
Abstract
Oligosaccharides and polysaccharides are comprised of complicated chemical structures owing to the structural variation of monosaccharide repeating units and the differences in the regio- and stereo-arrangements of the glycosidic linkages in their saccharide chains. Glucan phosphorylase (GP, EC 2.4.1.1) catalyzes consecutive enzymatic glycosylation in a manner similar to enzymatic polymerization employing α-d-glucose 1-phosphate (Glc-1-P) and maltooligosaccharide as a glycosyl donor and acceptor (or monomer and primer), respectively, to produce a well-defined α(1→4)-glucan polymer, that is, amylose, while liberating inorganic phosphate (Pi). After understanding the principal reaction mechanism and specificity of GP catalysis, the present review focuses on the enzymatic synthesis of unnatural oligosaccharides and polysaccharides linked through strictly controlled α(1→4)-glycosidic linkages by GP catalysis. Due to the weak specificity of the recognition of substrates by GP, unnatural oligosaccharides having different monosaccharide units at the nonreducing end have been precisely obtained by GP-catalyzed glycosylation using analog substrates of Glc-1-P, i.e., nonnative monosaccharide 1-phosphates. Highly branched α(1→4)-glucans have been employed as polymeric glycosyl acceptors and primers for GP-catalyzed enzymatic glycosylation and polymerization to obtain unnatural amphoteric and hydrogel materials. Thermostable GP catalyzes consecutive enzymatic glycosylation using α-d-glucosamine and α-d-mannose 1-phosphates as glycosyl donors. By removing Pi, consecutive reactions were accelerated, and enzymatic polymerization occurred, resulting in the synthesis of several unnatural α(1→4)-linked polysaccharides with well-defined structures.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
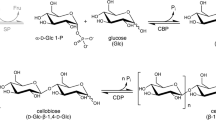
Oligosaccharides and polysaccharides are widely present in nature and play vital roles based on their chemical structures as suppliers of energy, structural materials, and key materials for specific biological and vital functions [1, 2]. Because of the structural variation of monosaccharide units and various regio- and stereo-type glycosidic linkages, oligosaccharides and polysaccharides typically exhibit complicated structures [3]. Therefore, it is well known that a subtle change in the kind of repeating unit and type of glycosidic linkage profoundly affects their properties and functions. Structurally well-defined oligosaccharides and polysaccharides are chemically synthesized by forming regio- and stereocontrolled glycosidic linkages among desired monosaccharide residues. The reaction is called glycosylation, and it occurs between a glycosyl donor and glycosyl acceptor. Accordingly, it has attracted increasing attention in the development of new carbohydrate-based functional materials [4,5,6]. The elongation of saccharide chains is theoretically induced by the repetition of glycosylation using appropriate substrates. However, this approach does not typically provide oligosaccharides and polysaccharides with the desired structure and a sufficient degree of polymerization. This is because it is difficult to perfectly control regio- and stereoselectivities, and complicated procedures are required, including the protection/deprotection of the hydroxy groups in substrates. Compared with the general chemical glycosylation approach, the enzymatic method has been recognized as an efficient approach to synthesize oligosaccharides and polysaccharides with well-defined structures. This is because, even without the protection of the hydroxy groups in substrates, it takes place in a highly regulated fashion in regio- and stereo-glycosidic arrangements according to each enzyme’s specificity [7,8,9,10]. Of the seven main classes of enzymes, glycosyl transferase (GT) and glycosyl hydrolase (GH) have been widely employed as efficient catalysts in the practical preparation of structurally controlled oligosaccharides and polysaccharides by enzymatic glycosylations [11,12,13]. GT is the enzyme that catalyzes the transfer of a sugar moiety from an activated glycosyl donor, typically activated by phosphate esters at the C-1 (anomeric) position, to a glycosyl acceptor to construct a glycosidic linkage (Fig. 1a) [14]. GH catalyzes the hydrolysis of natural oligosaccharides and polysaccharides under ambient conditions in aqueous media (Fig. 1b) [15]. When the formation of a glycosyl–enzyme complex is realized in the reaction using an activated nonnative substrate as the glycosyl donor, GH also catalyzes enzymatic glycosylation in vitro to form a glycosidic linkage. For example, β(1→4)-glucan, that is, cellulose, is successfully synthesized by the enzymatic polymerization of a β-cellobiosyl fluoride monomer by cellulase, which is a GH, via consecutive glycosylation (Fig. 1c) [16]. Interestingly, some enzymes exhibit weak specificity for the recognition of substrates; therefore, they can be employed as catalysts for the preparation of unnatural oligosaccharides and polysaccharides. Accordingly, enzymatic synthesis of unnatural β(1→4)-glucan derivatives has also been achieved by cellulase catalysis [17, 18].

General reaction schemes catalyzed by (a) glycosyl transferase (GT) and (b) glycosyl hydrolase (GH) and (c) GH (cellulase)-catalyzed enzymatic polymerization to obtain cellulose
Phosphorylases are GTs, and they have been employed as catalysts for practical enzymatic glycosylation to synthesize well-defined oligosaccharides and polysaccharides [19, 20]. Phosphorylases principally catalyze in vivo phosphorolysis of glycosidic linkages at the nonreducing end of each specific saccharide chain in the presence of inorganic phosphate (Pi), i.e., phosphorolytic cleavage, to yield the 1-phosphate of a corresponding monosaccharide residue. Simultaneously, a saccharide chain with one less degree of polymerization (DP) is generated (Fig. 2, from left to right). Phosphorylase-catalyzed enzymatic phosphorolysis progresses strictly according to regio- and stereo-specificities by either retention or inversion to form 1-phosphate esters of monosaccharide residues linked through the same or opposite anomeric stereo-arrangement as the cleaved glycosidic bond.

