
Abstract
The effects of tetrahydrofuran (THF) on the physical properties of poly(methyl methacrylate) at (PMMA)/silica nanoparticle interfaces in polymer nanocomposites were investigated via differential scanning calorimetry (DSC), Fourier transform infrared (FT-IR) spectroscopy, and X-ray diffraction (XRD) analysis. Amorphous structures containing PMMA chains with large numbers of trans-gauche conformers were observed in the annealed samples. The amorphous structures in the samples cast from THF suspensions contained PMMA chains rich in trans–trans conformers. These amorphous structures differed from those of neat PMMA prepared without silica. Although the polymer chain mobility was reduced due to interactions between PMMA and the silica nanoparticles, trans–trans-enriched PMMA chains within the amorphous structures were less affected by these interactions. When the amorphous structures were heated above the glass transition temperature of PMMA, the proportion of trans–gauche conformers increased.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Poly(methyl methacrylate) (PMMA) is one of the most important and widely used thermoplastic polymers due to its outstanding transparency, processability, and durability [1]. PMMA is thus frequently used as an organic glass or as a covering for lighting fixtures. It is also commonly found in optical devices, gratings, and waveguides [2, 3]. Recently, the demand for PMMA with enhanced performance in industry and academia has increased [4]. Incorporating nanoparticles into PMMA to form nanocomposites is an effective strategy for meeting this demand. Nanocomposites are often prepared by dissolving a polymer in an organic solvent. It is thus necessary to investigate the effects of solvents on the chain mobility and physical properties of PMMA [5, 6]. The structure of PMMA is thought to be affected by both the solvent and the nanofiller in polymer/nanoparticle composites prepared via solution casting. It is therefore imperative to elucidate the influence of solvents on the properties of PMMA and the interfaces between PMMA and nanoparticles.
The effects of solvents on PMMA are usually investigated using PMMA thin films cast onto solid substrates [7, 8]. The chain mobility of a solvent-cast PMMA film depends on the solvent used. Bulk syndiotactic poly(methyl methacrylate) (st-PMMA) has an amorphous structure. However, crystallization can be induced by ketone solvents such as chloroacetone and diethyl ketone. Kusuyama et al. [7] reported that crystallization in st-PMMA films cast from chloroform solutions was induced by solvent. Kamei et al. [8] reported that the crystallization of several polymers, including PMMA, could be induced by exposure to a suitable solvent or thermal conditions.
The properties of PMMA at interfaces with inorganic materials differ from those of bulk PMMA [9, 10]. Keddie et al. [11] measured the glass transition temperatures (Tgs) of at-PMMA thin films on native silicon oxide substrates using spectroscopic ellipsometry. They found that the restriction of the chain mobility due to the strong interactions between at-PMMA and the native silicon oxide of the substrate increased the Tg. For thin films, it has been reported that changes in Tg are caused by the effects of the free surface, chain confinement, interfacial interactions, and molecular weight [12,13,14,15]. The structures and dynamics of PMMA chains at solid interfaces have also been studied by using PMMA thin films. PMMA chains aggregate and form a high-density layer at the interface with a solid [16,17,18]. Segment motion is restricted in this layer, and its Tg is significantly higher than the bulk Tg [19]. Most of these studies were performed using polymer thin films on solid substrates. Forrest et al. [20] reported that the Tg values of a free-standing ultrathin polystyrene (PS) film and a supported PS film were lower than that of bulk PS. In addition, the decrease in the Tg of the free-standing ultrathin PS film relative to the bulk Tg was much larger than the decrease in the Tg of a supported film relative to the bulk. This indicates that the free surface of an ultrathin polymer film is an important factor that also affects its Tg. Various factors affect the Tgs of polymer thin films, and thin films differ from bulk samples in terms of their surfaces, interfacial effects, and entanglements. These results indicate that in addition to the solvent, the surface of a thin film and its interface with the substrate can significantly affect the structure of the PMMA.
