relaxation time increases considerably on approaching the glass transition temperature (Tg). Due to the very strong T-dependence of the time constant for α-relaxation, it displays non-Arrhenius behavior and is hence well expressed by the Vogel–Fulcher–Tammann (VFT) expression: [21,22,23]
In Eq. (1), T0 is the temperature at which the time constant of the glass forming system diverges, A is a pre-exponential constant on the logarithmic scale, and B is an activation parameter. The shapes of the absorption spectra for this α-process exhibit non-Debye characteristics except for monohydroxy alcohols [24, 25].
In addition to the primary relaxation (α-relaxation), additional relaxation processes usually referred to as secondary processes (β- and γ-) were observed in absorption curves at the higher frequency side relative to the main relaxation process (α-process). This process is more pronounced below Tg, and the average time constant follows an Arrhenius-type temperature dependence. The β-process generally arises in molecular glass formers, which have distinct chemical structures and unique properties in contrast to the structural relaxation (or α-) process. In polymeric glass formers, secondary relaxation is caused by restricted motion of the polymer backbone [26], but the exact mechanism is not well understood. In molecular glass formers, both inter- and intramolecular relaxation modes were detected below Tg. The faster intramolecular relaxation processes are not related to the α-process, hence the phenomenon of a glass transition. Another type of intermolecular secondary relaxation is caused by small angle reorientation of the whole molecule in a cage produced by neighboring molecules and is present even in systems with perfectly rigid molecular structures, as identified by Johari and Goldstein [27, 28]. Because of the strong connection to primary relaxation, the intermolecular process usually known as Johari-Goldstein (JG) relaxation [27, 28] and appears to operate universally in all glass-forming materials, including polymers [12, 29], molecular liquids [30], and plastic crystals [31, 32]. In many glass-forming materials, it has been observed that above Tg, the average time constants for α- and JG β- processes are frequently merged at the temperature TB at which the mean time constant of the α- process is ~100 ns [33, 34]. Identification of intermolecular or JG relaxation is a challenging task; therefore, extensive research has been carried out in recent years [12, 13, 29,30,31,32, 35, 36] to show that the βJG–process truly exists in all types of glasses formed by supercooling monomeric liquids or polymers. Several criteria, such as pressure sensitivity and physical aging [12, 13], have been used to identify this process. Aside from the pressure sensitivity of the process, concurrence of the time constant for the JG process and the primitive relaxation time anticipated by the coupling model given by Ngai and coresearchers [35, 37,38,39,39] is another rigorous criterion universally agreed upon for identifying the βJG–process. For example, Casalini and Roland [40] discovered that under the influence of high pressure, an extra secondary relaxation process developed toward the high-frequency side of the structural α-process in the absorption spectrum of tri-propylene glycol (3PG). The time constant for this newly discovered relaxation process was consistent with the calculated primitive time constant, which suggested that the relaxation was a true JG- process [37]. The same behavior was also noticed for the absorption curves of di-propylene glycol (2PG) under the influence of pressure. However, in 2PG, the excess wing was not so prominent [29]. Such characteristics of the secondary process for poly(propylene glycols) [PPGs] might be explained by its substantial dependence on hydrogen bonds produced predominantly by terminal hydroxyl groups [41].
In this context, we have chosen a derivative of poly(propylene glycol) [PPG] in which one terminal hydrogen atom has been replaced by a butyl group, poly(propylene glycol) monobutyl ether (PPGMBE). The present article is devoted to a thorough analysis of different relaxation processes observed for poly(propylene glycol) monobutyl ether, Mw = 340 g/mol, over broad temperature and frequency ranges at atmospheric pressure. The goal of this study is to show how relaxation processes are affected if one terminal hydrogen atom has been replaced by one butyl group in poly(propylene glycol) and to reveal the omnipresent JG or β-relaxation, which has not been clearly identified in poly(propylene glycol) derivatives of almost similar molecular weight. In addition, we would also like to clarify the anomalous behavior of the average time constant for the γ-relaxation process near and above Tg in the poly(propylene glycol) family.
Experimental section
Poly(propylene glycol) monobutyl ether, Mn = 340 g/mol (PPGMBE, purity ≥99.6%), was obtained from Sigma‒Aldrich Co., USA and used as received.
To measure ε′ and ε′′, two dielectric setups were used to cover the wide range of frequencies (ν) from 1 mHz to 1 GHz. For the frequency range (ν) 1 mHz–10 MHz, a Novocontrol alpha analyzer, a broadband dielectric spectrometer was used, and for a higher frequency range, viz. 1 MHz–1 GHz, a radio frequency impedance analyzer 4192B from Agilent was used. For the lower range frequency measurements, the polymer sample was kept between two gold-polished parallel circular electrodes with diameters of 30 mm. In the higher frequency range, the polymer sample was placed between two gold-polished parallel circular electrodes with diameters of 10 mm. In both cases, Teflon strips with 50 μm widths were used to specify the thickness of the polymer sample. Before studying the sample with a high-frequency analyzer, basic calibration was performed by employing the terminators as open, short, 50 Ω resistance and low-loss air capacitors. Each terminator was calibrated automatically by using the Novocontrol software. Isothermal frequency sweeps were carried out during heating from 120 K to 310 K with increments of 5 K after cooling the polymer sample at a rate of 10 K/min. The temperature of the polymer sample was controlled during this measurement to within ±0.1 K. It is worth mentioning that the amplitudes of the dielectric results obtained using the high-frequency analyzer differed significantly from the amplitudes of the dielectric results recorded using a low-frequency analyzer at the same temperature. Therefore, the amplitudes of those high-frequency dielectric results have been adjusted to the lower-frequency dielectric findings based on the overlapping frequency range. Similar criteria have been applied earlier [30] by several researchers working with glass-forming materials.
