
Abstract
The catalytic coupling and polymerization of CO2 and epoxides has been studied for over 50 years. While traditionally dominated by catalytic systems containing cobalt, chromium, and zinc, the use of iron catalysts has emerged in the past 10 years. This review provides an overview of the homogeneous iron-catalyzed transformations of carbon dioxide and epoxides to yield cyclic and/or polycarbonates. It is important to note the potential for cyclic carbonates to be used as monomers for polymer formation via transesterification or by ring-opening polymerization in some cases, e.g., cyclohexene carbonate. Typical catalytic systems are composed of a Lewis acidic iron center and an anionic nucleophilic source, either through an anionic group weakly bound to the metal center or the addition of an external cocatalyst, cooperatively described as a binary catalytic system. This review is divided into two sections: (1) iron catalysts for cyclic carbonates and (2) iron catalysts for polycarbonates. At the end of each section, a table summarizes each catalytic system and the reaction conditions utilized in an attempt to provide a clearer comparison across the literature. Focus is given to highlighting differences in product selectivity, reaction conditions, and relative amounts of cocatalyst used.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
The drive to develop a sustainable, carbon neutral economy continues to be present in media and in government policy. Climate change threatens not only our economic stability but also our environment, including ecosystems and biodiversity, on a global scale. The increase in climate change over recent decades has a direct correlation with human activity and has accelerated since the industrial revolution. While we as a society are largely responsible for climate change, we also have the potential to help to reverse the effects and work towards a solution through green energy initiatives and other forms of sustainable development [1]. It is important to note that while the use of CO2 as a building block alone cannot mitigate CO2 emissions or significantly reduce atmospheric levels, it is one small component that could collectively have a positive impact and make a difference. Scientists are working towards the development of polymers that incorporate CO2 with mechanical and thermal properties similar to those of petroleum-derived materials. As they perform this research, scientists must keep several key points in mind. Polymerization reactions should be performed under mild temperature and pressure conditions if possible. High temperatures and pressures require significant energy inputs and thus lead to an overall net increase in CO2 emissions. Catalyst design remains a critical aspect in this regard to allow reactions to occur at ambient conditions in the hopes of achieving a close-to-carbon neutral process. In addition, the study of polymers, especially polycarbonates, from an environmental persistence standpoint still requires much work.
Since the industrial revolution, the amount of carbon dioxide (CO2) in the atmosphere has risen nearly 50%, with current levels approaching 412 parts per million. However, the use of CO2 in a renewable manner (i.e., C-1 feedstock) has been a growing area of research but still poses challenges to chemists [2,3,4]. CO2 is extremely stable from a thermodynamic standpoint, and thus, researchers must often employ harsh reaction conditions and/or reactive substrates to overcome these barriers. In particular, reactive epoxides are often coupled with CO2, leading to cyclic, and/or polycarbonate formation [5,6,7]. First reported in 1969 [8], this area has grown extensively over the past 50 years and hence is quite well understood. However, catalyst design remains crucial in the selective formation of one product, i.e., polycarbonate rather than cyclic carbonate or polyether. A general reaction scheme for the metal-catalyzed coupling or copolymerization of CO2 and cyclohexene oxide (CHO) is shown in Scheme 1. The reaction begins with ring opening of the metal coordinated epoxide by a suitable nucleophile (i.e., X−). This can occur through one of three possible initiation steps. Following this, the metal-bound alkoxide can either undergo CO2 insertion to form a metal-bound carbonate or repetitive epoxide insertion, leading to polyether formation. If the rate of CO2 insertion is faster than that of epoxide insertion, carbonate linkages will be favored. This can be controlled by the nature of the catalyst along with the reaction conditions. If CO2 insertion occurs, the metal-bound carbonate can either ‘backbite’ to form cyclic carbonates or continue to undergo repeated epoxide/CO2 insertion, leading to polycarbonate formation [9, 10].

General reaction scheme for the metal-catalyzed coupling of CO2 and cyclohexene oxide to form cyclic carbonates, polycarbonates, and polyethers
The nature and amounts of the cocatalyst relative to those of the catalyst can also influence the mechanism and product selectivity. While in Scheme 1, X− implies an anionic cocatalyst, neutral cocatalysts have also proven effective [3, 10]. Essentially, the main role of the cocatalyst is to provide a nucleophile to initiate ring opening of the epoxide in the first step of the mechanism. This nucleophile can either come from the catalyst itself (i.e., a bifunctional catalytic system) or be added externally to the reaction mixture. Some of the most commonly used cocatalysts are shown in Fig. 1. These are discussed in more detail in the sections that follow.

Commonly used cocatalysts in reactions of CO2 and epoxides
In terms of commonly used epoxides, propylene oxide (PO), and CHO have been the most widely studied for their reactions with CO2 [11]. The ring-opening copolymerization (ROCOP) of CO2 and CHO has been extensively studied from a practical and mechanistic standpoint employing a variety of metal-based catalysts. The majority of research has focused on chromium-, cobalt-, zinc-, and aluminum-containing homogeneous catalysts [10]. The reaction of PO and CO2 often leads to backbiting and formation of the corresponding cyclic carbonate. However, the major drawback of these epoxides lies in the fact that they are currently petroleum-sourced for large-scale production, hence hindering the renewable aspect of the final product. In recent years, there has been motivation to move towards the use of renewable epoxides; however, there are still limited examples in comparison to PO and CHO [11]. A selection of commonly employed epoxides is shown in Fig. 2.

Selection of commonly used epoxides in coupling and polymerization reactions with CO2
While these transformations have been driven by metal-based catalysts centered on Cr, Co, Zn, and Al, in the past decade, the use of iron-based catalysts has begun to emerge [10]. The concept of using iron as a catalyst in organic transformations is not new; for decades, it has been widely employed within catalysts in the Haber process to produce ammonia. Iron is an attractive alternative to heavier transition metals due to its high earth abundance, relatively low cost and long-term sustainability. In addition, iron is present in many biological/metabolic processes in nature. Iron can exist in oxidation states ranging from −2 to +5, which contributes to its wide range of applications (i.e., lower oxidation states lead to a more nucleophilic nature, while higher oxidation states result in a higher Lewis acidity and electrophilicity).
Along with its use in cross-coupling, oxidation, hydrogenation, and cyclization, iron has played a key role in polymerization catalysts, with one of the most famous being the Brookhart–Gibson catalysts for olefin polymerization [12,13,14]. In addition, an emerging class of ‘switchable’ catalysts revolving around the incorporation of a ferrocene unit in the ligand backbone or iron serving as the active catalytic center have recently been reported by several groups (an example is shown in Fig. 3), and these have been particularly effective in ROCOP reactions [15,16,17,18,19,20,21,22].