Phosphorylase-catalyzed enzymatic phosphorolysis and glycosylation
Based on the comparable bond energy of a phosphate ester of the phosphorolysis product to that of a glycosidic linkage in the substrate, the reversible nature of phosphorylase-catalyzed reactions is observed, indicating that the reactions can be conducted as enzymatic glycosylations (Fig. 2, from right to left) [21, 22]. In glycosylation, GP catalyzes the transfer of a monosaccharide residue from a monosaccharide 1-phosphate (glycosyl donor) to the nonreducing end of a specific saccharide chain (glycolyl acceptor) to construct a glycosidic bond in strict regio- and stereocontrolled arrangements, liberating Pi. Among the various phosphorylases isolated to date, glucan phosphorylase (GP), which belongs to the GT35 family (EC 2.4.1.1, also called starch phosphorylase or glycogen phosphorylase), is the most well known. It catalyzes enzymatic glycosylation to synthesize oligosaccharides and polysaccharides consisting of α(1→4)-glycosidic linkages by retention [23,24,25,26]. Because of its weak specificity for the recognition of substrates, GP recognizes nonnative monosaccharide 1-phosphates as glycosyl donors in enzymatic glycosylation to yield α(1→4)-linked unnatural oligosaccharides and polysaccharides [27,28,29,30]. Based on the above background, GP-catalyzed enzymatic glycosylation and polymerization using nonnative substrates are herein overviewed to precisely synthesize well-defined unnatural oligosaccharides and polysaccharides. In addition to the studies summarized in previous articles [27,28,29,30], this review presents the recent progress of the enzymatic synthesis of unnatural polysaccharides by GP (isolated from thermophilic bacteria, Aquifex aeolicus VF5)-catalyzed polymerization and related reactions.
Characteristic features of GP-catalyzed enzymatic reactions
In biological systems, GP catalyzes the phosphorolysis of α(1→4)-glucan polymers, such as glycogen and starch, at the nonreducing end by the action of Pi to simultaneously generate α-d-glucose 1-phosphate (Glc-1-P) and α(1→4)-glucan with one less DP (Fig. 3a) [19, 20]. Because of the reversibility of phosphorolysis, GP-catalyzed glycosylation can be performed using the following substrates: the Glc-1-P donor and α(1→4)-glucan acceptor, under appropriate conditions (Fig. 3b) [23,24,25,26]. In this glycosylation, the transfer of a Glc residue from Glc-1-P to the nonreducing end of α(1→4)-glucan occurs with the controlled construction of an α(1→4)-glycosidic bond to obtain a well-defined α(1→4)-glucan containing one more Glc unit while liberating Pi. The reaction mechanism depends upon the close contact of the phosphate group in the covalently linked cofactor pyridoxal 5-phosphate to either Pi (toward phosphorolysis) or the phosphate group of Glc-1-P (toward glycosylation) [31,32,33,34]. In particular, GPs have the lowest recognized DP of glycosyl acceptors at the nonreducing end, in accordance with their sources. Therefore, α(1→4)-oligoglucans, known as maltooligosaccharides, with DPs greater than those of GPs, must be employed as glycosyl acceptors in GP-catalyzed enzymatic glycosylation. Maltotetraose (Glc4) is the smallest glycosyl acceptor recognized by GP isolated from potatoes (the most extensively studied) [19]. It was found that the lowest DP of the glycosyl acceptor recognized by thermostable GPs (isolated from thermophilic bacteria sources) is maltotriose (Glc3), one less than that recognized by potato GPs [35,36,37,38]. GPs have been found to recognize some analog substrates of Glc-1-P (nonnative monosaccharide 1-phosphates) as glycosyl donors, depending on their sources. Accordingly, the extension of GP-catalyzed glycosylation has been studied by employing 1-phosphate donors with different monosaccharide residues to yield unnatural oligosaccharides with the corresponding monosaccharide units at the nonreducing end with strictly controlled α(1→4)-linked chains [27,28,29,30].

Glucan phosphorylase (GP)-catalyzed (a) phosphorolysis, (b) glycosylation, and (c) polymerization
In the presence of an excess of Glc-1-P donor relative to the maltooligosaccharide acceptor, GP induces consecutive glycosylation in a manner similar to enzymatic polymerization to yield the well-defined α(1→4)-glucan polymer, amylose (Fig. 3c) [39,40,41,42]. The GP-catalyzed polymerization of the Glc-1-P monomer takes place via chain-growth polymerization because of the occurrence of initiation strictly at the nonreducing end of the acceptor followed by propagation from the nonreducing end of the elongating α(1→4)-glucan chain. Therefore, the glycosyl acceptor is also called a “primer” of polymerization. Amylose is a natural polysaccharide present in starch along with other components, such as amylopectin. Because it is difficult to completely separate amylose and amylopectin from starch, GP-catalyzed polymerization has been employed to obtain pure amylose samples. As chain transfer and termination reactions are excluded, GP-catalyzed polymerization proceeds analogously to living polymerization. Hence, the molecular weights of the enzymatically produced amyloses are controlled by monomer/primer feed ratios while retaining narrow polydispersity [43].
Amylose synthesized enzymatically by GP-catalyzed polymerization is gradually precipitated in aqueous reaction media owing to the spontaneous formation of a left-handed double helical assembly. In the double helix formation, interstrand stabilization occurs without any steric conflict and involves the occurrence of the O(2)…O(6) type of hydrogen bonding [44, 45]. Owing to the lack of participation of the reducing end of the maltooligosaccharide primer in glycosylation, GP-catalyzed polymerization can be carried out in the presence of modified maltooligosaccharide primers, which are linked covalently to other materials at the reducing ends, such as polymeric materials [46,47,48,49,50]. When polymeric primers with multiple nonreducing ends of α(1→4)-glucan chains are used in GP-catalyzed polymerization, amylose-branched and amylose-grafted polymeric materials are produced. For example, glycogen, which is a highly branched water-soluble polysaccharide comprised of α(1→4)-glucan chains, is additionally interlinked by α(1→6)-glycosidic branching points. Owing to the presence of numerous nonreducing α(1→4)-glucan chain ends, it was employed as a polymeric primer in the GP-catalyzed polymerization of Glc-1-P (Fig. 4) [51], which was conducted in acetate buffer at 40 °C for 24 h. The reaction solution was subsequently left standing at room temperature for 24 h, resulting in its hydrogelation. Hydrogelation is reasonably explained by the construction of network structures through the double-helix formation from the elongated amylose chains among glycogen molecules, which act as cross-linking points.