In a thin film, the effects of both the polymer-substrate interface and the molecular mobility of the polymer on the thin film surface are large. In this study, we prepared PMMA nanocomposites with high contents of silica nanoparticles. These samples are much less affected by surface effects than the thin film samples, and they accurately reflect only the physical properties of the PMMA-silica interfacial regions. In this paper, we investigated the molecular mobility of PMMA in the interfacial region using a PMMA/silica nanocomposite and the effects of tetrahydrofuran (THF) on the PMMA chain mobility via differential scanning calorimetry (DSC), X-ray diffraction (XRD) analysis, and Fourier transform infrared (FT-IR) spectroscopy.
Experimental procedure
Materials
At-PMMA was purchased from Aldrich (St. Louis, MO, USA). The average molecular weight (Mw), Tg, and density of at-PMMA were ~120,000, 105 °C and 1.188 g cm−3, respectively. Spherical silica nanoparticles (ρ = 2.2 g cm−3) with diameters ranging from 18.8 to 83.6 nm were purchased from Kanto Chemical (Tokyo, Japan).
Sample preparation
PMMA (0.1 g mL−1) was dissolved in THF (bp 66 °C) in sample vials. A polytetrafluoroethylene (PTFE) sheet was used as the substrate on which the solution was dropped. Since the interactions between PMMA and PTFE are very weak, we expected that the substrate would not affect the structure of the PMMA during the drying process. In addition, since the solvent evaporates from the surface of the cast solution of the polymer on the substrate during the drying process [21, 22], the residual solvent tends to remain near the substrate. Therefore, the sample was first dried for 24 h in air at room temperature, then turned over and further dried for another 24 h under the same conditions. The THF solutions were cast onto PTFE sheets to prevent interactions with the substrates at room temperature and allowed to air-dry for 24. Since the solvent molecules were expected to be concentrated at the interfaces between the polymer and the substrate [21, 22], the cast samples were peeled from the PTFE sheets, turned face down, and dried for an additional 24 h. The cast samples were then dried under vacuum for at least 7 days. These samples were referred to collectively as THF-cast samples, and they included neat PMMA (THF) and PMMA/SiO2 (THF). The amounts of residual solvent in the THF-cast samples were determined from the difference in the weight of the sample dropped on the substrate before and after drying.
Mixed samples containing PMMA and silica nanoparticles were prepared by dispersing silica nanoparticles in PMMA solutions in THF (0.1 g mL−1). The samples collectively referred to as THF-cast samples were prepared from pure PMMA in THF and PMMA/SiO2 in THF. The THF-cast samples were heated for 1 h at 180 °C, which was above the Tg of PMMA, and then allowed to cool slowly to room temperature. These samples were referred to collectively as melt samples, and they included neat PMMA (melt) and PMMA/SiO2 (melt).
To investigate the effects of the residual solvent on the molecular mobility of PMMA, heat-dried samples, in which the solvent was removed by heating, were prepared in addition to a THF-cast sample. The solvent was removed by heating a THF-cast sample at 120 °C. This temperature is higher than the Tg of PMMA and the boiling point of THF. The amount of residual solvent was changed by changing the heating time. These samples are called heat-dried samples.
Measurements
Differential scanning calorimetry (DSC)
The Tgs of the PMMA/silica nanoparticle mixtures were measured using DSC. Each sample (8–10 mg) was packed into an aluminum pan, and DSC curves were obtained using a DSC-6200 calorimeter (Hitachi High-Tech Science Corporation, Tokyo, Japan). The nitrogen gas flow rate, heating rate, and measurement temperature range were 30 mL min−1, 10 °C min−1, and −40 °C to 150 °C, respectively.
FT-IR spectroscopy
FT-IR spectra were obtained using an FT/IR-480 spectrometer (JASCO, Tokyo, Japan). A total of 128 scans with a resolution of 2 cm−1 were acquired for each sample over the spectral range from 400 to 4000 cm−1.
XRD analysis
XRD was performed using a MiniFlex 600 XRD instrument (Rigaku, Tokyo, Japan) equipped with a CuKα radiation source. The tube voltage, tube current, scan range, and scanning speed were 40 kV, 15 mA, 3–40° (2θ), and 2° min−1, respectively.