Redox-switchable iron catalysts reported by Byers et al. for chemoselective ring opening of cyclic diesters (purple, iron(II) system) and epoxides (red, iron(III) system) [16]
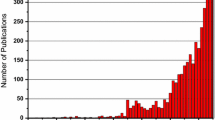
Iron catalysts in CO2 chemistry have been reported for CO2 reduction and hydroformylation reactions; however, the use of iron catalysts in CO2/epoxide chemistry has been less explored. Early patents reported the ability of iron halides to promote polymerization of PO and double metal cyanides composed of zinc hexacyanoferrate and zinc ferricyanide to copolymerize CO2 and epoxides; [23,24,25,26] in the latter examples, the epoxide is activated by the zinc center rather than iron. Most advancements in this area have occurred in the past ~10 years (Fig. 4).

Number of articles (journal, review, patents) published since 1969–present. (Note: the search was conducted using SciFinder using the keywords carbon dioxide, epoxide, and iron catalyst)
This review attempts to highlight the major advancements in reactions of CO2/epoxide transformations by iron-containing catalyst systems for either cyclic or polycarbonate formation in chronological order. The cyclic products can be used as monomers via transesterification or anionic ring-opening polymerization (ROP) reactions and are therefore relevant to researchers in the field of polymer chemistry. In 2018, Capacchione et al. published an excellent review of homogenous iron catalysts for these transformations with a strong focus on the importance and classification of the ligand structure [27]. In this review, we attempt to complement the review mentioned above by organizing examples based on (1) product selectivity, (2) reaction conditions, and (3) the importance of and nature of the cocatalyst (if one was employed). In addition, we highlight the important advancements made over the past 2 years in this rapidly moving field.
Iron catalysts for cyclic carbonate formation
Cyclic carbonates are commercially applied as green solvents, additives to gasoline and in the preparation of electrolyte solutions for lithium-ion batteries. In addition, cyclic carbonates can be used as monomers for polymerizations either through ROP (Scheme 2) or transesterification reactions. Although they have much potential, cyclic carbonates are normally formed on an industrial scale using harsh reaction conditions. Thus, the drive to develop sustainable efficient catalysts to allow their production under milder conditions is desired [28, 29]. In terms of their use as starting materials for polycarbonate synthesis, the results reported in the scientific literature are somewhat limited and tend to be dominated by larger cyclic carbonate reagents (6- and 7-membered rings). The 6-membered cyclic carbonates can be prepared via catalytic addition reactions of CO2 with oxetanes (4-membered ring analogs of epoxides), and as far as we are aware, this reaction has not been performed using iron catalysts. However, the ROP of trans-cyclohexene carbonate, a 5-membered cyclic carbonate, as a monomer has been reported and has yielded polymers with moderate molecular weights upon anionic polymerization using potassium tert-butoxide as an initiator [30]. Nevertheless, it is important to note that most 5-membered cyclic carbonates (such as propylene carbonate) are not suitable monomers for anionic ROP. In 2010, Carpentier, Guillaume et al. reported the synthesis and ROP of 7-membered cyclic carbonates (4-methyl- and 5-methyl 1,3-dioxepan-2-one; α-Me7CC and β-Me7CC, respectively), Scheme 2b [31]. The polymerization of these organic carbonates was studied with well-established Al-, Zn-, and Y-based catalysts alongside several organocatalysts. Polymers could be obtained with modest molecular weights and glass transition temperatures, Tg, ranging from –11 to 36 °C depending on the substrate used and obtained molecular weight. Prior to this report, previous examples focused on the ROP of the related unsubstituted 7-membered cyclic monomer.

Anionic ring opening of cyclic carbonates as monomers to form polycarbonates and select examples in the literature
Last year, Satoh, Isono et al. demonstrated the abilities of trimethyl glycine, a naturally sourced product, as an effective catalyst for ROP of the 6-membered cyclic monomer trimethylene carbonate (Scheme 2c) [32]. This system showed high catalytic activity, yielding polymers with narrow dispersities (Đ = 1.2) and moderate molecular weights (~4000 gmol−1). A novel and promising aspect of this study involved the use of functional catalytic initiators, leading to materials with various potential industrial applications. For example, the use of 6-azide-1-hexanol, which contains a terminal azido group, as an alcohol initiator led to a polymer material that can be further functionalized via ‘click’ chemistry, leading to the production of block copolymers and macromolecular architectures.
Finally, to demonstrate the potential applications of these materials, Cao, Chen et al. reported a nitric oxide functionalized polycarbonate (Scheme 2d) [33]. The cyclic monomer could be ring-opened in the presence of a polymeric alcohol initiator (mPEG-OH). This polymer, in the presence of doxorubicin (DOX), a commonly employed chemotherapeutic drug, formed a self-assembled biodegradable polymer/DOX micelle. Incorporation of NO donors was shown to improve drug release under physiological conditions. This example highlights one of the many potential applications for polymers produced via ROP of cyclic carbonate monomers.
The first report of an iron catalyst for cyclic carbonate formation came from a patent in 1994 from the Texaco Chemical Company [34]. Complex 1 (Fig. 5) was active for the coupling of PO and CO2, yielding propylene carbonate without the use of a cocatalyst. Good conversions were obtained (~80%), but the reaction rates were slow (Turnover Frequency, TOF = 58 h−1), and harsh reaction conditions were required (180 °C, ~100 bar CO2). Following this, He et al. reported a series of metal phthalocyanines as catalysts for the coupling of CO2 and terminal epoxides [35]. The presence of a base, tributylamine, was needed in excess (4.5 equiv. per Fe) for the reaction to proceed. Similar to a previous report, harsh reaction conditions (140 °C, 43 bar CO2) were used. In 2007, Jing et al. reported an iron porphyrin complex 3 for the coupling of PO and CO2 [36]. This paper mainly focused on analogous cobalt porphyrin complexes for the same reaction, but an iron analog was synthesized for comparison. The systems were active for cyclic carbonate formation at 25 °C and 7 bar CO2 in the presence of 2 equiv. phenyltrimethylammonium tribromide (TPAT). However, in comparison to the cobalt analogs, the iron complexes were far less reactive, yielding a conversion of only 10% after 3 h.

Early iron catalysts for the production of cyclic and polycarbonates
A major breakthrough in the field of iron-catalyzed CO2/epoxide chemistry came from the Williams group in 2011 [37] and focused on polycarbonate formation, which is discussed in detail in the sections that follow. Shortly after this, Rieger et al. reported tetraamine and diimine–diamine iron(III) complexes active towards the coupling of CO2 and PO to form propylene carbonate (Fig. 6) [38]. Complexes 4.1 and 4.2 were active for cyclic carbonate formation without the addition of an external cocatalyst but showed higher conversion with the addition of 1 equiv. of TBAB. Complex 5 was inactive alone but could obtain conversions >80% with the addition of TBAB. Kinetic studies on these systems, utilizing in situ IR spectroscopy, revealed a second-order dependence on the iron concentration, and thus, a bimetallic reaction pathway involving two separate iron centers was proposed. For example, in the absence of a cocatalyst, the mechanism for complex 4.1 begins by the dissociation of a weakly bound Cl−, resulting in a vacant binding site for epoxide coordination. Next, Cl− from a second catalyst molecule ring-opens the coordinated epoxide. CO2 insertions occur at the iron-alkoxide bond, followed by backbiting to yield cyclic carbonate and regenerate the active catalytic species.