GP-catalyzed polymerization to produce glycogen hydrogel
Synthesis of unnatural oligosaccharides by GP-catalyzed enzymatic glycosylations
As mentioned above, GPs have been found to exhibit weak specificity for the recognition of donor substrates [27,28,29,30]. Therefore, the synthesis of several unnatural oligosaccharides with different monosaccharide units at the nonreducing ends has been achieved by GP-catalyzed glycosylation employing nonnative monosaccharide 1-phosphates as glycosyl donors. For example, potato GP catalyzes enzymatic glycosylation using α-d-mannose 1-phosphate, α-d-xylose 1-phosphate, α-d-glucosamine 1-phosphate, and N-formyl-α-d-glucosamine 1-phosphate (Man-1-P, Xyl-1-P, GlcN-1-P, and GlcNF-1-P) as nonnative glycosyl donors with the Glc4 acceptor to yield well-defined unnatural α(1→4)-mannosylated, α(1→4)-xylosylated, α(1→4)-glucosaminylated, and N-formyl-α(1→4)-glucosaminylated oligosaccharides, that is, pentasaccharides (Fig. 5) [52,53,54,55]. The conditions for the respective reactions are shown in Fig. 5. The above results indicate weak recognition specificities of potato GP for the structures at the C-2 and C-6 positions in monosaccharide 1-phosphates. The (1→4)-glucosaminylated product, yielded by enzymatic glucosaminylation employing GlcN-1-P, is a functional material because it is a basic (cationic) oligosaccharide bearing a reactive amino group at the C-2 position of a nonreducing GlcN unit.

Potato GP-catalyzed glycosylations using nonnative glycosyl donors (monosaccharide 1-phosphates) with a maltotetraose (Glc4) acceptor
Furthermore, an attempt was made to synthesize another interesting functional oligosaccharide, that is, an acidic (anionic) oligosaccharide, by means of potato GP-catalyzed enzymatic glycosylation (glucuronylation) employing α-d-glucuronic acid 1-phosphate (GlcA-1-P) containing a carboxylate group at the C-6 position as the glycosyl donor. This GP, however, did not recognize GlcA-1-P. On the other hand, thermostable GP (from Aquifex aeolicus VF5) was found to recognize GlcA-1-P as the glycosyl donor. As a result, enzymatic glucuronylation was catalyzed; it was conducted in sodium acetate buffer at 50 °C for 48 h, and Glc3 was used as the glycosyl acceptor to yield an acidic oligosaccharide, that is, tetrasaccharide, containing a GlcA unit at the nonreducing end (Fig. 6) [56]. These findings illustrate thermostable GP’s higher tolerance in the recognition specificity for monosaccharide 1-phosphates compared to potato GP.

Thermostable GP-catalyzed enzymatic glucuronylation using -d-glucuronic acid 1-phosphate (GlcA-1-P) as a glycosyl donor with a maltotriose (Glc3) acceptor
Maltooligosaccharides with carboxylate groups at both chain ends (carboxylate-terminated maltooligosaccharides) were synthesized by the extension of the above enzymatic glucuronylation [57]. The introduction of a carboxylate group at the reducing end of maltoheptaose was first conducted by chemical oxidation in the presence of sodium hypoiodite (I2/NaOH). Afterward, thermostable GP-catalyzed glucuronylation employing GlcA-1-P with the produced maltooligosaccharide containing a carboxylate group was conducted in sodium acetate buffer at 50 °C for 3 days to precisely yield carboxylate-terminated maltooligosaccharides at both chain ends with varying DS values (Fig. 7a). During glucuronylation, disproportionation of the oligosaccharide chain occurred simultaneously to yield different-DP products. The obtained carboxylate-terminated maltooligosaccharides were used as the cross-linker for condensation. Water-soluble chitin (corresponding to 50% N-acetylated chitin, obtained by the partial deacetylation of chitin [58]) was used with a condensing agent (N-hydroxysuccinimide (NHS)/1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC)) to obtain network chitin (Fig. 7b). The reaction solution completely converted into a hydrogel as the cross-linking progressed. Pressing of the hydrogel resulted in a film.

(a) Synthesis of carboxylate-terminated maltooligosaccharides by thermostable GP-catalyzed glucuronylation using GlcA-1-P as a glycosyl donor with oxidized maltoheptaose as a glycosyl acceptor and (b) their use for cross-linking with water-soluble chitin to produce network chitin
Extension of GP-catalyzed enzymatic glycosylations to produce amphoteric branched polysaccharides
The abovementioned thermostable GP-catalyzed enzymatic glucuronylation/glucosaminylation has been extensively employed to produce amphoteric polysaccharides with highly branched structures having both acidic GlcA and basic GlcN units at the nonreducing ends. A highly branched cyclic dextrin, referred to as a glucan dendrimer (GD), was employed as the polymeric glycosyl acceptor with numerous nonreducing α(1→4)-glucan chain ends. This polysaccharide is a water-soluble dextrin that is obtained by cyclization of amylopectin by branching enzyme (EC 2.4.1.18, Bacillus stearothermophilus) catalysis [59, 60]. The thermostable GP-catalyzed enzymatic glucuronylation employing GlcA-1-P with GD (Mn = 1.25 × 105, the number of nonreducing ends = 59) was conducted in sodium acetate buffer at 55 °C for 8 h to produce an acidic α-glucan [61]. The subsequent thermostable GP-catalyzed enzymatic glucosaminylation employing GlcN-1-P in the presence of the acidic product in sodium acetate buffer at 55 °C for 8 h afforded dendritic amphoteric α-glucan with both GlcA and GlcN units at the nonreducing ends [62]. The order of the two enzymatic reactions was determined based on the different reactivities of GlcA-1-P and GlcN-1-P, in which the glucuronylation employing the less reactive GlcA-1-P should be carried out earlier than the glucosaminylation using the more reactive GlcN-1-P. Such successive reaction mechanisms in various GlcA-1-P/GlcN-1-P feed ratios afford amphoteric products with controlled GlcA/GlcN ratios. Their inherent isoelectric points were evaluated by ζ potential measurements, which changed in accordance with the GlcA/GlcN ratios in the products.
Furthermore, thermostable GP-catalyzed successive enzymatic glucuronylation/glucosaminylation using glycogen as a polymeric glycosyl acceptor under the same conditions as above also gave amphoteric branched polysaccharides [63]. Thermostable GP-catalyzed enzymatic glucuronylation employing GlcA-1-P in the presence of glycogen was first carried out to yield acidic glycogen (Fig. 8a), which was used for the subsequent thermostable GP-catalyzed enzymatic glucosaminylation employing GlcN-1-P to produce amphoteric glycogen (Fig. 8b). Elongation of amylose chains on the produced amphoteric glycogen was successfully achieved by the GP-catalyzed polymerization of Glc-1-P from pure nonreducing α(1 → 4)-glucan chain ends without functionalization by GlcA or GlcN units in sodium acetate buffer at 40 °C for 24 h (Fig. 8c). Double-helical assemblies from the amylose chains were formed among the amphoteric glycogen, which acted as cross-linking points for hydrogelation. The obtained amphoteric hydrogel exhibited a pH-responsive property.