Results and discussion
Figure 1 shows Tg as a function of solvent content for neat PMMA samples dried at room temperature or at 120 °C. The solid lines correspond to the linear fit of the data. When the neat PMMA samples were dried at room temperature, the Tg increases linearly with decreasing residual solvent. However, even if the residual solvent is extrapolated to 0 wt%, the Tg is significantly lower than the bulk Tg despite no residual solvent. The Tg values of the heat-dried samples treated at 120 °C also increase linearly with decreasing residual solvent content. However, Tg was significantly different depending on the drying temperature even with the same amount of residual solvent. In addition, when the amount of residual solvent in the heat-treated sample was extrapolated to 0 wt%, the Tg was the same as that of neat PMMA (melt). Although the residual solvent could not be completely moved from the samples in this study, the PMMA samples dried at room temperature have a distinctly different molecular mobility from those in the heated samples. Therefore, when the solvent was removed at room temperature, a structure distinct from bulk PMMA was formed due to the effects of the solvent, and this structure was maintained even after the solvent was removed.

Glass transition temperature as a function of solvent content for neat PMMA samples dried at different temperatures: solid lines correspond to linear fit of the data
The DSC curves of the neat PMMA and PMMA/SiO2 melt samples are shown in Fig. 2. The numbers in parentheses indicate the weight percentages of the nanoparticles in the samples. For example, the PMMA/SiO2 (30) sample contained 30 wt% SiO2. The curves in the figure were normalized by polymer weight [23, 24]. The DSC curves in Fig. 2 are all 1st scan data. Based on the measured weight of residual solvent, the melt samples in Fig. 2 contain no residual solvent, and the Tg of annealed neat PMMA was the same as that of PMMA without solvent casting. The DSC curve of the neat PMMA melt sample indicated a glass transition at 98.5 °C. Glass transitions in the DSC curves of the PMMA/SiO2 (30) and PMMA/SiO2 (50) melt samples were indicated at 105.1 °C and 118.8 °C, respectively. Therefore, the Tgs of the PMMA/SiO2 melt samples increased with increasing silica nanoparticle content. The higher Tgs of the PMMA/SiO2 nanocomposites were attributed to the inhibition of polymer chain mobility by the strong interactions between the PMMA and the silica nanoparticle surfaces [25,26,27].

DSC curves of neat PMMA (melt) and PMMA/silica nanoparticle composites (melt)
The DSC curves of the neat PMMA and PMMA/SiO2 THF-cast samples are shown in Fig. 3. The DSC curves in Fig. 3 are all 1st scan data. The neat PMMA (THF) sample has a residual solvent content of approximately 2.3 wt%, and the PMMA/SiO2 (THF) sample has a lower residual solvent content. The Tg from the 2nd scan data of the THF-cast samples was almost the same as the Tg from the 1st scan data of the melt samples. That is, when the THF-cast sample was heated at a temperature sufficiently higher than Tg in the 1st scan, the residual solvent was evaporated, and the structure was relaxed. The DSC curve of neat PMMA (THF) showed a glass transition at 65.8 °C. The DSC curves of the PMMA/SiO2 (30) and PMMA/SiO2 (50) THF-cast samples indicated glass transitions at 66.9 °C and 68.7 °C, respectively, and the Tg increased only slightly with increasing silica nanoparticle content. The Tgs of the THF-cast samples were thus much lower than those of any of the melt samples. Ito et al. [28] completely dissolved PMMA in organic solvents, including butyl acetate and acetone, and obtained PMMA thin films via compression molding. They reported that the Tgs of the PMMA thin films were lower than the bulk Tg, which was due to the enhanced segmental motion of PMMA with residual solvent. This was attributed to the strong dipolar interactions between the solvent molecules and the carbonyl groups in PMMA, which reduced the number of interactions between the PMMA chains. Barone and Salerno [29] reported that the Tg of PMMA was strongly influenced by the solvent used for preparation because polar groups in the solvent could form hydrogen bonds with the polymer. PMMA interacted with the solvent, which hindered the intramolecular interactions within the PMMA chains [29, 30].