Results
To show the operation of various relaxation modes, Fig. 1 displays the T-dependence of real (ε′) and imaginary (ε′′) parts of the complex permittivity at a test frequency ν = 1.15 Hz. Apart from the well-resolved α-relaxation process, two secondary relaxation modes, the β- and γ-mode, below Tg can be identified in the temperature-dependent loss spectra, which are marked by arrows in decreasing temperature order. The spectral dependence of complex permittivities such as ε′ and ε′′ of the main (or α) relaxation process over a wide range of frequencies at various temperatures is depicted in Fig. 2. The relaxation strength of the structural α-process clearly diminished with increasing temperature, demonstrating the inverse temperature dependence. This was consistent with the dielectric behaviors of most glass-forming materials. Additionally, a rapid enhancement in the dielectric absorption curves [Fig. 2(b)] was observed toward the lower frequency side, which was ascribed to the pure contribution of dc conductivity, which showed inverse frequency dependency. This rise toward the low-frequency regions was not observed for the ε′ curves of the complex dielectric permittivity shown in Fig. 2(a), which implied that the conductivity was entirely ohmic [20]. For the data obtained in this study, the primary relaxation process (α-process) was analyzed by using the Havriliak–Negami function (HN) [42], which is given by:
where ε∞ is the high-frequency dielectric constant, Δε is the strength of the relaxation process, τ is the dielectric relaxation time constant, σdc is the dc-conductivity, and αHN, γHN are the HN-shape parameters, which indicate the asymmetry and broadening of the dielectric absorption curve with αHN γHN ≤ 1, αHN > 0.

T dependency of (ε′) and (ε′′) for poly(propylene glycol) monobutyl ether [PPGMBE] at ν = 1.15 kHz. The vertical arrows represent the positions of α-, β- and γ-relaxation processes. The left-hand y-axis corresponds to the ε′ curve shown with the solid black square symbols, while the right-hand y-axis is related to the ε′′ curve represented by the solid red circle symbols. The molecular structure of PPGMBE is also depicted in this figure

Frequency variation of the complex permittivity a ε′ and b ε′′ for supercooled PPGMBE at various temperatures. The data are presented for 5 K steps between 185 K and 225 K and for 10 K steps between 235 K and 295 K in both panels
The showcase example for fitting the primary relaxation mode is displayed in Fig. 3 for T = 195 K. A deviation can be seen in the fitting curve for the experimentally observed data on the high-frequency side. The deviations seen on the high-frequency side are commonly observed for low molecular weight glass-forming materials as well as polymers [43]. The presence of secondary relaxation modes below Tg may also contribute to these high-frequency variations, as identified in the inset of Fig. 3 for T = 195 K, where two secondary relaxation processes (β- and γ-process) along with the contribution of the primary relaxation process and dc-conductivity can be identified. Figure 4 displays the frequency-dependent absorption spectra for secondary relaxation processes at different temperatures between 120 and 165 K. Two clearly resolved dielectric secondary relaxation modes (β- and γ-process) can be seen in the dielectric absorption curves depicted in Fig. 4. From this figure, one can identify a gradual rise in the dielectric loss spectra with increasing temperature toward the glass transition point (Tg). Most likely, this is due to the presence of a high-frequency tail for the α-relaxation mode, which is active at these temperatures but at considerably lower frequencies. The β-relaxation mode was fitted by the Cole–Cole (CC) equation, which was achieved by setting the shape parameter γHN = 1 in Eq. (2), while the γ-process was well described with the HN- function below Tg. We also tried to fit the γ-process with the Cole-Cole equation, but the fits were poor. Representative examples for fitting of secondary relaxation processes at 120 K are depicted in Fig. 5. This demonstrates that utilizing the HN equation yielded better fitting results for the γ relaxation mode than the CC function in the glassy state of PPGMBE, that is, below Tg. Our analysis is consistent with the procedure applied by Grzybowska et al. [13, 44]. for poly(propylene glycol) [PPG400] [13] and tri-propylene glycol (3PG) [44]. Similar asymmetric behavior in the relaxation spectra of the secondary relaxation mode below Tg was also identified for plastic crystalline materials by the present author and his coauthors [31, 32]. The complete activation map for PPGMBE is depicted in Fig. 6, which compares the dielectric time constants for dc-conductivity (σdc), the structural relaxation mode (α), and both secondary (β and γ) relaxation modes. The relaxation times displayed in Fig. 6 for dc-conductivity were calculated by using Eq. (3) [20]:
where ε0 = 8.854 pF/m is the free space dielectric constant, εs is the static dielectric constant obtained from the low-frequency side plateau values of έ curves, and σdc is the dc conductivity. The T-dependent relaxation time due to dc-conductivity was well expressed by the single Vogel–Fulcher–Tammann (VFT) expression shown in Eq. (1), while the temperature (T) variation of the time constants for the structural α-mode was not well described with a single VFT equation because of the 11-decade frequency range. Therefore, two VFT equations have been used to completely express the T-dependence of the time constants for the α-process. This behavior is clearer if we see the T-variation of the relaxation times (τα) for the primary relaxation mode in the form of the Stickel [33, 45] plot, as displayed in Fig. 7. This figure suggests that two different VFT equations are necessary to express the entire T-dependence of the relaxation time constants for the α-process displayed in Fig. 6 with the solid and dashed lines. This also indicates that the time constants for the structural α-process convert from optimal fitting with one VFT equation to another in the vicinity of TB ≈ 140 K. The fitting results for both VFT equations are depicted in Table 1.

