
Abstract
Malignant cells upregulate distinct energy metabolism programs that support their proliferation, migration, and adaptation to the stressful tumor microenvironment (TME). Additionally, this exaggerated metabolic activity allows cancer cells to hijack essential nutrients and outcompete neighboring infiltrating immune cells, thereby impairing antitumor immunity. During recent years, there has been great interest in the field to understand the tumor-induced energy metabolism signals that regulate the function of immune cells in individuals with cancer. Accordingly, it is now well accepted that uncovering the mechanisms that instruct the metabolic behavior of cancer cells and tumor-associated immune cells is an indispensable strategy for the development of new approaches to overcome immune suppression in tumors. Thus, in this minireview, we briefly discuss the interaction between particular metabolic signaling pathways and immunosuppressive activity in different subsets of myeloid cells within the TME. Additionally, we illustrate potential central mechanisms controlling the metabolic reprogramming of myeloid cells in response to tumor-derived factors.
Similar content being viewed by others


Introduction
The transformation of normal cells into malignant populations is accompanied by a significant reorganization of master energy metabolic pathways that sustain pathological proliferation. This phenomenon has been termed “metabolic reprogramming,” and it is one of the hallmarks of the process of cancer progression.1,2,3 Tumor cells fuel their exaggerated appetite for nutrients by exhibiting particular metabolic signatures, including increased aerobic glycolysis (Warburg effect). Indeed, the elevated glucose dependency of cancer cells plays a fundamental role in tumor growth and metastasis, and has been recently proposed as a central pathway in the evasion of immune responses.4,5,6 Additionally, the activation of aerobic glycolysis and other key metabolic programs in cancer cells triggers the accumulation of metabolic by-products that increase the regulatory effects of the stressful tumor microenvironment (TME) on infiltrating immune cells.5,7 The notion that tumor cells scavenge nutrients to create an immunosuppressive milieu is well accepted in the field, but the mechanisms by which this process occurs remain poorly understood. Most solid tumors are heavily infiltrated by different populations of immune cells, with myeloid subsets being the most frequent group. Myeloid cells, in the form of myeloid-derived suppressor cells (MDSCs), macrophages, and dendritic cells (DCs), accumulate in the tumors of most individuals with cancer, play a primary role in the inhibition of protective antitumor T-cell immunity, and represent a major obstacle in the development of effective cancer immunotherapies.8,9 Notably, the accumulation of myeloid cells in tumors correlates with worse prognosis in patients with different forms of cancer and makes tumors “cold” and highly resistant to checkpoint inhibitors.10,11 Upon infiltration into tumors, myeloid cells remodel their metabolic landscape and energy expenditure to cope with an aggressive environment characterized by an unbalanced supply of oxygen and nutrients.12 Seminal reports demonstrated the direct correlation between the intrinsic metabolic signatures of myeloid cells and the function, differentiation, and survival of these cells.13,14,15 In addition, tumor-induced modulation of metabolism in myeloid cells plays a key role in the growth of tumors and the inhibition of protective immunity. In this minireview, we briefly discuss the energy-producing metabolic pathways adopted by tumor-infiltrating myeloid cells, focusing on the metabolism of glucose, lipids, and amino acids. In addition, we debate the role of these metabolic processes in the immunosuppressive phenotypes of myeloid cells.