Tetraamine and diimine–diamine iron(III) complexes for the coupling of CO2 and propylene oxide
Building on this work, Wang et al. reported similar tetraamine and diimine–diamine iron (III) complexes active towards the coupling of PO, ECH, and CHO with CO2 [39]. Both complexes were highly active catalysts, and unlike the previously reported Rieger complex 5, 7 was active without the addition of a cocatalyst, but conversions remained low. However, a direct comparison cannot be made, as the reaction conditions differ slightly. Regardless, in the presence of 1 equiv. of TBAB, high activity was achieved. Furthermore, these systems were active for the coupling of CO2 with ECH and CHO. For CHO, no evidence of copolymer formation was observed. Instead, the cis-isomer of CHC was exclusively formed in moderate conversions.
Zevaco et al. published a family of iron(III) complexes using N4 and N2O2 chelating ligand frameworks for the coupling and copolymerization of epoxides and CO2 (Fig. 7). The first complexes, 8.1 and 8.2, were screened as catalysts for the formation of propylene carbonate [40]. The complex 8.1, with chloride groups in the axial positions, was quite active (78% conv. after 20 h), while the derivative complex with acetate groups (8.2) yielded 8% conv. under identical conditions. Modification of the aryl backbone in the ligand framework gave rise to complexes 9.1–9.4, which were all highly active catalysts towards the coupling of PO and CO2 for propylene carbonate formation [41]. In 2013, Zevco et al. expanded to N2O2-iron(III) (10) and iron(II) (11) systems that were active catalysts for the coupling of PO and nine other substituted epoxides with CO2 to yield cyclic carbonates [42]. It was found that the iron(II) system (11) was only active in the presence of a cocatalyst (TBAB) and required higher catalyst loadings to achieve high conversions. On the other hand, the iron(III) complex, 10, was active without an external cocatalyst and required lower catalyst loadings to achieve similar conversions. This complex was also screened for reactivity with a range of other epoxides, demonstrating the diverse applicability of this system.

Iron(III) reported by Zevaco et al. for coupling and polymerization reactions of CO2 and epoxides
In 2012, Kleij et al. reported iron(III) amine triphenolate complexes for the coupling of epoxides and CO2 [43]. Following their initial report, they published a follow-up study elucidating the possibility of the complexes existing as monomeric or dimeric species (Scheme 3, top) [44]. The dimer formation ability was attributed to ortho-position substitution of the phenolate backbone, leading to stability in the case of dimer formation. That is, complexes bearing tert-butyl or phenyl groups in the ortho-position favored the monomeric structure, while hydrogen and methyl groups in the ortho-position favored the dimeric species. In all cases, the monomeric species were far more active as catalysts for coupling reactions. In addition, during reactions, higher temperatures and the use of a coordinating solvent favored the dissociation of the dimer complex into its monomeric form.

Proposed ring-closure mechanisms proposed by Kleij et al. for the inversion or stereo-retention of trans-2,3-epoxybutane
An interesting study on these complexes came a year later that demonstrated their ability to control the stereochemistry of the final product when 2,3-epoxybutane was used as the substrate [45]. It was found that when starting with either pure cis- or trans-2-BO, the stereochemistry of the final product was dependent on the relative amount of cocatalyst used. For example, when 13.2 was used as a catalyst with cis-2-BO as a substrate, >95% of the corresponding cis-product was achieved when 10–16 equiv. of TBAB was present as a cocatalyst. However, once less than 2.5 equiv. of TBAB was present in solution, the selectivity began to switch towards the trans-product, with 89% trans-product being produced in the presence of 0.5 equiv. TBAB. The authors explained this reactivity through two separate ring-closing mechanisms, leading exclusively to one product over the other (Scheme 3). The mechanism begins in a manner similar to those of other reported systems. That is, ring opening of the coordinated epoxide by an external nucleophile by SN2 attack results in inversion of the stereochemistry at this carbon atom. This is then followed by CO2 insertion, leading to a metal-bound carbonate species. In the ring-closing pathway leading to the cis-product (high TBAB concentrations), there is competition between the metal-bound carbonate and excess Br− anions for the vacant Fe binding site. This results in dissociation of the metal-bound carbonate, which can then backbite through an outer sphere ring-closure mechanism, leading to the trans-product. On the other hand, under low concentrations of TBAB, the carbonate species is more likely to be bound to the iron center, and hence, the metal-center controls the inversion of stereochemistry. The bound bromide anion that ring-opened the initial epoxide interacts with the vacant cis-coordination site on iron, and the partially positive sp2 carbon undergoes a pseudo-SN1-type ring-closure mechanism.
Recently, a bimetallic iron(III) thioether-triphenolate complex reported by the Capacchione group showed exceptional activity towards the coupling of PO and CO2 to form cyclic carbonates under neat conditions, producing a high TOF of 580 h−1 (Fig. 8) [46]. Upon optimization of the reaction conditions, the catalyst (0.025 mol%) with 2 equiv. TBAB was used as a cocatalyst at 100 °C and 20 bar CO2, and 87% conversion of PO to the cyclic product was achieved in only 6 h. Other tested cocatalysts, including PPNCl and 4-dimethylaminopyridine (DMAP), proved less active. The authors attribute the high reactivity of their system to the favored coordination of the epoxide to the iron center as a result of the soft donor sulfur atoms on the chelating ligand being weakly bonded. DFT studies shed light on the importance of the hemilabile Fe–S bond and demonstrated that only one iron center is reactive in the catalytic cycle. The functional group tolerance and substrate scope of the catalyst were also assessed by testing its activity against a series of epoxide substrates. When the methyl group of PO was replaced with a chloride or hydroxyl (substrates being epichlorohydrin and glycidol, respectively), very similar or higher conversions were obtained. In the case of CHO, only 13% conversion to the cyclic product was obtained, yielding the cis-isomer with no evidence of polycarbonate formation [46].