Representative dielectric loss spectrum for PPGMBE depicted at a fixed temperature of 195 K. The solid and dashed-dotted lines drawn along this curve represent individual fitting of the structural α-mode and dc-conductivity, respectively. The inset shows frequency-dependent dielectric absorption curves at various temperatures (195 K ≤ T ≤ 295 K) with 20 K steps. The solid line along the 195 K curve corresponds to the combined fits to α-, β-, γ-process, and dc-conductivity, whereas the solid lines along the rest of the curves correspond to the combined fits to α-process and dc-conductivity. The fitting parameters corresponding to the absorption curves drawn at different temperatures are: T = 195 K (α-process: Δε = 6.5, log τ = −2.57, αHN = 0.83, γHN = 0.55, σdc = 1.93 × 10−15 S/cm; β-process: Δε = 0.20, log τ = −6.36, αCC = 0.64; γ-process: Δε = 0.095, log τ = −7.7, αCC = 0.82); T = 215 K (α-process: Δε = 5.49, log τ = −5.53, αHN = 0.78, γHN = 0.60, σdc = 7.43 × 10−13 S/cm); T = 235 K (α-process: Δε = 4.94, log τ = −7.25, αHN = 0.77, γHN = 0.65, σdc = 1.85 × 10−11 S/cm); T = 255 K (α-process: Δε = 4.41, log τ = −8.25, αHN = 0.78, γHN = 0.67, σdc = 1.32 × 10−10 S/cm); T = 275 K (α-process: Δε = 3.73, log τ = −8.94, αHN = 0.79, γHN = 0.76, σdc = 4.49 × 10−10 S/cm); and T = 295 K (α-process: Δε = 3.48, log τ = −9.41, αHN = 0.79, γHN = 0.73, σdc = 9.85 × 10−10 S/cm)

Frequency-dependent dielectric absorption (ε׳׳) curves for glassy PPGMBE at various temperatures in the sub-Tg region. Two relaxation processes, viz. the β- and γ-processes, are clearly visible in the absorption spectra

Comparison of the spectral analyses adopted for the dielectric absorption spectrum of PPGMBE at 120 K. The solid lines drawn along the data points represent combined fits to the dielectric loss curves of the β- and γ-relaxation modes. The β-relaxation mode was clearly fitted by the Cole-Cole equation and is represented by dotted lines in both panels. The dashed-dotted nonlinear curves represent fits to the γ-process. In a, both β- and γ-processes are fitted by a combination of two C-C functions. In b, the γ-relaxation mode is well represented by fitting with the HN equation and the β-process by using the C–C function. Clearly, the HN function gives a better approximation of the peak loss frequency for the γ-process compared to the Cole–Cole function

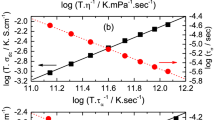
Activation plot for the time constants of different relaxation processes for glassy PPGMBE. Progressing from the slow to the fast process, the relaxation times are for dc-conductivity (τσ), structural relaxation (τα), and two secondary modes (τβ and τγ). The temperature dependence of τσ was well represented by a single VFT equation, while the structural relaxation time (τα) was well estimated by using two VFT expressions (Eq. (1)), both above (dashed line) and below (solid line) TB. The lines drawn along the τβ and τγ symbols represent Arrhenius fits (Eq. 4). The respective fitting parameters are given in Table 1. The star symbols represent τJG (Eq. 10), which was estimated by using the CM criterion above the glass transition temperature (Tg). Error bars along τγ and τβ above Tg represent the estimated uncertainties in the time constants for the γ- and β-processes, respectively

Stickel plot of the average time constants for the structural relaxation mode (τα) of PPGMBE. The solid line toward low-T and dashed line along the high-T data points represent linear fits to the Stickel function. The crossover temperature (TB) region is indicated by an arrow
The temperature dependences of both secondary relaxation peaks, i.e., β and γ, were described with the well-known Arrhenius equation [46] shown in Eq. (4):
where A is a logarithmic preexponential constant factor, R is the gas constant, T is the temperature in degrees K, and E is the activation energy corresponding to the average time constant. The fitting results for the β and γ- relaxation processes are also displayed in Table 1.
Discussion
Primary or α-relaxation process
The structural relaxation process, also known as the α-process, dominates the dielectric responses of polymers, just as it does in molecular glasses formed by supercooling monomeric liquids. As the temperature proceeds toward the glass transition point (Tg), the relaxation time increases dramatically. Similarly, in the present case, the main α-relaxation process commenced above Tg (see Fig. 2), and its average time constants increased by ~11 decades as the temperature decreased (see Fig. 6). It has been observed that the T- dependency of the mean time constants for the structural relaxation modes over 11 decades could not be satisfactorily expressed with only one VFT- equation. Therefore, the data have been replotted in terms of a derivative [45] of the VFT equation, i.e., [d(log10 τα)/d(1/T)]−1/2 versus 1000/T (see Fig. 7). With this method, any VFT dependency of τα would become a linear function with respect to inverse temperature. We found a crossover temperature at ~TB ≈ 240 K, where the conversion of the structural relaxation time (τα) from one VFT dependency to another took place. One important aspect of this crossover temperature was that the fitting parameters (T < TB) gave a more reliable value for the glass transition temperature than the VFT fit, which covered the entire temperature range. Hence, the glass transition temperature (Tg), at which τα = 100 s, was calculated based on VFT parameters (T < TB) with the following expression [47]:
As a result of the calculation with Eq. (5), the glass transition temperature (Tg) was found to be 179.3 K, which was slightly less than the Tg value reported earlier by Singh et al. [10] and other researchers [13] for the case of pure poly(propylene glycol) with an approximately similar molecular weight. Moreover, when the crossover temperature (TB) value is compared to the glass transition temperature (Tg), it is clear that the dynamic crossover occurred at TB = 1.34 Tg, which is very close to those found in various glass-forming materials, i.e., in the range (1.2–1.4) Tg [35]. It is useful to mention that there is no universally accepted explanation for TB; therefore, the authors of research articles often refer to a narrow crossover zone around TB [33, 45]. The changes from using a single VFT equation may also be found by comparing the activation parameters (B) obtained from the high-temperature and low-temperature VFT fits [47]. As the ratio of these activation parameters obtained from the low-temperature and high-temperature VFT fits increases, the deviations from single VFT behavior become progressively pronounced, as in the case of glass-forming materials such as o-terphenyl [20, 35] and salol [35, 48]. In the present case, this ratio (see Table 1) was observed to be close to 1.4; therefore, the deviation from a single VFT dependence appeared to be subtle.