The “Warburg” battle between tumor and myeloid cells
After malignant transformation, tumor cells acquire an increased ability to consume glucose through aerobic glycolysis, which is a process whereby glucose is metabolized into lactate and the characteristic acidification of the TME occurs. This phenomenon is known as the Warburg effect.16 A recent report illustrated that the increased glycolysis in tumors can be explained by oscillations in the energetics and redox status of cancer cells compared to normal cells during the cell cycle.17 The main purposes of this metabolic polarization are to generate energy, especially under hypoxic conditions, and to provide carbon sources for synthesizing biomolecules that feed the robust tumor growth and expansion.18,19 Moreover, seminal studies showed that the pathological aerobic glycolysis in cancer cells and the subsequent accumulation of lactate in the tumor milieu blocked protective T-cell immunity in tumors and enhanced the immunoregulatory activity of tumor-associated myeloid cells.4,5 Upon infiltration into the TME, myeloid cells enter an environment in which cancer cells hijack the majority of glucose through heightened uptake.4,20,21 This disadvantageous glucose competition can ultimately influence signal transduction and gene expression in myeloid cells, leading to their metabolic reprogramming and the promotion of immunosuppressive and protumorigenic properties.21
Tumor-associated macrophages (TAMs) represent a major component of the myeloid infiltrate in solid tumors and play a vital role in cancer progression and metastasis.9,22 Macrophages can be polarized in vitro toward M1 or M2 phenotypes,23 which are also characterized by opposite methods of glucose utilization. M1 macrophages primarily metabolize glucose through glycolysis24,25 and display a proinflammatory signature mediated by the production of cytokines such as interleukin (IL)-1β and tumor necrosis factor-α.23,26 During polarization toward M1, macrophages upregulate an array of genes induced by hypoxia-inducible factor alpha (HIF-1α), such as GLUT1, PFKFB3, and PGK1, which mediate the uptake and catabolism of glucose.27 In agreement with this finding, a recent study revealed that succinate originating from the Krebs cycle induces the expression of IL-1β through the activity of the transcription factor HIF-1α.24 Moreover, M1 macrophages are characterized by the increased production of nitric oxide (NO), a factor that promotes glycolysis and inhibits oxidative phosphorylation (OXPHOS) by blocking the activity of complexes I, II, and IV of the electron transport chain.28 These results suggest the existence of multiple mechanisms regulating the glycolytic potential of M1 macrophages. On the other hand, M2 macrophages are induced upon activation by cytokines associated with chronic inflammation, such as IL-4, IL-10, IL-13, and transforming growth factor-β, and show low glucose uptake and glycolytic activity. Instead, M2-polarized cells preferentially produce energy through increased lipid catabolism and OXPHOS.14 Upregulation of OXPHOS and lipid metabolism in M2 cells occurs through metabolic reprogramming mediated by the transcription factor peroxisome proliferator-activated receptor gamma (PPAR-γ) along with the master regulators of mitochondrial biogenesis, PPAR-γ-coactivator 1 (PGC1) α and β.29 Due to the dynamic and heterogeneous nature of the TME, it can be predicted that TAMs acquire the M1 phenotype in the early stages of tumor development, followed by a shift to the M2 phenotype after the tumor is established. However, such a hypothesis remains to be confirmed in preclinical models and tumors from patients. Another pivotal consideration is that a clear distinction between macrophage phenotypes may not exist in vivo, either in cancer or in other chronic inflammatory diseases.30 In support of this argument, a recent report showed that glucose metabolism also contributes to M2 activation by sustaining fatty acid synthesis and OXPHOS in a manner dependent on the activation of the mammalian target of rapamycin complex 2 (mTORC2). Accordingly, the absence of mTORC2 signaling in macrophages delays tumor growth but diminishes immunity against parasitic nematodes.31 Therefore, the TME may instead be infiltrated with a wide spectrum of TAM subsets,32 including both monocyte-derived and embryonically derived macrophages.33,34
The metabolic activity of tumor-associated DCs remains poorly characterized. Seminal studies show that under physiological conditions, DCs undergo increased glycolysis as they mature into professional antigen-presenting cells. This process appears to be related to the induction of anabolic pathways that support the expansion of the endoplasmic reticulum (ER) and the Golgi apparatus.35,36 Although hypoxia enhances the maturation of DCs through a shift toward glycolysis,37 the majority of tumor-infiltrating DCs are paradoxically immature and show immunoregulatory phenotypes.38 This incongruity can be explained by the fact that some tumor-secreted metabolites, such as adenosine and lactate, negatively impact the immunogenic activity of DCs.39,40 In addition, glucose deprivation in the TME could force DCs to transition to OXPHOS, thereby promoting immunosuppression. Consistent with this postulation, an increased accumulation of lipids and elevated levels of lipid oxidation products have been reported to be major regulators of the immunosuppressive activity of tumor-associated DCs.41,42 Thus, similar to TAMs, tumor-associated DCs undergo metabolic reprogramming toward lipid oxidation, a switch that serves as a key driver of their tolerogenic potential.