DSC curves of neat PMMA (THF) and PMMA/silica nanoparticle composites (THF)
A PMMA thin film prepared from PMMA dissolved in an organic solvent will have a lower Tg than the bulk Tg because the intramolecular interactions are weakened by contact or interactions with the solvent. Zhang et al. [22] reported that strong interactions between the polymer, solvent, and substrate decreased the mobility of PMMA chains and the Tg. These phenomena are observed when residual solvent is present in a PMMA thin film. The residual solvent is located primarily between the PMMA thin film and the substrate. However, the THF-cast sample prepared in this study had ~2 wt% residual solvent, and the PTFE sheets used as substrates did not interact with the PMMA. It was thought that the high-mobility region resulting from strong interactions between PMMA and THF, referred to as the high-mobility amorphous region, remained after the residual solvent was removed.
The THF-cast samples had significantly lower Tgs than the bulk Tg, and their Tgs had broader distributions. Feng et al. [31] reported that the Tgs of surfactant-free PMMA nanospheres in aqueous dispersions decreased significantly with decreasing particle size and that the distributions of the Tgs broadened. Water absorbed by PMMA did not affect the Tg. They suggested that the density of the entangled chains on the PMMA surface was lower than that in the bulk, while the free volume and segment mobility were greater [32,33,34]. In addition, PMMA nanospheres with diameters of 50 nm dispersed in n-heptane had a Tg that was ~60 K lower than the bulk Tg. Similarly, the Tgs of the THF samples that did not interact with the PTFE substrates were lower than the bulk Tg. This was because interactions between most of the PMMA chains in these samples were weakened due to the interactions between the PMMA and THF. This reduced the density of the entangled chains and enhanced segment mobility.
Each of the PMMA/SiO2 samples had a lower Tg than the melt samples. The presence of silica nanoparticles in the melt and THF-cast samples restricted the movement of the polymer chains and increased the Tg. However, the magnitude of the increase in Tg was smaller for the THF-cast samples than for the melt samples. Eriksson et al. [35] investigated the dependence of Tg on the residual solvent in the PMMA nanocomposites prepared from solutions of PMMA in toluene or methyl ethyl ketone that contained dispersed silica. The Tgs of the PMMA nanocomposites were lower when they contained 3 wt% or more of residual solvent. They reported that interactions between the PMMA and solvent molecules counteracted the effects of the filler.
We considered the results of this study as follows. In the PMMA/SiO2/THF system, PMMA has high molecular mobility in the THF solution because THF is a good solvent. Therefore, the PMMA structure near the silica surface is not dramatically different from that in regions other than the interface (bulk region). When the solvent is evaporated at a temperature below its Tg, the PMMA structure changes during the evaporation process to fill the gaps where the solvent molecules had been. Thus, we believe that the resulting structure must reflect the PMMA structure in the THF solution because the drying temperature is below Tg.
On the other hand, when the THF-cast sample is heat treated at Tg or higher, it relaxes to a state closer to the equilibrium state. This was why the Tgs of the THF-cast samples were lower than those of the melt samples.
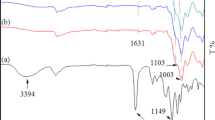
FT-IR spectroscopy was used to evaluate the physicochemical interactions between PMMA and the silica nanoparticle surfaces and the effect of THF on the PMMA/silica nanocomposites. The FT-IR spectra of the neat PMMA, THF-cast, and melt samples from 4000 to 500 cm−1 are shown in Fig. 4. The characteristic THF peaks at 2975, 2865, 1070, and 912 cm−1 were absent in the spectra of the THF-cast samples. In the spectra of the melt and THF samples, no new peaks appeared at these wavenumbers, and no peak shifts were observed. This indicated that the residual solvent contained in the sample is below the detection limit of FT-IR spectroscopy and does not significantly affect the FT-IR spectrum of PMMA [30, 36]. The FT-IR spectra of the neat PMMA and PMMA/silica nanoparticle melt samples from 1770 to 1690 cm−1 are shown in Fig. 5. The peak at ~1732 cm−1 was attributed to the stretching vibration of C=O in carbonyl groups [37]. When the spectra of the neat PMMA and PMMA/silica nanoparticle melt samples were compared, the peaks in the spectra of the PMMA/silica nanoparticle melt samples were shifted to lower wavenumbers. This shift is observed when carbonyl groups are influenced by hydrogen bonding [38] or acid–base interactions [39]. Fowkes et al. [40] reported that the carbonyl absorption band shifted to a lower frequency due to interactions between the PMMA carbonyl groups and the hydroxyl groups (silanol groups [41]) on the surfaces of SiO2 nanoparticles.