Similar behavior was also analyzed by calculating the temperature dependence of Kohlrausch William Watts (βKWW) parameters, which were obtained by applying the criterion given by Alvarez et al. [49], who showed that if the shape parameters αHN, γHN of the HN function given in Eq. (1) were constrained according to the following expression,
The frequency-domain HN function has a one-to-one connection with the time domain Kohlrausch William Watts (KWW) [50] equation, \(\varphi _{KWW} = \exp \left[ { - \left( {t/\tau _{KWW}} \right)^{\beta _{KWW}}} \right],\) where βKWW and τKWW are non-Debye shape parameters and the characteristic relaxation time, respectively. The temperature dependence of the non-Debye shape parameter (βKWW) is depicted in Fig. 8. In this figure, one can identify a step-like change in the region close to the crossover temperature (TB). Below the crossover temperature (TB), the shape parameter showed a nearly constant value, but it increased gradually with increasing temperature above the crossover temperature (TB). It has been observed by several researchers [35, 47] that glass-forming liquids those have smaller values of non-Debye shape parameter (βKWW) or a higher value of n = 1−βKWW at Tg, show a larger difference between low-T VFT and high-T VFT behavior. The current result supports the coupling model (CM) [35, 37,38,39] explanation for the crossover regions of the glass formers. According to the CM, the T-dependence change of the structural time constant (τα) from the low-T VFT to the high-T VFT dependency at the crossover temperature (TB) was caused by the analogous change in the T-dependence of the non-Debye shape parameter (βKWW). Based on the VFT parameters obtained for (T < TB), the fragility index [51, 52] was also calculated by using the following expression:

T-dependence of the Kohlrausch William Watts (βKWW) parameter for the α-process of PPGMBE. The crossover temperature (TB) is indicated by an arrow
This shows how the slope of the relaxation rates varies at Tg. Generally, the fragility index varies from the lowest reported value of 16 to as high as 191 for strong and highly fragile glasses, respectively. Using Eq. (7), the fragility index (m) of PPGMBE was found to be 73.9. This suggested that PPGMBE shows moderate fragile characteristics similar to those observed by various researchers for pure PPG [10, 13].
Finally, by taking the value of the Kohlrausch William Watts parameter (βKWW) at Tg, the fragility index was also calculated with the following master equation [51]:
As a result of the calculation with Eq. (8), the lowest and highest fragility indices (m) were found to be 107.2 K and 47.2, respectively. The fragility index obtained from Eq. (7) was between the values calculated with Eq. (8). Hence, as with other glass formers, Eq. (8) works well in the case of PPGMBE.
Dielectric spectroscopy is widely used for providing simultaneous measurements of the structural time constant (τα) and dc conductivity (σdc) under the influence of an electric field across a wide frequency range. σdc is usually detected toward the low-frequency regime at a higher temperature, which originates from the translational movement of ions. On the other hand, the rotational movements of dipoles are responsible for the structural relaxation mode. When these two physical quantities are connected, a well-known relationship is established between the T-dependence of τα and σdc for glass-forming liquids, as σdc (T) τα(T) = constant, which is often known as the Stokes–Einstein–Debye relation. However, at the glass transition temperature (Tg), the Stokes–Einstein–Debye relation (SED) frequently fails [53,54,55]. Therefore, to completely describe the experimental results, a fractional SED relation has been widely used and is given by
where ζ defines the fractional exponent, the value of which lies between 0 and 1. To see this relationship, a double logarithmic plot of σdc versus τα for PPGMBE is displayed in Fig. 9. The negative slope of unity is shown by the dashed line. Upon closer inspection of Fig. 9, one notices that Eq. (9) describes the data with a change in the exponent, i.e., ζ = −0.70 and −0.85 above and below the crossover temperature (TB), respectively. These exponent values agree with the values frequently observed by the researcher, which are in the range of 0.70 to 0.91 [53,54,55]. As a consequence, it is possible to deduce that upon approaching Tg, there is an increase in translational ionic motion relative to orientational molecular movements for PPGMBE.