Neutrophils mainly depend on glycolysis and the pentose phosphate pathway (PPP) for their maturation, function, and chemotaxis.43 The microbicidal activity of neutrophils relies on NADPH generated from the PPP as a precursor for NADPH oxidase 2. In addition, the assembly of neutrophil extracellular traps (NETs) requires the glycolytic pathway.44 Recent studies show that two populations of tumor-associated neutrophils accumulate in individuals with cancer: anti-tumorigenic PMNs and protumorigenic PMNs, which exhibit elevated immunosuppressive activity.45,46 Further results show that immunoregulatory PMNs (PMN-MDSCs) undergo active ER stress and express the endoplasmic stress-related marker lectin-type oxidized low-density lipoprotein receptor-1 (LOX-1).47 However, the metabolic checkpoints at which PMNs acquire pro- or antitumor potential remain largely unknown.
The accumulation of MDSCs in tumor-bearing hosts is an important mechanism underlying the suppression of protective T-cell responses and is a major obstacle to cancer immunotherapy. The inhibitory activity of MDSCs has been attributed to several pathways, including the release of reactive oxygen species (ROS) and peroxynitrite, the depletion of various amino acids, and the induction of regulatory T cells (Tregs). Despite the evident role of MDSCs in tumor-induced anergy, the therapeutic inhibition of pathways that regulate MDSCs has thus far resulted in a limited restoration of protective T-cell immunity and weak antitumor effects. Reports that unravel the effect of glycolysis in the expansion and suppressive capacity of MDSCs are few and are often contradictory. A dynamic metabolic flux analysis revealed that a high glycolytic flux is needed for the maturation of MDSCs from bone marrow precursors and suggested an indirect mechanism by which the consumption of carbon sources by MDSCs results in the suppression of effector T cells.48 Previous results showed that glycolysis promotes MDSC survival in tumor-bearing animals by preventing ROS-mediated apoptosis via the antioxidant activity of the glycolytic intermediate phosphoenolpyruvate.49 Conversely, SIRT1 reprogrammed MDSCs into inflammatory M1 macrophages by shifting them to HIF-1α-dependent glycolysis.50 In this context, activation of the glucocorticoid receptor regulated the tolerogenic function of MDSCs by blocking HIF-1α and glycolysis.51 Furthermore, the Warburg effect in cancer cells has been described to modulate MDSC accumulation and function. The activity of the enzyme lactate dehydrogenase A in pancreatic tumor cells increased the infiltration and suppression of MDSCs in mice challenged with tumors. Accordingly, a ketogenic diet resulted in lower levels of lactate production by tumor cells and slower tumor growth, which was associated with a decrease in MDSC numbers.7
Unbalanced lipid metabolism induces impaired and tolerogenic myeloid cells in tumors
In addition to metabolizing glucose, tumor cells obtain additional energy sources by using lipolysis to burn lipids acquired from the surrounding adipocytes and adipocyte-like fibroblasts.52 In addition, aggressive and invasive cancers undergo de novo fatty acid synthesis, which leads to the release of high amounts of fatty acids into the TME.53,54 These findings suggest that the TME is enriched in lipids and lipid derivatives, including lipoproteins and oxidized forms of lipids. Tumor-infiltrating myeloid cells tend to scavenge lipid species as an alternative fuel for energy metabolic pathways. In agreement with this observation, DCs within the tumor milieu can take up and accumulate lipids through upregulation of the transporter MSR1.41 Notably, DCs with high lipid content are unable to present antigens and acquire a suppressive phenotype.41,55 Moreover, nutrient limitation in the TME can promote the incompetence of DCs by triggering ER stress. A study conducted by Cubillos-Ruiz et al.56 revealed the role of XBP1 activation by lipid peroxides in blocking the capability of DCs to promote antitumor T-cell responses.