FT-IR spectra of the neat PMMA samples (melt and THF)

FT-IR spectra of neat PMMA (melt) and the PMMA/silica nanoparticle composites (melt). The peaks are normalized to the peak areas from 1770 to 1690 cm−1 in each spectrum
Although the structural changes in PMMA induced by interactions at the interfaces with silica nanoparticles were not analyzed in detail in this study, the shifts in the FT-IR spectra in Fig. 5 suggested the restriction of carbonyl groups through interactions with the surfaces of the silica nanoparticles. Table 1 shows the positions of the absorption bands in the FT-IR spectra of the melt samples. The peaks shifted to lower wavenumbers as the silica nanoparticle content increased. Although the peak shift was slight, it was reproducible.
The FT-IR spectra of the neat PMMA melt and the THF-cast samples from 1300 to 1000 cm−1 are shown in Fig. 6. The peaks at 1195, 1150, and 1060 cm−1 were attributed to anti-symmetric C–O–C vibrations, C–O stretching vibrations coupled with skeletal vibrations, and skeletal vibrations in the main chain, respectively [42, 43]. The intensity of the peak at 1060 cm−1 did not depend on conformation of the PMMA main chains, so the spectra were normalized to the peak at 1060 cm−1. The intensity of the absorption peak at 1150 cm−1 increases with decreasing proportion of trans–trans conformers, which is accompanied by an increase in the proportion of trans–gauche conformers. The intensities of the 1150 cm−1 peaks in the spectra of the THF-cast samples were lower than those of the 1150 cm−1 peaks in the spectra of the melt samples. Therefore, the THF-cast samples contained larger proportions of chains with trans–trans conformations.

FT-IR spectra of the neat PMMA samples (melt and THF)
The peak shifts due to the addition of SiO2 in the spectrum of each PMMA/SiO2 mixed sample are plotted in Fig. 7. The spectra of the THF-cast samples exhibited smaller peak shifts with increasing SiO2 nanoparticle content than those observed in spectra of the melt samples. These results indicated that the high-mobility amorphous regions created by interactions between PMMA and THF molecules inhibited the interactions between PMMA and the silica nanoparticles.

Peak shifts in the spectra of PMMA/SiO2 mixed samples due to the addition of SiO2. The dotted lines are included as guides
The XRD patterns of the neat PMMA and PMMA/silica nanoparticle composite melts are shown in Fig. 8. The patterns of the mixed samples were normalized by subtracting the nanoparticle pattern [44, 45]. The pattern of the neat PMMA melt sample contained a characteristic wide diffraction peak at (2θ) 13.1°, which indicated that the neat PMMA melt had an amorphous structure. The interplanar spacing calculated from this peak was 6.32 Å, which reflected the ordering of the PMMA main chains [46,47,48]. Peaks in the patterns of the PMMA/silica nanoparticle composites shifted to higher angles with increasing SiO2 nanoparticle content.

XRD patterns of neat PMMA (melt) and PMMA/silica nanoparticle composites (melt)
The XRD patterns of the neat PMMA and PMMA/silica nanoparticle composite THF-cast samples are shown in Fig. 9. Diffraction peaks from (2θ) 10° to 20° in the pattern of the neat PMMA THF-cast sample were broader than those in the pattern of the neat PMMA melt. In contrast, the peaks near (2θ) 16° in the patterns of the PMMA/silica nanoparticle (THF) composites were slightly shifted to higher angles. The shift was greater in the patterns of the composites with higher SiO2 nanoparticle contents.

XRD patterns of neat PMMA (THF) and PMMA/silica nanoparticle composites (THF)
The peaks in the patterns of the THF-cast samples were broader than the peaks in the patterns of the melt samples. These results suggested that interactions between the PMMA and THF molecules weakened the interactions between the chains and that the packing of the PMMA chains was disturbed. Their structures remained disordered at room temperature even after the solvent was removed. Since the peak shift observed with increasing silica nanoparticle content was quite small, the chains in the structure formed by PMMA and the solvent did not interact significantly with the silica nanoparticles at the interfaces.