Stokes–Einstein–Debye plot for PPGMBE. The dashed line drawn in the figure has a slope of −1. The logarithmic plot of σdc versus τα data shows a transition at 240 ± 2 K from low-temperature to high-temperature behavior with a change in the exponent, ζ = −0.85 to −0.70
Secondary relaxation processes (β- and γ-process)
In recent years [12, 13, 29,30,31,32, 35,36,37,38,39], it has been shown that a unique category of secondary processes known as JG relaxation modes [27, 28] has essential relevance due to its close relationship with the structural relaxation process (α-process) observed for many common glass formers. This class includes secondary relaxations caused by dispersing a small amount of completely rigid molecules in nonpolar glass-forming materials as well as specialized secondary modes for several other types of glassy materials. The JG relaxation [27, 28], also known as primitive relaxation [37,38,39], was named to make it easier to refer to this unique class of secondary modes. The JG nomenclature also honors these two colleagues [27, 28], who discovered secondary modes in fully rigid glassy materials and dispelled the myth that all secondary modes were caused by mobility of the side groups present in molecular glassy materials. Because of the significant links between the structural α-process and JG-process, many methodologies have been developed for distinguishing the JG-process from other secondary modes unrelated to the structural α-mode. As briefly highlighted in the earlier section, for the case of PPGMBE, we observed two well-resolved secondary modes, viz., γ- and β-relaxations below Tg. The relaxation spectra of the β-process are well expressed by the CC equation over the entire range of temperatures. The origin of this β-process has been evaluated in terms of its Johari-Goldstein (JG) character based on the Coupling Model (CM) [35, 37,38,39]. While the coupling model (CM) cannot account for microscopic details, it can predict the activation pattern of a JG mode, which is considered to be a good indicator of the secondary relaxation process. According to the Coupling Model, for T > Tg, the time constant for the JG relaxation process (τJG) can be predicted based on the time constant for the primary relaxation mode (τα) by using the following relation [35, 37,38,39]:
where τc (= 2 ps) is the primitive time constant and βKWW is the coupling exponent defined by Eq. (6). Using Eq. 10, the prediction for the primitive relaxation time (τJG) above the glass transition temperature (Tg) was indicated with stars in the activation map shown in Fig. 6. The values predicted for τJG by using CM criteria (Eq. (10)) were very close to the values observed for the time constant of the β-process (τβ). The Coupling Model also suggested predicting the energy of activation for JG relaxation below Tg by using the following relation: [35, 37,38,39]
where τg = τα (Tg) = 100 s. It is very well known that below Tg, the JG process exhibits an Arrhenius dependence, while above Tg, it shows non-Arrhenius characteristics. The current results for the β-process follow this behavior, as shown in Fig. 6. However, a slight deviation of the time constant for the β-relaxation mode can be observed in a small region above Tg, where the time constants for the β- and γ-relaxation processes were not properly estimated due to the closely spaced β- and γ-processes. The activation energy estimated with Eq. (11) was 30.7 kJ/mol, which was very close to the Eβ value obtained by fitting the experimental data for the β-process with Eq. (4) (see Table 1). Nevertheless, it is interesting to observe that when extrapolated above the glass transition temperature (Tg), the Arrhenius T-dependence of the τβ value for the β-process intersects with the time constant (τα) of the α-process at a temperature close to TB (see Fig. 6), as is often found with glass-forming systems. As mentioned by Kaminski et al. [12] and Gainaru et al. [57], in poly(propylene glycol) oligomers, the observed β-process is due to the presence of residual water. The reported activation energy of this process was ~6400 K or 54 kJ/mol, which is commonly observed in various aqueous glass-forming polymers [10]. Additionally, they noted that the strength of this process decreased notably with heat treatment. These results suggest that the β-process is not an inherent property of poly(propylene glycols). However, in PPGMBE, the Eβ value for the β-process was estimated to be 27.8 kJ/mol (see Table 1), which is approximately half of the activation energy observed for PPG oligomers. Hence, the influence of water present in the atmosphere was probably negligible in our measurement of the PPGMBE sample. To ensure that the observed β-process for PPGMBE was not influenced by the moisture present in the environment, we heated our fresh polymer PPGMBE sample to 395 K and kept the temperature there for approximately 90 minutes under an N2 gas atmosphere. After cooling the sample, the absorption spectrum was recorded at a temperature far below Tg, i.e., at 120 K. The remeasured absorption spectrum was approximately similar to the spectrum obtained without heat treatment, as shown in Fig. 10(a). This gave us confidence and suggested that the relaxation dynamics we determined for the PPGMBE sample did not include an observable effect from the moisture present in the environment. Furthermore, after recording the dielectric spectrum at 120 K, we kept the sample at this low temperature for approximately 14 hours to look for aging effects. After 14 h of aging at 120 K, we recorded the absorption spectrum. Interestingly, the β-relaxation process was more affected than the γ-relaxation mode, as shown in panel b of Fig. 10. The peak loss frequency for the β-process was shifted approximately 1.08 decades, while the peak location for the γ-process was displaced by ~0.2 decades toward the lower frequency side. The dielectric strengths of both secondary modes also decreased slightly with aging. This was expected because the glass densified spontaneously as its structural integrity was recovered. The groundbreaking investigations of Williams [58] and Johari [59] were among the first to show that densification brought on by aging or pressure application reduced the dielectric strengths of the secondary processes. This behavior, along with the consistency of the above results determined for PPGMBE by using the CM criteria, confirmed that the detected β-relaxation process was intermolecular, that is, a true JG-type relaxation process.