The Th2 cytokine IL-4 polarizes macrophages into a M2 phenotype through a mechanism involving the STAT6–PGC1β axis, which in turn reprograms metabolism toward fatty acid oxidation (FAO).29 Tumor-associated MDSCs, which share some similarities with M2 macrophages, were shown to utilize FAO to fuel their energy needs.57 MDSCs in the TME increased fatty acid uptake, upregulated key enzymes required for FAO, and consumed more oxygen. Pharmacological approaches that interfered with FAO inhibited the suppressive activity of MDSCs and delayed tumor growth. In addition, blocking FAO enhanced the therapeutic outcomes of chemotherapy and adoptive cellular therapy (ACT). Furthermore, LOX-1 has been used as a biomarker to differentiate between suppressive, low-density PMN-MDSCs, and high-density neutrophils in non-small-cell lung carcinoma patients.47 Interestingly, a whole-genome analysis showed that PMN-MDSCs displayed an elevated ER stress signature compared to neutrophils from healthy individuals. This observation was further endorsed when healthy neutrophils were converted into immunosuppressive cells in response to the induction of ER stress. Despite the importance of these results, the mechanisms by which ER stress contributes to the overall metabolism and function of PMN-MDSCs in tumors remain obscure.58
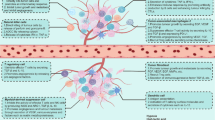
The central mechanisms that govern FAO and lipid metabolism in tumor-associated MDSCs are still unclear. Recent research by Al-Khami et al.59 suggested the role of the tumor-derived factors granulocyte colony-stimulating factor and granulocyte–macrophage colony-stimulating factor and subsequent signaling through STAT3 and STAT5 in the lipid uptake and FAO metabolic reprogramming that occurs in tumor-related MDSCs (Fig. 1). Another potential mediator of this effect is the AMP-activated protein kinase (AMPK).60 The decreased availability of glucose and other nutrients in tumors can reduce the intracellular levels of ATP, thereby leading to the activation of AMPK. To cope with starvation-related stress, AMPK (in the form of α1–α2/β1γ1 heterotrimers) promotes cellular catabolic pathways such as FAO and mitochondrial biogenesis.61,62 AMPK signaling drives mitochondrial biogenesis and induces the expression of an array of metabolic genes associated with FAO, such as carnitine palmitoyltransferase 1 (CPT1), CD36, 3-hydroxyacyl-CoA dehydrogenase (HADHA), and acyl-CoA dehydrogenase (ACADM), through direct activation of the PGC1β/PPARγ axis. In addition, AMPK phosphorylates and deactivates acetyl-CoA carboxylase (ACC1),63 an allosteric inhibitor of CPT1, the key limiting enzyme of FAO. Moreover, AMPK phosphorylates ULK1 and MFF, an action that supports mitochondrial quality by initiating mitochondrial fission and enhancing the recycling of damaged mitochondria.64,65 Notably, knockdown of AMPK promoted the accumulation of mitochondrial ROS, thereby stabilizing HIF-1α and pushing cells into aerobic glycolysis.66 Thus, AMPK could play a major role in the metabolic polarization of myeloid cells.67 However, most of the metabolic studies investigating the function of AMPK have been completed in tumor cell lines cultured in vitro. Thus, although the role of AMPK has been well established in cancer cells,62 the endogenous effect of AMPK on the modulation of tumor-associated myeloid cells remains unclear. Initial reports showed that pharmacological activation of AMPK blocked MDSC function in tumors,68,69 while another study showed that AMPK signaling promoted MDSC activity in doxorubicin-resistant tumors.70 In models of autoimmune and metabolic disease, AMPK activity in macrophages triggered an anti-inflammatory phenotype by supporting FAO and a shift away from aerobic glycolysis.71,72,73