The effect of THF on PMMA in interfacial regions was investigated using PMMA/silica composites. DSC, FT-IR, and XRD were used to analyze the melted and solvent-cast neat PMMA and PMMA/silica nanoparticle composites. The structure of PMMA was clearly different from that of the melt sample after removing the solvent at room temperature. This structure is maintained at room temperature even if the residual solvent is removed. The Tgs of the PMMA/silica composites were higher than that of neat PMMA. The results confirmed that interactions between PMMA and the silica surfaces restricted the movement of the PMMA chains. The Tgs of the THF-cast samples were lower than those of the melt samples. This was due to the interactions between PMMA and THF, which weakened the interactions between the PMMA chains and increased chain mobility. This suggested that the interactions between PMMA and THF were stronger than the interactions between PMMA and silica. The FT-IR results indicated that the interactions between the PMMA and silica involved carbonyl groups in the PMMA and silanol groups on the silica nanoparticles. The XRD patterns of the solvent-cast samples contained broader peaks than the patterns of the melt samples. The PMMA chains were thought to be randomly arranged because the interactions between them were weakened by their interactions with THF. PMMA chain packing remained random at room temperature. Collectively, the results showed that PMMA and THF formed an amorphous structure that differed from that of bulk PMMA. PMMA interacted with silica nanoparticles, but the interactions between the highly mobile chains in the amorphous region and the silica nanoparticles were limited. We propose for the first time that THF affects the interfacial regions of PMMA/silica nanoparticles. The effects of solvents on the morphologies of the polymer chains in the PMMA/silica interfacial regions will be explored in more detail in a future study.
References
Borges AMG, Benetoli LO, Licínio MA, Zoldan VC, Santos-Silva MC, Assreuy J, et al. Polymer films with surfaces unmodified and modified by non-thermal plasma as new substrates for cell adhesion. Mater Sci Eng C Mater Biol Appl. 2013;33:1315–24.
Giacon VM, da Silva Padilha GS, Bartoli JR. Fabrication and characterization of polymeric optical by plasma fluorination process. Optik. 2015;126:74–6.
Hamdy MS, Alfaify S, Al-Hajry A, Yahia IS. Optical constants, photo-stability and photo-degradation of MB/PMMA thin films for UV sensors. Optik. 2016;127:4959–63.
Chen S, Yang Z, Wang F. Investigation on the properties of PMMA/reactive halloysite nanocomposites based on halloysite with double bonds. Polymers. 2018;10:919.
Okolieocha C, Beckert F, Herling M, Breu J, Mülhaupt R, Altstädt V. Preparation of microcellular low-density PMMA nanocomposite foams: influence of different fillers on the mechanical, rheological and cell morphological properties. Compos Sci Technol. 2015;118:108–16.
Meth JS, Zane SG, Chi C, Londono JD, Wood BA, Cotts P, et al. Development of filler structure in colloidal silica-polymer nanoocomposites. Macromolecules. 2011;44:8301–13.
Kusuyama H, Takase M, Higashihata Y, Tseng H, Chatani Y, Tadokoro H. Structural change of st-PMMA on drawing, absorption and desorption of solvents. Polymer. 1982;23:1256–8.
Kamei D, Ajiro H, Hongo C, Akashi M. Solvent effects on isotactic poly(methyl methacrylate) crystallization and syndiotactic poly(methacrylic acid) incorporation in porous thin films prepared by stepwise stereocomplex assembly. Langmuir. 2009;25:280–5.
Jones RAL, Richards RW. Polymers at surface and interface. Cambridge: Cambridge University Press; 1999.
Karim A, Kumar S. Polymer surface, interface and thin films, World scientific publishing Co. Pte. Ltd, Toh Tuck. Singapore: Link; 2000.
Keddie JL, Jones RAL, Cory RA. Interface and surface effects on the glass-transition temperature in thin polymer films. Faraday Disc. 1994;98:219–30.