Thermostable GP-catalyzed (a) glucuronylation and (b) subsequent glucosaminylation to yield amphoteric glycogen and (c) following enzymatic polymerization to yield amphoteric glycogen hydrogel
Synthesis of unnatural amylose analogs by GP-catalyzed enzymatic polymerization
The abovementioned investigations on potato GP-catalyzed glycosylation employing nonnative glycosyl donors have suggested that after transferring the monosaccharide residue from the substrate to the nonreducing end of the Glc4 acceptor, further glycosylation does not take place. This is because the resulting nonreducing end structure, which differs from the native Glc unit, is no longer recognized as a glycosyl acceptor by potato GP.
However, as GP is known to exhibit more tolerance at the C-2 position in weak specificity than at the C-6 position for the recognition of substrates [19], it has been reported to induce consecutive glycosylations using some C-2 analog substrates [52], such as 2-deoxy-α-d-glucose 1-phosphate (2dGlc-1-P) [64,65,66]. Potato GP-catalyzed consecutive glycosylation using 2dGlc-1-P with maltooligosaccharide as a glycosyl acceptor, that is, enzymatic polymerization, was achieved by its in situ formation from 1,2-dideoxy-d-glucose (d-glucal) in the presence of Pi in Tris-acetate buffer at 30 °C for several days to produce a well-defined 2-deoxyamylose according to the following mechanism [52, 64]. A 2dGlc unit is enzymatically produced in the presence of GP or chemically transferred from d-glucal to the nonreducing end of maltooligosaccharide at the C-4 position in a 1,2-addition reaction with α-stereo-arrangement assisted by Pi (Fig. 9a). The GP-catalyzed enzymatic mechanism is reasonably explained for the regio- and stereoselective reactions in the 1,2-addition reaction rather than the general chemical mechanism. Subsequently, the 2dGlc residue at the nonreducing end of the produced oligosaccharide is liberated by GP-catalyzed phosphorolysis to produce 2dGlc-1-P in situ (Fig. 9b), which acts as the monomer in the enzymatic polymerization to produce 2-deoxyamylose (Fig. 9c). The difference in the potato GP-catalyzed enzymatic glycosylation using 2dGlc-1-P and GlcN-1-P/Man-1-P, that is, consecutive and one-step reactions, respectively, is attributed to the distance of the structure of the former substrate to Glc-1-P compared to the latter. The enzymatically synthesized 2-deoxyamylose spontaneously formed a specific crystalline structure, resulting in precipitation in the reaction media; the crystalline structure was absolutely different from the well-known native double helix of amylose. Powder X-ray diffraction (XRD) measurements and molecular dynamics simulations of the 2-deoxyamylose crystal suggested that the isolated polymeric chains spontaneously assemble into a novel double helix arising from building blocks with controlled hydrophobicity via pyranose ring stacking (Fig. 10) [67].

(a, b) Two-step production of 2-deoxy-α-d-glucose 1-phosphate (2dGlc-1-P) in the presence of inorganic phosphate (Pi) and potato GP and c potato GP-catalyzed polymerization to produce 2-deoxyamylose

Crystalline structure of 2-deoxyamylose suggested by molecular dynamics simulations
By means of the in situ production of 2dGlc-1-P, the thermostable GP-catalyzed copolymerization of d-glucal with Glc-1-P using the Glc3 primer was conducted in Tris-acetate buffer at 40 °C for 24 h to obtain unnatural heteropolysaccharides composed of α(1→4)-linked 2dGlc/Glc residues, called partially 2-deoxygenated amyloses (Fig. 11) [68]. XRD analysis of the heteropolysaccharides showed that the crystalline structures followed the majority rule, where the heteropolysaccharides comprising higher ratios of 2dGlc units and Glc units than others constructed the same crystalline structures as those of the respective homopolysaccharides, that is, 2-deoxyamylose and amylose. Conversely, the heteropolysaccharide with an adapted unit ratio showed an amorphous nature because of the random sequence of the 2dGlc/Glc residues, resulting in film formation by the casting method (Fig. 11).

Thermostable GP-catalyzed copolymerization of d-glucal/Glc-1-P using the Glc3 primer
Considering the different recognition behaviors of thermostable GP (from Aquifex aeolicus VF5) and potato GP [56], the former enzyme catalyzes different glycosylation mechanisms from the latter when GlcN-1-P and Man-1-P, which also have different structures at the C-2 position from the native Glc-1-P, are used as glycosyl donors [8, 12, 30]. As mentioned earlier, potato GP-catalyzed glycosylation using these donors with Glc4 produced pentasaccharides with a GlcN or Man residue at the nonreducing end, but enzymatic polymerization did not occur [52, 54]. As seen in the enzymatic glucuronylation using GlcA-1-P [56], thermostable GP appears to have more tolerance for the recognition of substrates than potato GP. Consequently, consecutive glucosaminylations/mannosylations took place in thermostable GP-catalyzed enzymatic glycosylations using Glc3 and excess molar ratios of GlcN-1-P and Man-1-P in sodium acetate buffer at 40 °C for 4 days to obtain unnatural heterooligosaccharides containing α(1→4)-linked oligo-GlcN/Man chains (Fig. 12) [69]. In the matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectra of the produced heterooligosaccharides in a donor/acceptor feed ratio of 10:1, several peaks assignable to the molecular masses of tetra–octa-saccharides bearing one to five GlcN or Man residues with Glc3 were detected However, even at a relatively high donor/acceptor feed ratio, Pi, which was produced by the glycosyl donors, inhibited the production of relatively long chains because Pi is a native substrate for chain cleavage (phosphorolysis) by GP catalysis.