a Comparison of the PPGMBE relaxation spectra recorded at 120 K prior to (red solid circles) and after heat treatment at 295 K in an N2 gas environment (black solid squares). b Comparison of the relaxation spectra for PPGMBE recorded at 120 K after heat treatment at 295 K under an N2 gas environment (black solid squares) and for a sample aged for ~14 h (blue open circles). The vertical arrows in b show the shift of peak loss frequencies before and after aging the sample at 120 K for ~14 h
Now, we focused on the outcomes of the γ-relaxation process, which showed asymmetric behavior below Tg and was hence well expressed with the HN-function (Eq. (2)) [see Fig. 5]. However, around Tg, the γ-mode was fitted properly with the Cole-Cole function (see the inset of Fig. 3 at T = 195 K). This behavior of the γ-mode was consistent with that observed by Grazybowska et al. [13, 29, 44] for di-, tri-, and poly(propylene glycol). Similar characteristics were also observed by Singh et al. [10, 31, 32] for poly(propylene glycol) [10] as well as for several plastic crystalline materials [31, 32]. In PPGMBE, we have not seen any anomalies in the time constant for the γ-process (τγ) above Tg, as observed for di-, tri- and poly(propylene glycol) [13, 29, 44]. This could be due to the presence of clearly resolved β- or JG-processes in PPGMBE, which was not clearly observed with the abovementioned PPG family. This suggests that the unusual behavior of τγ observed for the abovementioned propylene glycol family was caused by encroachment of the time constant for the β-process (τβ) or JG-process at temperatures near and above Tg. Our results support the explanation given by Ngai et al. [56] for the anomalous behavior of the γ-process (τγ) observed with the PPG family. The ratio of the primitive time constant (τJG) to the time constant for the β-process (τβ) at Tg was found to be 2.97, while the (τJG/τγ) ratio was estimated to be 19.30. The high value of (τJG/τγ) suggested that the γ-process observed in PPGMBE was intramolecular in nature, that is, related to reorientational motions of the local groups rather than to JG- or primitive relaxation. Our results are consistent with those observed for poly(propylene glycol) [13, 29, 40] as well as di-propylene glycol [44]. In PPGMBE, the Eγ value of the γ-process was estimated as 19.77 kJ/mole (see Table 1), which is slightly less than those observed for pure poly(propylene glycol) [13, 29]. This suggests that the end group, viz. butyl group, present in PPG influences the average time constant for the γ-relaxation mode. Moreover, the hydrogen bonding capability was reduced by the presence of the butyl group in PPGMBE, and hence, as a result, its activation energy was decreased. The current results support the findings of Grzybowska et al. [44] for di-propylene glycol, who pointed out that the strength of the γ-relaxation mode was suppressed by the decreased ability to form hydrogen bonds, and this process was completely absent for the modified sample, which cannot form hydrogen bonds. They determined that the γ- relaxation mode was either weakly dependent or insensitive to applied pressure. As a result, it was determined that the γ-mode in di-propylene glycol, as well as the whole glycol family, is linked to hydrogen bonding and is intramolecular in nature. According to the present aging experiments on PPGMBE, as depicted in Fig. 10(b), the peak frequency for the γ-relaxation process had a more negligible influence on annealing compared to the β-relaxation process. This behavior is consistent with the effect of applied pressure on γ-relaxation in poly(propylene glycol) oligomers [56, 57] as well as intramolecular relaxation occurring in other glass formers [58, 60]. Kaminski et al. [12] also identified that annealing did not affect the peak frequency of the γ-process in amino-terminated poly(propylene glycol). Hence, based on the above discussion as well as on our current physical aging experiment with PPGMBE, we infer that the detected γ-relaxation mode is intramolecular in nature and may be connected to the reorientation motions of hydrogen-bonded structures.
Summary and conclusion
Poly(propylene glycol) monobutyl ether (PPGMBE) has been thoroughly investigated over a wide temperature (120–310 K) and frequency band (10 mHz–1.0 GHz) using wideband dielectric spectroscopy. In addition to the structural relaxation mode (α-) above Tg, two well-resolved secondary modes (β- and γ-processes) were discovered in the sub-Tg region. The β-relaxation mode was clearly fitted by the Cole-Cole equation and identified as a true Johari-Goldstein (JG) relaxation on the basis of the CM criterion and the current physical aging experiment. The absorption spectra for the γ-process showed asymmetric behavior below Tg, and hence were well described by the Havrialiak–Negami equation and did not show any of the peculiar behavior seen with many PPG families. Our results suggest that the unusual behavior of the relaxation time for the γ-process (τγ) observed with the poly(propylene glycol) family was due to encroachment of the time constant for the β-process (τβ) or JG-process at temperatures near and above Tg. Furthermore, this process was weakly dependent on physical aging, and hence, the nature of this process (γ-process) has been attributed to reorientational motions of the local groups, i.e., the process is of intramolecular origin. The T-dependence of τα was not well described with a single VFT equation over the entire temperature range; hence, two VFT equations have been used. The derivative plot showed a crossover temperature of 240 ± 2 K. Similar behavior was also identified for the temperature-dependent non-Debye shape parameter (βKWW), which showed different slopes below and above the crossover temperature (TB). The present experimental findings revealed that a logarithmic plot of σdc versus τα showed a nonlinear dependence and hence could be characterized by the fractional SED relation with the exponent changing from −0.85 to −0.70 on going from low-T (<240 K) to high-T (>240 K).
References
Wang CH, Fytas G, Lilge D, Dorfmüller T. Laser light beating spectroscopic studies of dynamics in bulk polymers: poly(propylene glycol). Macromolecules. 1981;14:1363–70.
Eckert-Kastner S, Meier G, Alig I. Phase behaviour of mixtures of polyethylene glycol and polypropylene glycol: Influence of hydrogen bond clusters on the phase diagram. Phys Chem Chem Phys. 2002;4:3743–49.