Xu J, Liu Z, Lan Y, Zuo B, Wang X, Yang J, et al. Mobility gradient of poly(ethylene terephthalate) chains near a substrate scaled by the thickness of the adsorbed layer. Macromolecules. 2017;50:6804–12.
White RP, Price CC, Lipson JEG. Effect of interfaces on the glass transition of supported and freestanding polymer thin films. Macromolecules. 2015;48:4132–41.
Chen J, Li J, Xu L, Hong W, Yang Y, Chen X. The glass-transition temperature of supported PMMA thin films with hydrogen bond/plasmonic interface. Polymers. 2019;11:601.
Burroughs MJ, Napolitano S, Cangialosi D, Priestley RD. Direct measurement of glass transition temperature in exposed and buried adsorbed polymer nanolayers. Macromolecules. 2016;49:4647–55.
Scheutjens JMHM, Fleer GJ. Statistical theory of the adsorption of interacting chain molecules. 1. Partition function, segment density distribution, and adsorption isotherms. J Phys Chem. 1979;83:1619–35.
Jiang N, Shang J, Di X, Endoh MK, Koga T. Formation mechanism of high-density, flattened polymer nanolayers adsorbed on planar solids. Macromolecules. 2014;47:2682–9.
Jiang N, Endoh MK, Koga T, Masui T, Kishimoto H, Nagao M, et al. Nanostructures and dynamics of macromolecules bound to attractive filler surfaces. ACS Macro Lett. 2015;4:838–42.
Tanaka K, Tateishi Y, Okada Y, Nagamura T, Doi M, Morita H. Interfacial mobility of polymers on inorganic solids. J Phys Chem B. 2009;113:4571–7.
Forrest JA, Dalnoki-Veress K, Stevens JR, Dutcher JR. Effect of free surfaces on the glass transition temperature of thin polymer films. Phys Rev Lett. 1996;77:2002–5.
Perlich J, Metwalli E, Schulz L, Georgii R, Müller-Buschbaum P. Solvent content in thin spin-coated polystyrene homopolymer films. Macromolecules. 2009;42:337–44.
Zhang X, Yager KG, Kang S, Fredin NJ, Akgun B, Satija S, et al. Solvent retention in thin spin-coated polystyrene and poly(methyl methacrylate) homopolymer films studied by neutron reflectometry. Macromolecules. 2010;43:1117–23.
Matsuura K, Matsuda Y, Tasaka S. Influence of tetrahydrofuran and alumina nanoparticle interface on the structure of syndiotactic poly(methyl methacrylate). Kobunshi Ronbunshu. 2018;75:275–9.
Fukatsu H, Kuno M, Matsuda Y, Tasaka S. Surface effect of silica nano-particles with different size on thermotropic liquid crystalline polyester composites. World J Nano Sci Eng. 2014;04:35–41.
Moll J, Kumar SK. Glass transitions in highly attractive highly filled polymer nanocomposites. Macromolecules. 2012;45:1131–5.
Zhang FA, Lee DK, Pinnavaia TJ. PMMA–mesocellular foam silica nanocomposites prepared through batch emulsion polymerization and compression molding. Polymer. 2009;50:4768–74.
Chinthamanipeta PS, Kobukata S, Nakata H, Shipp DA. Synthesis of poly(methyl methacrylate)–silica nanocomposites using methacrylate-functionalized silica nanoparticles and RAFT polymerization. Polymer. 2008;49:5636–42.
Ito A, Ayerdurai V, Miyagawa A, Matsumoto A, Okada H, Courtoux A, et al. Effects of residual solvent on glass transition temperature of poly(methyl methacrylate). Nihon Reoroji Gakkaishi. 2018;46:117–21.
Patra N, Barone AC, Salerno M. Solvent effects on the thermal and mechanical properties of poly(methyl methacrylate) cast from concentrated solution. Adv Polym Technol. 2011;30:12–20.
Bistac S, Schultz J. Solvent retention in solution-cast films of PMMA: study by dielectric spectroscopy. Prog Org Coat. 1997;31:31. 347–50
Feng S, Chen Y, Mai B, Wei W, Zheng C, Wu Q, et al. Glass transition of poly(methyl methacrylate) nanospheres in aqueous dispersion. Phys Chem Chem Phys. 2014;16:15941–7.