Thermostable GP-catalyzed consecutive enzymatic glucosaminylations/mannosylations employing GlcN-1-P/Man-1-P with a Glc3 acceptor in acetate buffer
When thermostable GP-catalyzed enzymatic glucosaminylation employing GlcN-1-P was carried out in ammonium buffer (0.5 M, pH 8.6) containing dissolved MgCl2, the liberated Pi was precipitated from the reaction media because Pi formed a water-insoluble salt with ammonium/magnesium ions [70]. As a result, the thermostable GP-catalyzed polymerization of GlcN-1-P using the Glc3 primer (30:1) in such a buffer solution was successfully conducted at 40 °C for 7 days to precisely obtain α(1→4)-linked unnatural aminopolysaccharide, which was named ‘amylosamine’; and the DP of the GlcN units was ~20 (Fig. 13a) [71]. This corresponds to the stereoisomer of chitosan, which was subjected to N-acetylation using acetic anhydride to successfully convert it into a chitin stereoisomer (Fig. 13b). Amylosamine is water soluble owing to the lack of formation of a regular crystalline assembly in aqueous media, unlike amylose, which forms a double helix. As amylosamine exhibits cationic nature due to of the presence of amino substituents, it forms a double helix with the anionic α(1→4)-linked GlcA polymer, another amylose analog called amylouronic acid, by ammonium/carboxylate electrostatic interactions under acidic conditions [72]. The reductive amination of amylosamine induced hierarchically produced controlled assemblies of different sizes depending on the reaction conditions, which contain both the hemiacetalic reducing end and amino substituents [73]. The reaction involves the conversion of a carbonyl group, such as an aldehyde, in the presence of a primary amine into a secondary amino derivative via an intermediate imine. Therefore, reductive amination is an efficient method for amino derivatization at the hemiacetalic reducing end of saccharides using amines because the reducing end structure is in equilibrium with an aldehyde in aqueous media. The reductive amination of amylosamine was investigated using NaBH3CN as a reductant in acetic acid aq. (0.1 mol/L) at 60 °C for 1 h. A relatively low feed ratio of the reductant/reducing end of amylosamine induced the formation of nanoparticles. Numerous amylosamine chains assembled with increasing feed ratios of the reductant/reducing end to form water-insoluble microaggregates and further macrohydrogels.

(a) Thermostable GP-catalyzed polymerization of GlcN-1-P from Glc3 with precipitation of Pi to produce amylosamine (chitosan stereoisomer) and (b) its conversion into stereoisomer of chitin by N-acetylation
The thermostable GP-catalyzed copolymerization of native Glc-1-P with nonnative GlcN-1-P and Man-1-P was achieved under the same operating conditions: in ammonia/MgCl2 buffer at 40 °C for 7 days while removing Pi, unnatural glucosaminoglucan consisting of α(1→4)-linked Glc/GlcN residues and mannoglucan containing α(1→4)-linked Glc/Man residues were obtained (Fig. 14a) [74, 75]. The thermostable GP-catalyzed polymerization of GlcN-1-P from the primer of a maltooligosaccharide-functionalized amylouronic acid (amylouronic acid with an α(1→4)-oligoglucan chain at the nonreducing end) was conducted under the same conditions as stated above. The reaction afforded an amphoteric block polysaccharide comprised of α(1→4)-linked GlcN (basic) and GlcA (acidic) blocks connected through the maltooligosaccharide chain (Fig. 14b) [76]. The block polysaccharides showed pH-responsive properties.

Structures of (a) unnatural glucosaminoglucan and mannoglucan and (b) amphoteric block polysaccharide synthesized by thermostable GP-catalyzed (co)polymerization
The occurrence of a one-step transfer reaction in the abovementioned GP-catalyzed glycosylation employing Xyl-1-P/GlcA-1-P indicates more importance at the C-6 position than at the C-2 position for recognition specificity at the nonreducing ends of the glycosyl acceptors. Thermostable GP-catalyzed enzymatic glycosylation was conducted using 6-deoxy-α-d-glucose 1-phosphate (6dGlc-1-P) as the nonnative glycosyl donor, whose structural similarity to Glc-1-P at the C-6 position was higher than that of Xyl-1-P and GlcA-1-P, with the Glc3 acceptor. Furthermore, the reaction was conducted in ammonia/MgCl2 buffer at 40 °C for 4 days, and Pi was removed. Consequently, consecutive reactions (oligomerization) took place to mainly produce a tetrasaccharide with one 6dGlc residue at the nonreducing end, together with a minor oligosaccharide with two 6dGlc units, that is, pentasaccharide (6-deoxygenated α(1→4)-oligoglucans) (Fig. 15) [77]. Furthermore, thermostable GP-catalyzed cooligomerization from the Glc3 primer using both 6dGlc-1-P and the native substrate Glc-1-P afforded 6-deoxygenated α(1→4)-oligoglucans with variations in DPs.