Bimetallic iron(III) thioether-triphenolate catalyst for propylene carbonate formation
In 2016, Repo et al. reported a bifunctional Schiff base iron(III) complex for cyclic carbonate formation in the absence of a cocatalyst [47]. Reactions proceeded at low pressures (10 bar CO2), and although active without an additional cocatalyst, small amounts of TBAB were shown to improve yields. In addition to PO, the bifunctional system was active towards the coupling of CO2 and ECH, SO, CHO and 1-hexene oxide. Kinetic studies were performed via in situ IR spectroscopy and revealed an overall first-order dependence on iron supporting the intramolecular catalytic process. The imidazole group of the ligand serves to activate CO2, producing an anionic carbonate species that can then ring open a coordination epoxide at the iron center (Scheme 4). Intramolecular ring-closing releases the final product while regenerating the active catalytic species.

Proposed intramolecular mechanism reported by Repo et al. for the synthesis of propylene carbonate
In 2016, our group reported a family of iron(III) complexes supported by tetradentate amino-bis(phenolate) ligands that were active towards the coupling of CO2 and a range of terminal epoxides in the presence of a suitable cocatalyst (16.1–16.5) [48]. Electron-withdrawing substituents on the phenolate rings led to complexes with the highest catalytic activity. However, during coupling reactions, we observed reaction mixtures that changed from intense purple to a red-orange color over the course of the reaction and postulated that an oxo-bridged species formed in situ during the reaction. Hence, in a follow-up report, we synthesized the corresponding oxo-species and tested them as catalysts [49]. Compared with their monometallic analogs, we found the oxo-species to be less active. Interestingly, through speciation studies, we determined that during a reaction catalyzed by the monometallic species, the catalyst can transform into the corresponding bimetallic oxo-species through an epoxide deoxygenation reaction (Scheme 5). The idea of potential competing epoxide deoxygenation reactions has been overlooked in the field of CO2-epoxide couplings and copolymerizations. In some cases, deoxygenation may occur to form the actual active catalytic species from a precatalyst. Following this result and the discovery of the epoxide deoxygenation process, we reported an in-depth study of iron complexes bearing amino-bis(phenolate) ligands in varying coordination geometries and found that only the complexes originally in a square pyramidal geometry could perform the epoxide deoxygenation reaction.

Monometallic and bimetallic iron(III) complexes for the coupling of CO2 and epoxides. In situ bimetallic species formed through an alternative epoxide deoxygenation pathway
Due to the range of reactivities exhibited by iron aminophenolate complexes during CO2-epoxide reactions, we chose to conduct a comprehensive study in an attempt to determine a structure–activity relationship with a collection of 17 different iron aminophenolate complexes with varying ligand electronics, sterics, and metal-center geometries (Fig. 9) [50]. We found that the reaction selectivity towards either cyclic or polycarbonate production was dependent on a range of factors, including the metal-center geometry, electronics of the phenolate substituents within the ligand and the ratio of iron to external cocatalyst used. The complexes that were active catalysts for cyclic carbonate formation are discussed below, and those selective for polycarbonates are discussed in the following sections. First, the complexes in a square pyramidal geometry (i.e., 16.1–16.5) were only able to produce cyclic carbonates (discussed above). The other complexes studied contained iron in a trigonal bipyramidal geometry with ligands containing various phenolate substituents and neutral donor arms (18, 19, 29, 30). In general, all complexes screened showed high activity for the coupling of PO and CO2 to form propylene carbonate. However, complexes with electron-withdrawing substituents on the phenolate ring (29.3) showed the highest reactivity. With 4 equiv. of PPNCl, at reaction conditions of 100 °C and 20 bar, CO2 propylene carbonate could be produced with a TOF of 1240 h−1. In addition, when the steric bulk of the phenolate neutral donor arm was increased, the (30.1 and 18.1–18.3) reactivity declined. Perhaps the most exciting aspect of this paper was the development of a structure–activity relationship for activity towards polycarbonate formation, which is discussed in a later section within this review.

Selection of recent iron catalysts reported for cyclic carbonate formation
In 2018, Garden et al. reported phenoxyimine iron(III) complexes containing varying ortho substituents in the ligand backbone (20.1–20.3) as catalysts for cyclic carbonate formation (Fig. 8) [51]. All complexes were found to be highly active for the selective coupling of PO and CO2 to yield propylene carbonate under relatively mild conditions (20 bar) and low catalyst loading (0.05 mol%) with excess TBAB. Complexes of ligands containing electron-withdrawing substituents (20.3) showed the highest activity, which was attributed to the increased Lewis acidity of the iron(III) center. These air-stable, robust systems were also active towards a selection of commonly employed epoxides, and in the case of CHO, exclusively the cis-cyclic product was obtained.
Building upon their previous report in 2015, Capacchione et al. reported the synthesis of mononuclear iron(III) complexes bearing sulfur atoms in the ligand backbone (21.1–21.4, Fig. 8) [52]. In terms of cyclic carbonate formation, these systems were very active even under very mild reaction conditions (35 °C, 1 bar CO2), with 21.4 proving to be the most active catalyst. Without the addition of a cocatalyst, no conversion was obtained; however, excess TBAB resulted in an efficient binary catalytic system. Kinetic studies revealed a first-order process with respect to iron concentration and suggested that the rate determining step was epoxide ring opening. These systems demonstrated a wide epoxide scope, and at 1 bar CO2, they demonstrated the highest initial TOF (290 h−1 in the case of PO) for a homogeneous catalytic system at this pressure. In addition, these systems were active for polycarbonate formation and are discussed in more detail below.
Salen, salan, and salalen ligands have long been studied in CO2/epoxide coupling and polymerization reactions, particularly with chromium- and cobalt-based systems. However, their complexation with iron for these transformations has been less explored. In 2018, Lamberti et al. reported a selection of such complexes (23.1–23.4) based on iron(III) (Fig. 9) [53]. In the case of PO, all complexes were active and selective towards the formation of propylene carbonate with good conversions. Of the four complexes screened, complex 23.4 showed the highest activity, followed closely by 23.1, 23.2, and 23.3. The authors attributed this trend in reactivity to an enhanced flexibility in the ligand backbone in 23.4 compared to 23.3 and highlighted the importance of a flexible backbone in the improvement in the catalytic activity. This may be due to the increased ability to coordinate and activate the requisite reagents for this catalyzed reaction.
To the best of our knowledge, there have been only two reports of iron catalysts for cyclic carbonate formation in 2019. The first came from Pescarmona and Otten, who reported six different formazanate ferrate(II) complexes bearing labile halide ligands that were active without the addition of a cocatalyst (Fig. 9) [54]. Changing the ancillary ligand (22.1–22.3) resulted in a reactivity trend from most active to least active catalyst with regards to the halide present: Br− > I− > Cl−. From a mechanistic viewpoint, this makes sense, as reactivity depends on the leaving group ability and thus access to a vacant site for epoxide coordination. In addition, the effect of substituents in the ligand backbone was investigated. Changing the aromatic group from a p-tol to the more electron-withdrawing C6F5 group or more electron-donating p-OMe (in an attempt to modify the Lewis acidity of the iron center) did not afford significant changes in reactivity, and the conversions remained similar.
Following this report, Jones et al. reported a collection of air-stable iron(III) acetate complexes bearing salan, salen and salalen ligand frameworks (Fig. 10) [55]. These complexes exhibited activity towards the coupling of CHO and CO2 to yield, in the majority of cases, exclusively cis-CHC. Comparing the diverse group of complexes, iron salan acetate complex 24.10 showed the highest activity. Increasing the flexibility of the aminopiperidine ligand backbone (i.e., 24.4–24.5) resulted in improved activity, which is consistent with the findings of Garden et al. discussed above [51]. In addition, 24.10 was screened as a catalyst against a range of terminal epoxides with varying electronic/steric effects, yielding moderate-to-high conversions in all cases. These systems were also active towards the ROP of rac-lactide but are not be discussed in detail here, as this is outside the scope of this review. Interestingly, the authors also noted a distinct color change from purple to red in the reaction mixtures over the course of the coupling reactions. Upon future investigation through UV–vis studies, this was attributed to the formation of a μ-oxo-bridged species formed through epoxide deoxygenation, which is consistent with our earlier studies on similar systems [49].