AMPK induces PGC1β/PPARγ to upregulate CPT1 (1). AMPK phosphorylates ULK1, which promotes mitophagy and enhances mitochondrial function (2). Additionally, AMPK induces PGC1α to block mtROS, which can shift MDSCs to glycolysis driven by HIF-1α (3). In addition, tumor-derived GM-CSF and G-CSF induce STAT3/STAT5 signaling, leading to an increase in lipid uptake through the translocase CD36, which activates oxidative metabolism and promotes the suppressive functions of MDSCs (4). Whether these signaling events occur simultaneously or are partitioned and occur in a particular order remains to be determined
Prostaglandin production regulates the activity of myeloid cells in tumors
Arachidonic acid metabolism and the formation of prostaglandins (PGs) impact the function of tumor-infiltrating myeloid cells. Additionally, in melanoma cells, the production of PGE2 mediated by COX-2 activity shifted the myeloid compartment toward the support of tumor growth and immune escape.74 Furthermore, cancer cell-derived PGE2 induced the expression of the immunosuppressive factor arginase I in tumor-infiltrating MDSCs.75 Accordingly, the genetic deletion of COX in tumors allows myeloid cells to mature into immunogenic phenotypes characterized by type I interferon responses that prime T cells to eradicate tumors. Additionally, several groups explored the intrinsic role of PGs and COX enzymes in regulating the accumulation and immunoregulatory activity of MDSCs.70,76,77,78 These studies show the key role of intrinsic prostanoid production in the immunosuppressive activity of tumor-associated myeloid cells. However, the mechanistic connection between prostanoids and the polarization of energy metabolic processes in myeloid cells remains unknown.
Amino-acid starvation: a hallmark of tumor-associated myeloid cell-induced suppression
Human tumor cells, stromal fibroblasts, and infiltrating myeloid cells can degrade amino acids in their local environment.79,80,81 Arginine metabolism has emerged as a critical regulator of innate and adaptive immune responses. The key arginine-catabolizing enzymes linked to immune responses are the isoforms of arginase (arginase 1 and 2) and NO synthase (NOS 1–3). It is becoming increasingly clear that myeloid cells modulate immunity through the regulation of arginine. Polarized macrophages show different modalities of metabolizing the nonessential amino acid arginine.82,83 M1 macrophages break down arginine into NO and citrulline through the expression of NOS2.84,85 Controlled levels of NO can have antitumor effects by inducing direct cytotoxicity.86,87 Thus, the exclusive induction of NOS2 in tumor-infiltrating myeloid cells is associated with antitumor responses.88 On the other hand, M2 macrophages and TAMs utilize arginase I to convert arginine into ornithine and polyamines that promote tumor growth by enhancing cell differentiation, proliferation, and tissue remodeling.89,90 Although the processes seem contradictory, both mechanisms of arginine degradation can be active in the TME. Indeed, MDSCs block antitumor immunity by adopting both mechanisms of l-arginine metabolism.91,92 By upregulating arginase I, MDSCs block T-cell proliferation by limiting access to extracellular arginine.93 Furthermore, under limiting amounts of arginine, iNOS activity in MDSCs generates reactive nitrogen species that induce oxidative stress in T cells and block T-cell proliferation and interferon-γ production. The clinical importance of arginine availability has been suggested by reports that show a correlation between lower levels of arginine and poor T-cell function in patients with advanced tumors.94 In addition, injection of mice with arginine increased antitumor T-cell activity and improved the effect of ACT,95 indicating the primary role of arginine in T-cell function.