Thermostable GP-catalyzed oligomerization of 6-deoxy-α-d-glucose 1-phosphate (6dGlc-1-P) from the Glc3 Primer
Conclusions
This review has illustrated that GP-catalyzed glycosylation and polymerization employing nonnative monosaccharide 1-phosphates are powerful tools for the precise synthesis of α(1→4)-linked unnatural oligosaccharides and polysaccharides. Owing to weak specificity for the recognition of substrates by GP, several monosaccharide 1-phosphates have acted as glycosyl donors to enzymatically introduce the corresponding monosaccharide units at the nonreducing end of the maltooligosaccharide chains. Unnatural polysaccharides have been produced by thermostable GP (from Aquifex aeolicus VF5)-catalyzed glycosylation and polymerization using nonnative substrates and polymeric primers, owing to their tolerance for recognition specificity. As new functions and properties different from those of native materials can be expected from unnatural materials, enzymatically synthesized unnatural oligosaccharides and polysaccharides have been used as pH-responsive, cross-linking, self-assembling, and hydrogelling materials. The enzymatic approach, including GP-catalyzed reactions, has been recognized as a useful tool to obtain oligosaccharides and polysaccharides with strictly controlled structures based on high regio- and stereoselectivities. Further methodological development of enzymatic reactions will be attempted to synthesize new unnatural oligosaccharides and polysaccharides in the future. Such unnatural substrates have high applicability as practical functional materials in pharmaceutical, medicinal, and biological research fields.
References
Schuerch C Polysaccharides. In: Mark HF, Bilkales N, Overberger CG (eds) Encyclopedia of polymer science and engineering, 13. 2nd edn. John Wiley & Sons, New York, 1986; pp 87-162.
Berg JM, Tymoczko JL, Stryer L Biochemistry. 7th edn. WH Freeman, New York. 2012.
Yalpani M Polysaccharides: Syntheses, modifications, and structure/property relations. Studies in organic chemistry, 36. Elsevier, Amsterdam; New York. 1988.
Das R, Mukhopadhyay B. Chemical O-glycosylations: an overview. ChemistryOpen. 2016;5:401–33.
Williams R, Galan MC. Recent advances in organocatalytic glycosylations. Eur J Org Chem. 2017;2017:6247–64.
Nielsen MM, Pedersen CM. Catalytic glycosylations in oligosaccharide synthesis. Chem Rev. 2018;118:8285–358.
Kobayashi S, Makino A. Enzymatic polymer synthesis: An opportunity for green polymer chemistry. Chem Rev. 2009;109:5288–353.
Kadokawa J. Precision polysaccharide synthesis catalyzed by enzymes. Chem Rev. 2011;111:4308–45.
Shoda S, Uyama H, Kadokawa J, Kimura S, Kobayashi S. Enzymes as green catalysts for precision macromolecular synthesis. Chem Rev. 2016;116:2307–413.
Shoda S. Development of chemical and chemo-enzymatic glycosylations. Proc Jpn Acad Ser B. 2017;93:125–45.
Shoda S, Noguchi M, Li G, Kimura S Synthesis of polysaccharides I: Hydrolase as catalyst. In: Kohayasni S, Uyama H, Kadokawa J (eds) Enzymatic Polymerization towards Green Polymer Chemistry, https://doi.org/10.1007/978-981-13-3813-7_2. Springer, Heidelberg, 2019; 15-46.
Loos K, Kadokawa J Synthesis of polysaccharides II: Phosphorylase as catalyst. In: Kobayashi S, Uyama H, Kadokawa J (eds) Enzymatic Polymerization towards Green Polymer Chemistry, https://doi.org/10.1007/978-981-13-3813-7_3. Springer, Heidelberg, 2019; 47-87.
Kimura S, Iwata T Synthesis of polysaccharides III: Sucrase as catalyst. In: Kobayashi S, Uyama H, Kadokawa J (eds) Enzymatic Polymerization towards Green Polymer Chemistry, https://doi.org/10.1007/978-981-13-3813-7_4. Springer, Heidelberg, 2019; 89-104.
Renkonen O. Enzymatic glycosylations with glycosyltransferases. Carbohydr Chem Biol. 2008;2-4:647–61.
Shoda S, Izumi R, Fujita M. Green process in glycotechnology. Bull Chem Soc Jpn. 2003;76:1–13.
Kobayashi S, Kashiwa K, Kawasaki T, Shoda S. Novel method for polysaccharide synthesis using an enzyme—The 1st in vitro synthesis of cellulose via a nonbiosyntehtic path utilizing cellulase as catalyst. J Am Chem Soc. 1991;113:3079–84.
Okamoto E, Kiyosada T, Shoda S, Kobayashi S. Synthesis of alternatingly 6-O-methylated cellulose via enzymatic polymerization of a substituted cellobiosyl fluoride monomer catalyzed by cellulase. Cellulose 1997;4:161–72.
Izumi R, Suzuki Y, Shimizu Y, Fujita M, Ishihara M, Noguchi M, et al. Synthesis of artificial oligosaccharides by polycondensation of 2’-O-methyl cellobiosyl fluoride and mannosyl-glucosyl fluoride catalyzed by cellulase. In: Kadokawa J (ed) Interfacial researches in fundamental and material sciences of oligo- and polysaccharides. Transworld Research Network, Trivandrum, India, 2009; 45-67.
Kitaoka M, Hayashi K. Carbohydrate-processing phosphorolytic enzymes. Trends Glycosci Glycotechnol. 2002;14:35–50.
Puchart V. Glycoside phosphorylases: Structure, catalytic properties and biotechnological potential. Biotechnol Adv. 2015;33:261–76.
Nakai H, Kitaoka M, Svensson B, Ohtsubo K. Recent development of phosphorylases possessing large potential for oligosaccharide synthesis. Curr Opin Chem Biol. 2013;17:301–9.
O’Neill EC, Field RA. Enzymatic synthesis using glycoside phosphorylases. Carbohydr Res. 2015;403:23–37.
Seibel J, Beine R, Moraru R, Behringer C, Buchholz K. A new pathway for the synthesis of oligosaccharides by the use of non-Leloir glycosyltransferases. Biocatalysis Biotransformation. 2006;24:157–65.