Iron(III) acetate complexes reported by Jones et al. for the selective coupling of CO2 and several terminal epoxides
Table 1 provides a summary of the systems discussed in the sections above for the coupling of CO2 and epoxides to yield cyclic carbonates. In most cases, the addition of an external cocatalyst is needed to obtain high activity. In the majority of cases, high reaction temperatures and moderate CO2 pressures are employed. To date, PO has been the most explored substrate for these transformations, but in recent years, researchers have expanded the substrate scope to other terminal and more challenging internal epoxides.
Iron catalysts for polycarbonate formation
ROP and ROCOP of epoxides, cyclic carbonates, and other cyclic monomers are generally driven by a reduction in the bond and/or angle strain of the cyclic monomer. Polycarbonates are used in many industrial applications due to their desirable properties, including low weight, durability, transparency, and high-impact resistance. The majority of industrially produced polycarbonates today are petroleum-derived and not sustainable, i.e., the carbonate group is provided through the use of phosgene or other reagents. The synthesis of polycarbonates via the copolymerization of epoxides with CO2 leads to potentially renewable incorporation of CO2. While this has been studied widely in recent years, the majority of examples focus on chromium and cobalt; iron has been less explored, with limited examples reported to date (Fig. 11).

Iron catalysts used for CO2/epoxide copolymerization
A major breakthrough in iron catalysis for both selective polycarbonate and cyclic carbonate formation was reported by Buchard et al. in 2011 [37]. The system was based on an air-stable di-iron(III) complex, 25, in which the selective formation of either poly(cyclohexene carbonate) or cis-cyclohexene carbonate could be controlled by the amount of bis(triphenylphosphine)iminium chloride (PPNCl) present in the reaction (Scheme 6). In terms of polycarbonate formation, CHO could be copolymerized with CO2 under neat conditions with a catalyst loading of 0.1 mol% at 80 °C and CO2 pressures <10 atm. Although active at 1 atm CO2, the produced polycarbonate contained only 66% carbonate linkages. However, when the pressure was increased to 10 atm CO2, polycarbonates with >99% carbonate linkages could be produced within 5 h.