The immunomodulatory enzyme indoleamine 2,3-dioxygenase (IDO) is expressed by cancer cells and various subsets of myeloid cells infiltrating the TME.81 The activity of IDO has been associated with poor clinical outcomes.96,97,98 IDO catabolizes tryptophan (Trp), an essential amino acid, generating catabolites known collectively as kynurenines (Kyns). The enzymatic activity of IDO suppresses T-cell responses and supports Treg formation99 through either the depletion of Trp or the production of Kyns. Mechanistically, the general control nonderepressible 2 (GCN2) kinase in T cells was shown to be involved in mediating T-cell anergy resulting from Trp consumption.100 Additional studies showed the role of the mammalian target of rapamycin 2 as an IDO target mediator.101 Moreover, Kyns generated through Trp catabolism arrest T-cell proliferation, induce T-cell apoptosis, and expand Treg cohorts by binding to the aryl hydrocarbon receptor.102,103,104 Tolerogenic DCs in tumor-draining lymph nodes were noted for their ability to upregulate IDO and promote tumor growth.105 In a later study, IDO-competent DCs were shown to activate Tregs through a GCN2-dependent mechanism.106
Tumor cells expressing IDO attract MDSCs to their local environment, a process that is orchestrated by the presence of Tregs.107 Moreover, MDSCs upregulate IDO in various tumor models and hematological malignancies, and IDO governs the ability of MDSCs to block Th1 polarization and promote Treg development.108,109,110,111 Tumor-associated factors such as IL-6 and PGs were found to be required for the upregulation of IDO in MDSCs.110,112 An interesting study illustrated the role played by IDO in the regulation of IL-6 production, which directly correlated with MDSC infiltration and tumor aggressiveness and metastasis in an oncogenic KRAS-induced lung carcinoma model.113 The downstream mechanisms by which IDO mediates IL-6 expression remain to be determined. However, one study shows that GCN2 enhances the expression of IL-6 upon stimulation with the TLR4 ligand lipopolysachharide in macrophages experiencing amino-acid deprivation.114
Although glutamine is not an essential amino acid, glutamine addiction has been recognized in tumors.115 Tumors take up glutamine and rapidly convert it to glutamate through the activity of the glutaminase enzyme.116 Glutamine is utilized not only as a source of nitrogen for the production of nucleotides but also for the maintenance of mitochondrial biogenesis and integrity. Moreover, glutamine is required for the assembly of glutathione (GSH), which is involved in redox homeostasis, thereby suggesting a key role for glutamine in the adaptation of cells undergoing oxidative stress in the TME.117 Cancer patients show high plasma levels of glutamate.118 The targeting of glutamine catabolism was found to be effective in inhibiting the development of acute myeloid leukemia and in synergizing with therapeutic approaches such as BCL-2 and FLT3 blockade.119,120,121 The upregulation of enzymes involved in glutaminolysis has been associated with M2-polarized macrophages and TAMs.5 However, the relevance of this metabolic behavior and its impact on the suppressive functions of myeloid cells in tumor settings remain elusive. Taken together, these observations highlight glutamine metabolism as a novel target in cancer therapy.
Summary and conclusions
One of the foremost challenges facing the field is to decipher the mechanisms by which tumor and immune cells share metabolites (Fig. 2). Unfortunately, there is no win–win situation within the TME. In most scenarios, tumor cells succeed in hijacking essential nutrients and redirecting them away from immune cells, a tactic that exacerbates the existing immunosuppressive environment. Metabolic waste products regulate the immune activity of the tumor infiltrates and alter their metabolic profile. Some metabolites can serve both as substrates that fuel metabolic pathways and as signaling molecules that impact the function, polarization, trafficking, and epigenetic reprogramming of immune cells. Nutrient competition is not the only factor that determines the metabolic fate of the cell. Inflammatory cytokines, redox status, mitochondrial fitness, and systemic metabolism can also influence the cellular immunometabolism. Indeed, much remains to be learned about the mechanisms by which these elements act together or individually to shape the overall metabolic configuration.