Seibel J, Jordening HJ, Buchholz K. Glycosylation with activated sugars using glycosyltransferases and transglycosidases. Biocatalysis Biotransformation. 2006;24:311–42.
Kadokawa J. Precision synthesis of functional polysaccharide materials by phosphorylase-catalyzed enzymatic reactions. Polymers. 2016;8:138 https://doi.org/10.3390/polym8040138
Kadokawa J. α-Glucan phosphorylase: A useful catalyst for precision enzymatic synthesis of oligo- and polysaccharides. Curr Org Chem. 2017;21:1192–204.
Kadoakwa J Enzymatic synthesis of non-natural oligo- and polysaccharides by phosphorylase-catalyzed glycosylations using analogue substrates. In: Cheng HN, Gross RA, Smith PB (eds) Green polymer chemistry: Biobased materials and biocatalysis. vol 1192. ACS Symposium Series 1192; American Chemical Society, Washington, DC, 2015; 87-99.
Kadokawa J. Synthesis of non-natural oligosaccharides by α-glucan phosphorylase-catalyzed enzymatic glycosylations using analogue substrates of α-D-glucose 1-phosphate. Trends Glycosci Glycotechnol 2013;25:57–69.
Kadokawa J α-Glucan phosphorylase-catalyzed enzymatic reactions using analog substrates to synthesize non-natural oligo-and polysaccharides. Catalysts. 2018; 8: https://doi.org/10.3390/catal8100473.
Kadokawa J α-glucan phosphorylase-catalyzed enzymatic reactions to precisely synthesize non-natural polysaccharides. In: Sustainability & green polymer chemistry Volume 2: Biocatalysis and biobased polymers, vol 1373. ACS Symposium Series, vol 1373. American Chemical Society, 2020; 31-46.
Palm D, Klein HW, Schinzel R, Buehner M, Helmreich EJM. The role of pyridoxal 5’-phosphate in glycogen phosphorylase catalysis. Biochemistry 1990;29:1099–107.
Browner MF, Fletterick RJ. Phosphorylase: a biological transducer. Trends Biochemical Sci. 1992;17:66–71.
Johnson LN. Glycogen phosphorylase: control by phosphorylation and allosteric effectors. FASEB J. 1992;6:2274–82.
Schinzel R, Nidetzky B. Bacterial α-glucan phosphorylases. FEMS Microbiol Lett. 1999;171:73–9.
Ubiparip Z, Beerens K, Franceus J, Vercauteren R, Desmet T. Thermostable α-glucan phosphorylases: characteristics and industrial applications. Appl Microbiol Biotechnol. 2018;102:8187–202.
Boeck B, Schinzel R. Purification and characterisation of an α-glucan phosphorylase from the thermophilic bacterium Thermus thermophilus. Eur J Biochem. 1996;239:150–5.
Takaha T, Yanase M, Takata H, Okada S. Structure and properties of Thermus aquaticus α-glucan phosphorylase expressed in Escherichichia coli. J Appl Glycosci. 2001;48:71–8.
Yanase M, Takata H, Fujii K, Takaha T, Kuriki T. Cumulative effect of amino acid replacements results in enhanced thermostability of potato type L α-glucan phosphorylase. Appl Environ Microbiol. 2005;71:5433–9.
Ziegast G, Pfannemüller B. Linear and star-shaped hybrid polymers. Phosphorolytic syntheses with di-functional, oligo-functional and multifunctional primers. Carbohydr Res. 1987;160:185–204.
Fujii K, Takata H, Yanase M, Terada Y, Ohdan K, Takaha T, et al. Bioengineering and application of novel glucose polymers. Biocatalysis Biotransformation. 2003;21:167–72.
Yanase M, Takaha T, Kuriki T. α-Glucan phosphorylase and its use in carbohydrate engineering. J Sci Food Agriculture. 2006;86:1631–5.
Ohdan K, Fujii K, Yanase M, Takaha T, Kuriki T. Enzymatic synthesis of amylose. Biocatalysis Biotransformation. 2006;24:77–81.
Kitamura S Starch polymers, natural and synthetic. In: Salamone C (ed) The Polymeric materials encyclopedia, Synthesis, properties and applications, vol 10. CRC Press, New York, 1996; 7915-22.
Imberty A, Chanzy H, Perez S, Buleon A, Tran V. The double-helical nature of the crystalline part of A-starch. J Mol Biol. 1988;201:365–78.
Imberty A, Perez S. A revisit to the three-dimensional structure of B-type starch. Biopolymers 1988;27:1205–21.
Kadokawa J Synthesis of amylose-grafted polysaccharide materials by phosphorylase-catalyzed enzymatic polymerization. In: Smith PB, Gross RA (eds) Biobased monomers, polymers, and materials. vol 1043. ACS Symposium Series 1105; American Chemical Society, Washington, DC, 2012; 237-55.
Kadokawa J Synthesis of new polysaccharide materials by phosphorylase-catalyzed enzymatic α-glycosylations using polymeric glycosyl acceptors. In: Cheng HN, Gross RA, Smith PB (eds) Green polymer chemistry: Biocatalysis and materials II. vol 1144. ACS Symposium Series 1144; American Chemical Society, Washington, DC, 2013; 141-61.
Kadokawa J. Chemoenzymatic synthesis of functional amylosic materials. Pure Appl Chem. 2014;86:701–9.
Nishimura T, Akiyoshi K Amylose engineering: phosphorylase-catalyzed polymerization of functional saccharide primers for glycobiomaterials. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016 https://doi.org/10.1002/wnan.1423.
Kadokawa J. Enzymatic preparation of functional polysaccharide hydrogels by phosphorylase catalysis. Pure Appl Chem. 2018;90:1045–54.
Izawa H, Nawaji M, Kaneko Y, Kadokawa J. Preparation of glycogen-based polysaccharide materials by phosphorylase-catalyzed chain elongation of glycogen. Macromol Biosci. 2009;9:1098–104.
Evers B, Thiem J. Further syntheses employing phosphorylase. Bioorg Medicinal Chem. 1997;5:857–63.
Nawaji M, Izawa H, Kaneko Y, Kadokawa J. Enzymatic synthesis of α-D-xylosylated maltooligosaccharides by phosphorylase-catalyzed xylosylation. J Carbohydr Chem. 2008;27:214–22.