Eckert-Kastner S, Meier G, Alig I. Phase behaviour of mixtures of polyethylene glycol and polypropylene glycol: Influence of hydrogen bond clusters on critical composition fluctuations. Phys Chem Chem Phys. 2003;5:3202–11.
Connor TM, Blears DJ, Allen G. Proton spin-lattice relaxation in polypropylene oxides. Trans Faraday Soc. 1965;61:1097–109.
Vogel M, Torbrugge T. Nonexponential polymer segmental motion in the presence and absence of ions: 2H NMR multitime correlation functions for polymer electrolytes poly(propylene glycol)-LiClO4. J Chem Phys. 2007;126:204902.
Barlow AJ, Erginsav J. Viscoelastic properties of poly(propylene glycols). Polymer. 1975;16:110–14.
Cochrane J, Harrison G, Lamb J, Phillips DW. Creep, creep recovery and dynamic mechanical measurements of a poly(propylene glycol) oligomer. Polymer. 1980;21:837–44.
Smith SW, Freeman BD, Hall CK. Pressure-dependent photon correlation spectroscopic investigation of poly(propylene oxide) near the glass transition. Macromolecules. 1997; 30: 2052–7.
Moon IK, Jeong YH, Furukawa T. Enthalpy and dielectric relaxation in the glass transition region of polypropylene glycol. Thermochim Acta. 2001;377:97–104.
Singh LP, Cerveny S, Alegría A, Colmenero J. Dynamics of water in supercooled aqueous solutions of Poly(propylene glycol) as studied by broadband dielectric spectroscopy and low-temperature FTIR-ATR spectroscopy. J Phys Chem B. 2011;115:13817–27.
Pawlus SS, Hensel-Bielowka S, Grzybowska K, Zioło J, Paluch M. Temperature behavior of secondary relaxation dynamics in tripropylene glycol. Phys Rev B. 2005;71:174107.
Kaminski K, Kipnusu WK, Adrjanowicz K, Mapesa EU, Iacob C, Jasiurkowska M et al. Comparative study on the molecular dynamics of a series of polypropylene glycols. Macromolecules. 2013;46:1973–80.
Grzybowska K, Grzybowski A, Zioło J, Paluch M, Capaccioli S. Dielectric secondary relaxations in polypropylene glycols. J Chem Phys. 2006;125:044904.
Engberg D, Schüller J, Strube B, Sokolov AP, Torell LM. Brillouin scattering and dielectric relaxation in PPG of different chain lengths and end groups. Polymer. 1999;40:4755–61.
Beevers MS, Elliott DA, Williams G. Molecular motion in melt samples of poly (propylene glycol) studied using dielectric and Kerr effect relaxation techniques. Polymer. 1980;21:13–20.
Mattsson J, Bergman R, Jacobsson P, Borjesson L. Chain-length-dependent relaxation scenarios in an oligomeric glass-forming system: from merged to well-separated α and β loss peaks. Phys Rev Lett. 2003;90:075702.
Dyre J, Olsen NB. Minimal model for beta relaxation in viscous liquids. Phys Rev Lett. 2003;91:155703.
Schonhals A, Schlosser E. Relation between main- and normal-mode relaxation. A dielectric study on poly (propyleneoxide). Prog Colloid Polym Sci. 1993;91:158–61.
Schonhals A, Stauga R. Broadband dielectric study of anomalous diffusion in a poly(propylene glycol) melt confined to nanopores. J Chem Phys. 1998;108:5130.
Kremer F, Schonhals A. Eds. Broadband dielectric spectroscopy. Springer: Berlin; 2003.
Vogel H. Das. Temperaturabhaengigkeitsgesetz der Viskositaet von Fluessigkeiten. Phys Z. 1921; 22:645.
Fulcher GS. Analysis of recent measurements of the viscosity of glasses. J Am Ceram Soc. 1925;81:339–55.
Tammann G, Hesse W. Die Abhängigkeit der Viscosität von der Temperatur bie unterkühlten Flüssigkeiten. Z Anorg Allg Chem. 1926;156:245–57.
Johari GP, Danhauser W. Dielectric study of the pressure dependence of intermolecular association in isomeric octyl alcohols. J Chem Phys. 1968;48:5114.
Singh LP, Richert R. Watching hydrogen-bonded structures in an alcohol convert from rings to chains. Phys Rev Lett 2012;109:167802.
Cerveny S, Bergman R, Schwartz GA, Jacobsson P. Dielectric α- and β-relaxations in uncured styrene butadiene rubber. Macromolecules. 2002;35:4337–42.
Johari GP, Goldstein M. Viscous liquids and the glass transition. II. Secondary relaxations in glasses of rigid molecules. J Chem Phys. 1970;53:2372.
Johari GP, Goldstein M. Viscous liquids and the glass transition. III. Secondary relaxations in aliphatic alcohols and other nonrigid molecules. J. Chem. Phys. 1971;55:4245.
Grzybowska K, Pawlus S, Mierzwa M, Paluch M, Ngai KL. Changes of relaxation dynamics of a hydrogen-bonded glass former after removal of the hydrogen bonds. J Chem Phys. 2006;125:144507.
Singh LP, Alba-Simionesco C, Richert R. Dynamics of glass-forming liquids. XVII. Dielectric relaxation and intermolecular association in a series of isomeric octyl alcohols. J Chem Phys. 2013;139:144503.
Singh LP, Murthy SSN. Study of secondary relaxation in disordered plastic crystals of isocyanocyclohexane, cyanocyclohexane, and 1-cyanoadamantane. J Chem Phys. 2008;129:094501.