The high consumption rate of glucose, lipids, and amino acids, which is associated with a massive release of metabolites and waste by-products, shapes the metabolic landscape of myeloid cells within the TME. Ultimately, this shaping leads to the expansion of suppressive myeloid cells, which are incapable of presenting antigens and priming effective T-cell responses against tumors
Cancer immunotherapies are designed to elicit immune responses against tumors; however, the TME stands as a strong barrier against the development of more effective and durable therapies. Among the different mechanisms by which cancer cells evade the immune effectors is the metabolic editing of tumor-associated cell populations. Thus, in order to develop successful immunotherapeutic strategies, it is important to understand the correlation between the aberrant metabolism and the impaired antitumor function of immune cells. Approaches to alter or block particular metabolic pathways are being tested for their ability to augment antitumor immune responses. Nonetheless, a caveat against these approaches is that they can nonspecifically target all cells in the tumor bed, which may negatively impact antitumor effectors. Considering this possibility, the development of novel technologies for targeting metabolic pathways uniquely utilized by protumorigenic immune cells is pivotal. Importantly, the majority of tumor-infiltrating myeloid cells adopt an oxidative metabolism that promotes their tolerogenic phenotypes. Therefore, switching the metabolic preference of these inhibitory subpopulations can imprint them with an immunogenic signature, possibly through the induction of epigenetic modifications that will upregulate host defense genes rather than suppressive mediators.
In summary, very little is currently known about the differentiation and metabolic landscape of myeloid populations in tumors, particularly in humans. In addition, integrating all of the tumor-associated cellular metabolic pathways into one matrix that can allow us to understand the immunological outcomes remains one of the main challenges in the field. Ultimately, cancer may not only be the unruly proliferation of cells under the influence of oncogenes, but it could also be the perfect interaction between pathological metabolic alterations and mutation loads in cells.
References
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Ward, P. S. & Thompson, C. B. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21, 297–308 (2012).
Boroughs, L. K. & DeBerardinis, R. J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 17, 351–359 (2015).
Chang, C. H. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241 (2015).
Colegio, O. R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014).
Calcinotto, A. et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 72, 2746–2756 (2012).
Husain, Z., Huang, Y., Seth, P. & Sukhatme, V. P. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 191, 1486–1495 (2013).
Murdoch, C., Muthana, M., Coffelt, S. B. & Lewis, C. E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 8, 618–631 (2008).
Qian, B. Z. & Pollard, J. W. Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010).
Gajewski, T. F., Schreiber, H. & Fu, Y. X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013).
DeNardo, D. G. et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 1, 54–67 (2011).
Biswas, S. K. Metabolic reprogramming of immune cells in cancer progression. Immunity 43, 435–449 (2015).
Biswas, S. K. & Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 15, 432–437 (2012).
Buck, M. D., Sowell, R. T., Kaech, S. M. & Pearce, E. L. Metabolic instruction of immunity. Cell 169, 570–586 (2017).
Mills, E. L., Kelly, B. & O’Neill, L. A. J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 18, 488–498 (2017).
Warburg, O., Wind, F. & Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 (1927).
Moreira, J. D. et al. The redox status of cancer cells supports mechanisms behind the Warburg effect. Metabolites 6, pii: E33 (2016).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Keibler, M. A. et al. Metabolic requirements for cancer cell proliferation. Cancer Metab. 4, 16 (2016).
Al-Khami, A. A., Rodriguez, P. C. & Ochoa, A. C. Metabolic reprogramming of myeloid-derived suppressor cells (MDSC) in cancer. Oncoimmunology 5, e1200771 (2016).
Al-Khami, A. A., Rodriguez, P. C. & Ochoa, A. C. Energy metabolic pathways control the fate and function of myeloid immune cells. J. Leukoc. Biol. 102, 369–380 (2017).
Biswas, S. K., Allavena, P. & Mantovani, A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin. Immunopathol. 35, 585–600 (2013).
Biswas, S. K. & Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 11, 889–896 (2010).
Tannahill, G. M. et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–242 (2013).
Rodriguez-Prados, J. C. et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J. Immunol. 185, 605–614 (2010).
Biswas, S. K., Sica, A. & Lewis, C. E. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J. Immunol. 180, 2011–2017 (2008).
Burke, B. et al. Hypoxia-induced gene expression in human macrophages: implications for ischemic tissues and hypoxia-regulated gene therapy. Am. J. Pathol. 163, 1233–1243 (2003).