Proposed reaction intermediates leading to the exclusive formation of cis-cyclohexene carbonate
The ability of this system to selectively form the cis-cyclic product is quite impressive. In general, cyclic cyclohexene carbonate is more difficult to produce than polycarbonate, and when it is formed, it is often the trans-isomer produced due to the backbiting mechanism. To produce the cis-isomer, a double inversion of the stereochemistry must occur at the chiral centers of CHO. The authors observed that by increasing the amount of anionic cocatalyst (PPNCl in this case) to two equiv. with respect to the catalyst, cis-cyclohexene carbonate was selectively produced. They suggested that the presence of excess Cl− favors the formation of anionic carbonate species (Scheme 6, bottom right). This species is more nucleophilic than the carbonate bound to the Fe center in the initial steps and would then undergo an intramolecular nucleophilic substitution (SN2-like), leading to an inversion in the stereochemistry and producing the cis-product. In addition, this catalyst was active for the cyclization of PO and styrene oxide under 1 atm CO2, yielding cyclic carbonates at conversions of 91% and 98%, respectively.
The first report of PO/CO2 iron-catalyzed copolymerization was reported by the Nozaki group in 2013 [56]. Their system was composed of an iron-corrole complex, 26, with bridging oxo axial ligands, combined with PPNCl as a cocatalyst. The copolymerization of PO with CO2 was achieved at 60 °C and 20 bar CO2 pressure with a catalyst loading of 0.05 mol% iron and 0.5 equiv. PPNCl with respect to iron. These conditions gave an impressive TOF of 1004 h−1; however, in all cases, the % carbonate linkages in the obtained copolymer were quite low, never reaching values >30%. Increasing the amount of PPNCl decreased the selectivity, and large amounts of propylene carbonate were observed. When the bimetallic iron species was replaced with a monomeric iron complex with chloride as the axial ligand, the catalytic activity diminished. When an iron(III) species supported by tetraarylporphyrin ligands was used, only the cyclic carbonate product was produced. This demonstrated that the combination of bimetallic iron species supported by a corrole ligand was essential in producing poly(propylene carbonate).
In 2013, Kleij, Pescarmona and coworkers reported iron amino triphenolate catalysts that were active for cyclic carbonate (discussed above) and polycyclohexene carbonate formation under supercritical CO2 (scCO2) conditions [57]. By carefully tuning the nature and relative amount of cocatalyst in relation to those of the catalyst, they were able to switch the selectivity of their system to produce either the cyclic or polymeric product when CHO was used as a substrate. They note that to control selectivity, the intermediate in the catalytic cycle where backbiting or further epoxide insertion can occur (similar to in Scheme 3 above) is very important. That is, backbiting to form the cyclic product is favored if the nucleophile X− is a good leaving group or if the metal-bound carbonate can easily dissociate and/or is displaced by another equivalent of X−, preventing further epoxide insertion leading to polycarbonate formation. The authors screened a series of both tetrabutylammonium halides and bis(triphenylphosphine)iminium halides and observed that in general, at higher ratios of cocatalyst to catalyst (10:1), formation of the cyclic product was favored, and only the cis-isomer was formed. This discovery was shown to be similar to work by Williams et al. as discussed previously, suggesting a displacement of the metal-bound carbonate before ring closure [29]. Comparing the three complexes studied, similar activity was observed in complexes bearing either methyl or tert-butyl substituents (27.1 and 27.2), suggesting that sterics had no major influence on the reaction mechanism. The chloride-substituted complex (27.3) showed lower activity, which was attributed to its lower solubility in scCO2. In all cases, the obtained polymers gave broad GPC traces that could be deconvoluted, resulting in two separate molecular weight fractions. The Tg of the polycarbonates were in the range of 70–80 °C, and 13C{1H} NMR analysis revealed that the polymer samples contained both isotactic and syndiotactic diads.
Following this report, Pescarmona et al. reported an iron(III)amino-bis(phenolate) complex (28) as a catalyst for the copolymerization of CHO and CO2 in scCO2 (60 °C, 80 bar CO2) in combination with a suitable cocatalyst (PPNCl or tetrabutylammonium salts—[Bu4N][X], where X = Cl, Br, or OAc) [58]. Fine-tuning the relative amounts of catalyst: cocatalyst yielded >99% selectivity towards cis-CHC, but for PCHC formation, conversions were good, but only up to 88% selectively for the polymeric product could be achieved. The polymers produced were of an oligomeric nature (740–1600 gmol−1) with narrow dispersities. When the substrate was switched from CHO to VCHO, the selectivity towards PVCHC increased to 98% under optimized conditions; however, the overall epoxide conversion remained low at 48%. The molecular weights also showed an overall increase up to 3800 gmol−1. The microstructure of these materials was also investigated. In the case of both CHO and VCHO 13C{1H}, NMR analysis revealed atactic polycarbonates. The obtained PVCHC could also be cross-linked using the radical initiators azobisisobutyronitrile (AIBN) and 1,3-propanedithiol, leading to an overall increase of 55 °C in the original polymer Tg. This cross-linking resulted in lower solubility in a range of organic solvents, and improved chemical resistance of the cross-linked materials. SEM images showed a distinct difference between the morphologies of the PVCHC and the corresponding cross-linked PVCHC, which was attributed to possible nucleation and particle growth differences in ethanol between the two materials.
The Zevaco group has published two reports of iron complexes containing an N,N-bis(2-pyridinecarboxamide)-1,2-benzene ligand framework (8 and 9) as catalysts for both cyclic carbonate and PCHC formation. In general, these complexes showed higher activity towards cyclic carbonates (discussed above); however, for reactions of CHO and CO2, PCHC was obtained with very low molecular weights ranging from 760 to 2700 gmol−1 [40, 41].
In 2018, Capacchione et al. reported [OSSO]-type iron (III) complexes (21.1–21.4) that were active for cyclic carbonate (discussed above) and polycarbonate formation in the case of CHO/CO2 copolymerization [52]. In all cases, bimodal molecular weight distributions were observed for the resulting polymers via GPC analysis, which was largely attributed to trace amounts of water in the reaction mixtures. In an attempt to minimize this, the epoxide was distilled twice over CaH2, which resulted in both improved catalytic activity and nearly unimodal molecular weight distributions. As mentioned above, in the case of cyclic carbonates, kinetic studies revealed a first-order process in iron. However, in the case of polycarbonate formation, kinetic studies revealed a second-order process in iron concentration, suggesting the involvement of two iron centers in the catalytic cycle. DFT studies revealed, in the case of CHO, that the energy barrier for chain propagation is lower than that of the ring-closing step, explaining polycarbonate selectivity in the case of CHO.