Singh LP, Murthy SSN. Dielectric and calorimetric investigation of an unusual two-component plastic crystal: cyclohexanol-neopentylglycol. Phys Chem Chem Phys. 2009;11:5110–18.
Hansen C, Stickel F, Berger T, Richert R, Fischer EW. Dynamics of glass-forming liquids. III. Comparing the dielectric α- and β-relaxation of 1-propanol and o-terphenyl. J Chem Phys. 1997;107:1086–93.
Richert R, Angell CA. Dynamics of glass-forming liquids. V. On the link between molecular dynamics and configurational entropy. J Chem Phys. 1998;108:9016–26.
Ngai KL. Relaxation and diffusion in complex systems. Springer: New York; 2011.
Casalini R, Snow AW, Roland CM. Temperature dependence of the Johari–Goldstein relaxation in poly(methyl methacrylate) and poly(thiomethyl methacrylate). Macromolecules. 2013;46:330–34.
Ngai KL, Paluch M. Classification of secondary relaxation in glass-formers based on dynamic properties. J Chem Phys. 2004;120:857.
Ngai KL, Capaccioli S. Relation between the activation energy of the Johari-Goldstein beta relaxation and T(g) of glass formers. Phys Rev E. 2004;69:031501.
Capaccioli S, Ngai KL. Relation between the alpha-relaxation and Johari-Goldstein beta-relaxation of a component in binary miscible mixtures of glass-formers. J Phys Chem B. 2005;109:9727–35.
Casalini R, Roland CM. Pressure evolution of the excess wing in a type-B glass former. Phys Rev Lett. 2003;91:015702.
Leon C, Ngai KL, Roland CM. Relationship between the primary and secondary dielectric relaxation processes in propylene glycol and its oligomers. J Chem Phys. 1999;110:11585.
Havriliak S, Negami SA. Complex plane analysis of α-dispersions in some polymer systems. J Polym Sci Part C. 1966;14:99–117.
Dixon PK, Wu L, Nagel SR, Williams BD, Carini JP. Scaling in the relaxation of supercooled liquids. Phys Rev Lett. 1990;65:1108–11.
Grzybowska K, Grzybowski A, Pawlus S, Hensel-Bielowka S, Paluch M. Dielectric relaxation processes in water mixtures of tripropylene glycol. J Chem Phys. 2005;123:204506.
Stickel F, Fischer EW, Richert R. Dynamics of glass-forming liquids. I. Temperature-derivative analysis of dielectric relaxation data. J Chem Phys. 1995;102:6251–57.
Hill NE, Vaughan WE, Price AH, Davies M. Dielectric properties and molecular behaviour. Van Nostrand Reinhold, London; 1969.
Richert R. Scaling versus Vogel-Fulcher type structural relaxation in deeply supercooled materials. Physica A. 2000;287:26–36.
Paluch M, Casalini R, Roland CM. Cohen-Grest model for the dynamics of supercooled liquids. Phys Rev E. 2003;67:021508.
Alvarez F, Alegria A, Colmenero J. Relationship between the time-domain Kohlrausch-Williams-Watts and frequency-domain Havriliak-Negami relaxation functions. Phys Rev B. 1991;44:7306–12.
Williams G, Watts DC. Non-symmetrical dielectric relaxation behavior arising from a simple empirical decay function. Trans Faraday Soc. 1970;66:80–85.
Bohmer R, Ngai KL, Angell CA, Plazek DJ. Nonexponential relaxations in strong and fragile glass formers. J Chem Phys. 1993;99:4201.
Angell CA. Why C1 = 16–17 in the WLF equation is physical—and the fragility of polymers. Polymer. 1997;38:6261–66.
Roland CM, Hansel-Bielowka S, Paluch M, Casalini R. Supercooled dynamics of glass-forming liquids and polymers under hydrostatic pressure. Rep Prog Phys. 2005;68:1405.
Corezzi S, Campani E, Rolla PA, Fioretto D. Changes in the dynamics of supercooled systems revealed by dielectric spectroscopy. J Chem Phys. 1999;111:9343.
Johari GP, Andersson O. On the nonlinear variation of dc conductivity with dielectric relaxation time. J Chem Phys. 2006;125:124501.
Ngai KL, Pawlus S, Grzybowska K, Kaminski K, Capaccioli S, Paluch M. Does the Johari–Goldstein β-relaxation exist in polypropylene glycols? Macromolecules. 2015;48:4151–57.
Gainaru C, Hiller W, Bohmer R. A dielectric study of Oligo- & poly(propylene glycol). Macromolecules 2010;43:1907–14.
Williams G. Complex dielectric constant of dipolar compounds as a function of temperature, pressure and frequency. Trans Faraday Soc. 1964;60:1548–55.
Johari GP. Effect of annealing on the secondary relaxation in glasses. J Chem Phys. 1982;77:4619–26.
Singh LP, Alegria A, Colmenero J. Broadband dielectric spectroscopy and calorimetric investigations of D-lyxose. Cabohydr Res. 2011;346:2165–72.
Acknowledgements
The author acknowledges Masaryk University, Brno, Czech Republic for a visiting research specialist grant. The author also acknowledges the support of the Central Instrumental Facility of the Department of Material Science and Engineering, IITB, India, and Dr. Priti Yadav for her critical reading and valuable comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Singh, L.P. Primary and secondary relaxation processes in poly(propylene glycol) monobutyl ether: a broadband dielectric spectroscopy investigation. Polym J 55, 141–151 (2023). https://doi.org/10.1038/s41428-022-00728-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-022-00728-7
- Springer Nature Limited