Fakhraai Z, Forrest JA. Measuring the surface dynamics of glassy polymers. Science. 2008;319:600–4.
Mansfield KF, Theodorou DN. Molecular dynamics simulation of a glassy polymer surface. Macromolecules. 1991;24:6283–94.
Itagaki H, Nishimura Y, Sagisaka E, Grohens Y. Glass transition of stereoregular poly(methyl methacrylate) at interfaces. Langmuir. 2006;22:742–8.
Eriksson M, Goossens H, Peijs T. Influence of drying procedure on glass transition temperature of PMMA based nanocomposites. Nanocomposites. 2015;1:36–45.
Patra N, Salerno M, Diaspro A, Athanassiou A. Effect of solvents on the dynamic viscoelastic behavior of poly(methyl methacrylate) film prepared by solvent casting. J Mater Sci. 2011;46:5044–9.
Vien DL, Colthup NB, Fateley WG, Grasselli JG. The handbook of infrered and raman characteristic frequencies of organic molecules. New York: Academic Press; 1991.
Tannenbaum R, Hakanson C, Zeno A, Tirrell M. Spectroscopic study of the chemistry at the Cr-PMMA interface. Langmuir. 2002;18:5592–9.
Grohens Y, Brogly M, Labbe C, Schultz J. Interfacial conformation energies of stereoregular poly(methyl methacrylate) by infra-red reflection abspectroscopy. Polymer. 1997;38:5913–20.
Fowkes FM, Kaczinski MB, Dwight DW. Characterization of polymer surface sites with contact angles of test solutions. 1. Phenol and iodine adsorption from methylene iodide onto PMMA films. Langmuir. 1991;7:2464–70.
Ioannis MK, Vassilikou-Dova Aglaia, Eugen RN. Dielectric characterization of poly(methyl methacrylate) geometrically confined into mesoporous SiO2 glasses. Mater Res Innov. 2001;4:322–33.
Tretinnikov ON, Ohta K. Conformation-sensitive infrared bands and conformational characteristics of stereoregular poly(methyl methacrylate)s by variable-temperature FTIR spectroscopy. Macromolecules. 2002;35:7343–53.
Fujii Y, Akabori K, Tanaka K, Nagamura T. Chain conformation effects on molecular motions at the surface of poly(methyl methacrylate) films. Polym J. 2007;39:928–34.
Matsuura K, Matsushita M, Matsuda Y, Tasaka S. Structure of polyacrylate/nanoparticle interfaces. Micro Nano Lett. 2017;12:667–9.
Matsuura K, Matsuda Y, Tasaka S. Metastable interface formation in isotactic poly(methyl methacrylate)/alumina nanoparticle mixtures. Polym J. 2018;50:375–80.
Abbas H, Iqbal S, Ahmad S, Arfin N. Synthesis and characterisation of poly(methyl methacrylate)-silica composites. Mater Res Express. 2018;5:085312.
Ahmad S, Ahmad S, Agnihotry SA. Synthesis and characterization of in situ prepared poly(methyl methacrylate) nanocomposites. Bull Mater Sci. 2007;30:31–5.
Fu HP, Hong RY, Zhang YJ, Li HZ, Xu B, Zheng Y, et al. Preparation and properties investigation of PMMA/silica composites derived from silicic acid. Polym Adv Technol. 2009;20:84–91.
Acknowledgements
This work was supported by Grant-in-Aid for JSPS Research Fellow Number JP19J14528. The XRD measurements were performed at the Ookayama Materials Analysis Division, Technical Department.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Matsuura, K., Kuboyama, K. & Ougizawa, T. Effect of tetrahydrofuran on poly(methyl methacrylate) and silica in the interfacial regions of polymer nanocomposites. Polym J 52, 1203–1210 (2020). https://doi.org/10.1038/s41428-020-0375-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-020-0375-0
- Springer Nature Limited
This article is cited by
-
Synthesis, optoelectronic and thermal characterization of PMMA-MWCNTs nanocomposite thin films incorporated by ZrO2 NPs
Journal of Materials Science: Materials in Electronics (2022)