We recently reported a family of iron(III) amino-bis(phenolate) complexes varying in phenolate substituents within the ligands and coordination geometries and our attempts to establish structure–activity relationships for CO2-epoxide reactions [50]. The complexes that were selective towards PCHC are discussed below, and those selective towards cyclic carbonate formation are discussed in the sections above. Complexes 29–30 were found to be active catalysts for the copolymerization of CHO and CO2, yielding polymers with >99% carbonate linkages. Reactions were performed at 60 bar CO2 and 60 °C at a catalyst loading of 0.5 mol% iron. Decreasing the CO2 pressure led to a decline in the carbonate linkages; however, even at 7 bar CO2 pressure, polymers could be obtained with >50% carbonate linkages. Several cocatalysts were screened, but PPNCl proved superior. The use of a neutral cocatalyst, DMAP, resulted in no overall conversion. Similar to other iron catalyst systems for these reactions, when the amount of cocatalyst was increased, the product selectivity switched from PCHC to cis-CHC.
In general, the reactivity of complexes towards PCHC formation was highly dependent on the nature of the pendant donor of the tripodal ligands. For complexes 29.1–29.4, (i.e., those containing a pyridyl donor), electron rich substituents on the phenolate rings gave slightly higher activities than those with electron-donating substituents. Changing the sterics of the substituents on the phenolate donor did not lead to significant changes in reactivity. However, when the hybridization of the pendent donor was changed from sp2 (29) nitrogen to sp3 (30), this trend in reactivity relating to phenolate substituents was reversed. In addition, as the steric bulk of the sp3 nitrogen donor group increased (18.1–18.3), the product selectivity switched from PCHC to cis-CHC. No complexes in a square-based pyramidal geometry were active for PCHC formation.
Table 2 summarizes the data for the systems discussed above, which are active towards the copolymerization of CO2 and epoxides to yield polycarbonates. In comparison to cyclic carbonate formation, there are fewer reports of iron catalysts for polycarbonate formation; however, this area has advanced significantly in recent years. In a comparison of the systems in Table 2, it is worth noting the difference in % CO3 linkages for the obtained polycarbonates. For example, while the Nozaki iron corrole system gave the highest molecular weight polymer to date, the degree of carbonate linkage incorporation was quite low (Table 2, entry 3). In addition, in the case of CHO, controlling selectivity towards either PCHC or the cyclic product is crucial and can often be controlled by the relative amount of cocatalyst to iron.
Conclusions and outlook
The reaction of CO2 and epoxides to yield either cyclic or polycarbonates has been dominated by metal-based catalysis in the literature in particular systems based upon Cr, Co, and Zn. The concept of iron-based catalysis has emerged in the past decade and poses a promising alternative to other transition metal-based systems. Especially during the past 5–6 years, promising iron systems for these transformations have emerged at a steady pace. Due to the ease of access to various oxidation states using iron systems, there is significant potential to build systems based on redox-switching activity in the future of polymerizations using CO2 as a building block. Research into ring-opening transesterification catalysis for the conversion of cyclic carbonates into polycarbonates has also emerged, and thus, new iron-containing and other sustainable catalyst systems for making a diverse range of cyclic carbonate monomers will be desirable moving forward. Several examples in the literature to date mention the ability to further modify the obtained polycarbonates through either cross-linking or the addition of a functional monomer or functional initiation/chain-transfer agent. This concept, while certainly promising for the production of materials finding broader industrial applications, is quite new in the field of CO2-epoxide derived polymers, with the majority of examples focused on CHO-based polycarbonates; therefore, it is ripe for further study. In addition, while outside the scope of this review, reports of metal-catalyzed block polymerizations and terpolymerizations incorporating CO2 and epoxides have been increasing and leading to new classes of materials with varying and often tailorable properties. Iron catalysts could surely be employed in this field, especially with their history in a wide range of catalytic transformations from polymerizations to hydroelementations. Finally, while the efficient catalyzed production of these materials is important, scientists should also be conducting depolymerization and degradation studies on these materials to determine their practicality and renewable aspects towards their placement in the consumer market.
References
Kätelhön A, Meys R, Deutz S, Suh S, Bardow A. Climate change mitigation potential of carbon capture and utilization in the chemical industry. Proc Natl Acad Sci USA. 2019;116:11187.
Cokoja M, Bruckmeier C, Rieger B, Herrmann WA, Kühn FE. Transformation of carbon dioxide with homogeneous transition-metal catalysts: a molecular solution to a global challenge? Angew Chem Int Ed. 2011;50:8510–37.
Liu Q, Wu L, Jackstell R, Beller M. Using carbon dioxide as a building block in organic synthesis. Nat Commun. 2015;6:5933.
North M, Styring P. Perspectives and visions on CO2 capture and utilisation. Faraday Discuss. 2015;183:489–502.
Aresta M, Dibenedetto A, Angelini A. Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem Rev. 2014;114:1709–42.
Aresta M. Activation of small molecules. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany, 2006. pp 1–41.
Artz J, Müller TE, Thenert K, Kleinekorte J, Meys R, Sternberg A, et al. Sustainable conversion of carbon dioxide: an integrated review of catalysis and life cycle assessment. Chem Rev. 2018;118:434–504.
Inoue S, Koinuma H, Tsuruta T. Copolymerization of carbon dioxide and epoxide. J Polym Sci. 1969;7:287–92.
Kember MR, Buchard A, Williams CK. Catalysts for CO2/epoxide copolymerisation. Chem Commun. 2011;47:141–63.
Kozak CM, Ambrose K, Anderson TS. Copolymerization of carbon dioxide and epoxides by metal coordination complexes. Coord Chem Rev. 2018;376:565–87.
Poland SJ, Darensbourg DJ. A quest for polycarbonates provided via sustainable epoxide/CO2 copolymerization processes. Green Chem. 2017;19:4990–5011.
Small BL, Brookhart M, Bennett AMA. Highly active iron and cobalt catalysts for the polymerization of ethylene. J Am Chem Soc. 1998;120:4049–50.
Ittel SD, Johnson LK, Brookhart M. Late-metal catalysts for ethylene homo- and copolymerization. Chem Rev. 2000;100:1169–204.
Britovsek GJP, Bruce M, Gibson VC, Kimberley BS, Maddox PJ, Mastroianni S, et al. Iron and cobalt ethylene polymerization catalysts bearing 2,6-bis(imino)pyridyl ligands: synthesis, structures, and polymerization studies. J Am Chem Soc. 1999;121:8728–40.
Qi M, Dong Q, Wang D, Byers JA. Electrochemically switchable ring-opening polymerization of lactide and cyclohexene oxide. J Am Chem Soc. 2018;140:5686–90.
Delle Chiaie KR, Biernesser AB, Ortuño MA, Dereli B, Iovan DA, Wilding MJT, et al. The role of ligand redox non-innocence in ring-opening polymerization reactions catalysed by bis(imino)pyridine iron alkoxide complexes. Dalton Trans. 2017;46:12971–80.
Biernesser AB, Delle Chiaie KR, Curley JB, Byers JA. Block copolymerization of lactide and an epoxide facilitated by a redox switchable iron-based catalyst. Angew Chem Int Ed. 2016;55:5251–4.
Wei J, Riffel MN, Diaconescu PL. Redox control of aluminum ring-opening polymerization: a combined experimental and DFT investigation. Macromolecules. 2017;50:1847–61.
Wei J, Diaconescu PL. Redox-switchable ring-opening polymerization with ferrocene derivatives. Acc Chem Res. 2019;52:415–24.
Wang X, Thevenon A, Brosmer JL, Yu I, Khan SI, Mehrkhodavandi P, et al. Redox control of group 4 metal ring-opening polymerization activity toward L-lactide and Ε-caprolactone. J Am Chem Soc. 2014;136:11264–7.