Nawaji M, Izawa H, Kaneko Y, Kadokawa J. Enzymatic α-glucosaminylation of maltooligosaccharides catalyzed by phosphorylase. Carbohydr Res. 2008;343:2692–6.
Kawazoe S, Izawa H, Nawaji M, Kaneko Y, Kadokawa J. Phosphorylase-catalyzed N-formyl-α-glucosaminylation of maltooligosaccharides. Carbohydr Res. 2010;345:631–6.
Umegatani Y, Izawa H, Nawaji M, Yamamoto K, Kubo A, Yanase M, et al. Enzymatic α-glucuronylation of maltooligosaccharides using α-glucuronic acid 1-phosphate as glycosyl donor catalyzed by a thermostable phosphorylase from Aquifex aeolicus VF5. Carbohydr Res. 2012;350:81–5.
Kadokawa J, Chigita H, Yamamoto K Chemoenzymatic synthesis of carboxylate-terminated maltooligosaccharides and their use for cross-linking of chitin. International Journal of Biological Macromolecules. 2020 https://doi.org/10.1016/j.ijbiomac.2020.05.082.
Kurita K. Chitin and chitosan: Functional biopolymers from marine crustaceans. Marine. Biotechnol. 2006;8:203–26.
Takata H, Takaha T, Okada S, Hizukuri S, Takagi M, Imanaka T. Structure of the cyclic glucan produced from amylopectin by Bacillus stearothermophilus branching enzyme. Carbohydr Res. 1996;295:91–101.
Takata H, Takaha T, Nakamura H, Fujii K, Okada S, Takagi M, et al. Production and some properties of a dextrin with a narrow size distribution by the cyclization reaction of branching enzyme. J Fermentation Bioeng. 1997;84:119–23.
Takemoto Y, Izawa H, Umegatani Y, Yamamoto K, Kubo A, Yanase M, et al. Synthesis of highly branched anionic α-glucans by thermostable phosphorylase-catalyzed α-glucuronylation. Carbohydr Res. 2013;366:38–44.
Takata Y, Shimohigoshi R, Yamamoto K, Kadokawa J. Enzymatic synthesis of dendritic amphoteric α-glucans by thermostable phosphorylase catalysis. Macromol Biosci. 2014;14:1437–43.
Takata Y, Yamamoto K, Kadokawa J. Preparation of pH-responsive amphoteric glycogen hydrogels by α-glucan phosphorylase-catalyzed successive enzymatic reactions. Macromol Chem Phys. 2015;216:1415–20.
Klein HW, Palm D, Helmreich EJM. General acid-base catalysis of α-glucan phosphorylases: Stereospecific glucosyl transfer from D-glucal is a pyridoxal 5′-phosphate and orthophosphate (arsenate) dependent reaction. Biochemistry. 1982;21:6675–84.
Evers B, Mischnick P, Thiem J. Synthesis of 2-deoxy-α-D-arabino-hexopyranosyl phosphate and 2-deoxy-maltooligosaccharides with phosphorylase. Carbohydr Res. 1994;262:335–41.
Evers B, Thiem J. Synthesis of 2‐deoxy‐maltooligosaccharides with phosphorylase and their degradation with amylases. Starch ‐ Stärke. 1995;47:434–9.
Uto T, Nakamura S, Yamamoto K, Kadokawa J Evaluation of artificial crystalline structure from amylose analog polysaccharide without hydroxy groups at C-2 position. Carbohydrate Polymers. 2020;240:116347.
Kadokawa J, Nakamura S, Yamamoto K Thermostable α-glucan phosphorylase-catalyzed enzymatic copolymerization to produce partially 2-deoxygenated amyloses. Processes. 2020; 8: https://doi.org/10.3390/pr8091070.
Shimohigoshi R, Takemoto Y, Yamamoto K, Kadokawa J. Thermostable α-glucan phosphorylase-catalyzed successive α-mannosylations. Chem Lett. 2013;42:822–4.
Borgerding J. Phosphate deposits in digestion systems. J Water Pollut Control Fed. 1972;44:813–9.
Kadokawa J, Shimohigoshi R, Yamashita K, Yamamoto K. Synthesis of chitin and chitosan stereoisomers by thermostable α-glucan phosphorylase-catalyzed enzymatic polymerization of α-D-glucosamine 1-phosphate. Org Bimol Chem. 2015;13:4336–43.
Yui T, Uto T, Nakauchida T, Yamamoto K, Kadokawa JI. Double helix formation from non-natural amylose analog polysaccharides. Carbohydr Polym. 2018;189:184–9.
Nakauchida T, Yamamoto K, Kadokawa J Hierarchically controlled assemblies from amylose analog aminopolysaccharides by reductive amination: from nano- to macrostructures. J Appl Polymer Sci. 2018; 135: https://doi.org/10.1002/app.45890.
Yamashita K, Yamamoto K, Kadoakwa J. Synthesis of non-natural heteroaminopolysaccharides by α-glucan phosphorylase-catalyzed enzymatic copolymerization: α(1->4)-linked glucosaminoglucans. Biomacromolecules 2015;16:3989–94.
Baba R, Yamamoto K, Kadokawa J. Synthesis of α(1->4)-linked non-natural mannoglucans by α-glucan phosphorylase-catalyzed enzymatic copolymerization. Carbohydr Polym. 2016;151:1034–9.
Nakauchida T, Takata Y, Yamamoto K, Kadokawa J. Chemoenzymatic synthesis and pH-responsive properties of amphoteric block polysaccharides. Org Biomol Chem. 2016;14:6449–56.
Kadokawa J, Lee LH, Yamamoto K. Thermostable α-glucan phosphorylase-catalyzed enzymatic chain-elongation to produce 6-deoxygenated α(1→4)-oligoglucans. Curr Org Chem. 2021;25:1345–52.
Acknowledgements
The author gratefully thanks Grants-in-Aid for Scientific Research from Ministry of Education, Culture, Sports, and Technology, Japan (Nos. 17K06001 and 21K05170) for financial support. The author also acknowledges the supply of thermostable glucan phosphorylase from Ezaki Glico Co. Ltd., Osaka, Japan. The author is indebted to the coworkers, whose names are found in references from his papers, for their enthusiastic collaborations. The author would also like to thank Editage (www.editage.com) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kadokawa, Ji. Glucan phosphorylase-catalyzed enzymatic synthesis of unnatural oligosaccharides and polysaccharides using nonnative substrates. Polym J 54, 413–426 (2022). https://doi.org/10.1038/s41428-021-00584-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-021-00584-x
- Springer Nature Limited