Quan SM, Wang X, Zhang R, Diaconescu PL. Redox switchable copolymerization of cyclic esters and epoxides by a zirconium complex. Macromolecules. 2016;49:6768–78.
Lai A, Hern ZC, Diaconescu PL. Switchable ring-opening polymerization by a ferrocene supported aluminum complex. ChemCatChem. 2019;11:4210–18.
Kruper WJ, Swart DJ. US Patent 4,500,704, 1985.
Kruper WJ, Dellar DV. Catalytic formation of cyclic carbonates from epoxides and CO2 with chromium metalloporphyrinates. J Org Chem. 1995;60:725–7.
Darensbourg DJ, Rainey P, Yarbrough J. Bis-salicylaldiminato complexes of zinc. examination of the catalyzed epoxide/CO2 copolymerization. Inorg Chem. 2001;40:986–93.
Darensbourg DJ, Adams MJ, Yarbrough JC, Phelps AL. Synthesis and structural characterization of double metal cyanides of iron and zinc: catalyst precursors for the copolymerization of carbon dioxide and epoxides. Inorg Chem. 2003;42:7809–18.
Della Monica F, Buonerba A, Capacchione C. Homogeneous iron catalysts in the reaction of epoxides with carbon dioxide. Adv Synth Catal. 2019;361:265–82.
Shibasaki Y, Sanada H, Yokoi M, Sanda F, Endo T. Activated monomer cationic polymerization of lactones and the application to well-defined block copolymer synthesis with seven-membered cyclic carbonate. Macromolecules. 2000;33:4316–20.
Suriano F, Coulembier O, Hedrick JL, Dubois P. Functionalized cyclic carbonates: from synthesis and metal-free catalyzed ring-opening polymerization to applications. Polym Chem. 2011;2:528–33.
Tezuka K, Komatsu K, Haba O. The anionic ring-opening polymerization of five-membered cyclic carbonates fused to the cyclohexane ring. Polym J. 2013;45:1183–7.
Brignou P, Priebe GM, Casagrande O, Carpentier J-F, Guillaume SM. Polycarbonates derived from green acids: ring-opening polymerization of seven-membered cyclic carbonates. Macromolecules. 2010;43:8007–17.
Saito T, Takojima K, Oyama T, Hatanaka S, Konno T, Yamamoto T, et al. Trimethyl glycine as an environmentally benign and biocompatible organocatalyst for ring-opening polymerization of cyclic carbonate. ACS Sustain Chem Eng. 2019;7:8868–75.
Gao L, Dong B, Zhang J, Chen Y, Qiao H, Liu Z, et al. Functional biodegradable nitric oxide donor-containing polycarbonate-based micelles for reduction-triggered drug release and overcoming multidrug resistance. ACS Macro Lett. 2019;8:1552–8.
Marquis ET, Sanderson JR. US Patent 5,283,356, 1994.
Ji DF, Lu XB, He R. Syntheses of cyclic carbonates from carbon dioxide and epoxides with metal phthalocyanines as catalyst. Appl Catal A-Gen. 2000;203:329–33.
Jin L, Jing H, Chang T, Bu X, Wang L, Liu Z. Metal porphyrin/phenyltrimethylammonium tribromide: high efficient catalysts for coupling reaction of CO2 and epoxides. J Mol Catal A-Chem. 2007;261:262–6.
Buchard A, Kember MR, Sandeman KG, Williams CK. A bimetallic iron(Iii) catalyst for CO2/epoxide coupling. Chem Commun. 2011;47:212–14.
Dengler JE, Lehenmeier MW, Klaus S, Anderson CE, Herdtweck E, Rieger B. A one-component iron catalyst for cyclic propylene carbonate synthesis. Eur J Inorg Chem. 2011;3:336–43.
Sheng X, Qiao L, Qin Y, Wang X, Wang F. Highly efficient and quantitative synthesis of a cyclic carbonate by iron complex catalysts. Polyhedron. 2014;74:129–33.
Adolph M, Zevaco TA, Walter O, Dinjus E, Döring M. Easy-to-handle ionic transition metal complexes in the formation of carbonates from epoxides and CO2: A N4-ligand system based on N,N-bis(2-pyridinecarboxamide)-1,2-benzene. Polyhedron. 2012;48:92–8.
Adolph M, Zevaco TA, Altesleben C, Walter O, Dinjus E. New cobalt, iron and chromium catalysts based on easy-to-handle N4-chelating ligands for the coupling reaction of epoxides with CO2. Dalton Trans. 2014;43:3285–96.
Fuchs MA, Zevaco TA, Ember E, Walter O, Held I, Dinjus E, et al. Synthesis of cyclic carbonates from epoxides and carbon dioxide catalyzed by an easy-to-handle ionic iron(III) complex. Dalton Trans. 2013;42:5322–9.
Whiteoak CJ, Martin E, Martinez Belmonte M, Benet-Buchholz J, Kleij AW. An efficient iron catalyst for the synthesis of five- and six-membered organic carbonates under mild conditions. Adv Synth Catal. 2012;354:469–76.
Whiteoak CJ, Gjoka B, Martin E, Martinez Belmonte M, Escudero-Adan EC, Zonta C, et al. Reactivity control in iron(III) amino triphenolate complexes: comparison of monomeric and dimeric complexes. Inorg Chem. 2012;51:10639–49.
Whiteoak CJ, Martin E, Escudero-Adán E, Kleij AW. Stereochemical divergence in the formation of organic carbonates derived from internal epoxides. Adv Synth Catal. 2013;355:2233–9.
Buonerba A, De Nisi A, Grassi A, Milione S, Capacchione C, Vagin S, et al. Novel iron(III) catalyst for the efficient and selective coupling of carbon dioxide and epoxides to form cyclic carbonates. Catal Sci Technol. 2015;5:118–23.
Al-Qaisi FaM, Nieger M, Kemell ML, Repo TJ. Catalysis of cycloaddition of carbon dioxide and epoxides using a bifunctional Schiff case iron(III) catalyst. ChemistrySelect. 2016;1:545–8.
Alhashmialameer D, Collins J, Hattenhauer K, Kerton FM. Iron amino-bis(phenolate) complexes for the formation of organic carbonates from CO2 and oxiranes. Catal Sci Technol. 2016;6:5364–73.
Andrea KA, Brown TR, Murphy JN, Jagota D, McKearney D, Kozak CM, et al. Characterization of oxo-bridged iron amino-bis(phenolate) complexes formed intentionally or in situ: mechanistic insight into epoxide deoxygenation during the coupling of CO2 and epoxides. Inorg Chem. 2018;57:13494–504.
Andrea KA, Butler ED, Brown TR, Anderson TS, Jagota D, Rose C, et al. Iron complexes for cyclic carbonate and polycarbonate formation: selectivity control from ligand design and metal-center geometry. Inorg Chem. 2019;58:11231–40.
Fazekas E, Nichol GS, Shaver MP, Garden JA. Stable Fe(III) phenoxyimines as selective and robust CO2/epoxide coupling catalysts. Dalton Trans. 2018;47:13106–12.
Della Monica F, Maity B, Pehl T, Buonerba A, De Nisi A, Monari M, et al. [OSSO]-type iron(III) complexes for the low-pressure reaction of carbon dioxide with epoxides: catalytic activity, reaction kinetics, and computational study. ACS Catal. 2018;8:6882–93.
Cozzolino M, Leo V, Tedesco C, Mazzeo M, Lamberti M. Salen, salan and salalen iron(III) complexes as catalysts for CO2/epoxide reactions and ROP of cyclic esters. Dalton Trans. 2018;47:13229–38.
Kamphuis AJ, Milocco F, Koiter L, Pescarmona P, Otten E. Highly selective single-component formazanate ferrate(II) catalysts for the conversion of CO2 into cyclic carbonates. ChemSusChem. 2019;12:3635–41.
Driscoll OJ, Hafford-Tear CH, McKeown P, Stewart JA, Kociok-Köhn G, Mahon MF, et al. The synthesis, characterisation and application of iron(III)–acetate complexes for cyclic carbonate formation and the polymerisation of lactide. Dalton Trans. 2019;48:15049–58.
Nakano K, Kobayashi K, Ohkawara T, Imoto H, Nozaki K. Copolymerization of epoxides with carbon dioxide catalyzed by iron–corrole complexes: synthesis of a crystalline copolymer. J Am Chem Soc. 2013;135:8456–9.
Taherimehr M, Al-Amsyar SM, Whiteoak CJ, Kleij AW, Pescarmona PP. High activity and switchable selectivity in the synthesis of cyclic and polymeric cyclohexene carbonates with iron amino triphenolate catalysts. Green Chem. 2013;15:3083–90.
Taherimehr M, Serta J, Kleij AW, Whiteoak CJ, Pescarmona PP. New iron pyridylamino-bis(phenolate) catalyst for converting CO2 into cyclic carbonates and cross-linked polycarbonates. ChemSusChem. 2015;8:1034–42.
Acknowledgements
We would like to thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for financial support, including a Discovery Grant (FMK). KAA is supported by a Vanier Canada Graduate Scholarship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Andrea, K.A., Kerton, F.M. Iron-catalyzed reactions of CO2 and epoxides to yield cyclic and polycarbonates. Polym J 53, 29–46 (2021). https://doi.org/10.1038/s41428-020-00395-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-020-00395-6
- Springer Nature Limited