Everts, B. et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120, 1422–1431 (2012).
Vats, D. et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell. Metab. 4, 13–24 (2006).
Murray, P. J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014).
Huang, S. C. et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity 45, 817–830 (2016).
Xue, J. et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288 (2014).
Ginhoux, F. & Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 44, 439–449 (2016).
Zhu, Y. et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 597 (2017).
Pearce, E. J. & Everts, B. Dendritic cell metabolism. Nat. Rev. Immunol. 15, 18–29 (2015).
Everts, B. et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat. Immunol. 15, 323–332 (2014).
Jantsch, J. et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 180, 4697–4705 (2008).
Scarlett, U. K. et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 209, 495–506 (2012).
Gottfried, E. et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107, 2013–2021 (2006).
Novitskiy, S. V. et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 112, 1822–1831 (2008).
Herber, D. L. et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 16, 880–886 (2010).
Cubillos-Ruiz, J. R., Bettigole, S. E. & Glimcher, L. H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 168, 692–706 (2017).
Jun, H. S., Weinstein, D. A., Lee, Y. M., Mansfield, B. C. & Chou, J. Y. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood 123, 2843–2853 (2014).
Rodriguez-Espinosa, O., Rojas-Espinosa, O., Moreno-Altamirano, M. M., Lopez-Villegas, E. O. & Sanchez-Garcia, F. J. Metabolic requirements for neutrophil extracellular traps formation. Immunology 145, 213–224 (2015).
Mantovani, A., Cassatella, M. A., Costantini, C. & Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 11, 519–531 (2011).
Singhal, S. et al. Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell 30, 120–135 (2016).
Condamine, T. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 1, pii: aaf8943 (2016).
Goffaux, G., Hammami, I. & Jolicoeur, M. A dynamic metabolic flux analysis of myeloid-derived suppressor cells confirms immunosuppression-related metabolic plasticity. Sci. Rep. 7, 9850 (2017).
Jian, S. L. et al. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 8, e2779 (2017).
Liu, G. et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. 74, 727–737 (2014).
Lu Y. et al. Glucocorticoid receptor promotes the function of myeloid-derived suppressor cells by suppressing HIF1alpha-dependent glycolysis. Cell. Mol. Immunol. (2017). https://doi.org/10.1038/cmi.2017.5.
Nieman, K. M. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 17, 1498–1503 (2011).
Nomura, D. K. et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140, 49–61 (2010).
Currie, E., Schulze, A., Zechner, R., Walther, T. C. & Farese, R. V. Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 18, 153–161 (2013).
Gao, F. et al. Radiation-driven lipid accumulation and dendritic cell dysfunction in cancer. Sci. Rep. 5, 9613 (2015).
Cubillos-Ruiz, J. R. et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161, 1527–1538 (2015).
Hossain, F. et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 3, 1236–1247 (2015).
Condamine, T. et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Invest. 124, 2626–2639 (2014).
Al-Khami, A. A. et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 6, e1344804 (2017).
Hammami, I. et al. Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC Cell Biol. 13, 18 (2012).
O’Neill, L. A. & Pearce, E. J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 213, 15–23 (2016).
O’Neill, L. A. & Hardie, D. G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493, 346–355 (2013).
Galic, S. et al. Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Invest. 121, 4903–4915 (2011).
Egan, D. F. et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 (2011).
Toyama, E. Q. et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281 (2016).
Rabinovitch, R. C. et al. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 21, 1–9 (2017).
Krawczyk, C. M. et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749 (2010).
Trikha, P. et al. Targeting myeloid-derived suppressor cells using a novel adenosine monophosphate-activated protein kinase (AMPK) activator. Oncoimmunology 5, e1214787 (2016).
Kim, S. H. et al. Phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J. Invest. Dermatol. 137, 1740–1748 (2017).
Rong, Y. et al. Doxorubicin resistant cancer cells activate myeloid-derived suppressor cells by releasing PGE2. Sci. Rep. 6, 23824 (2016).
Steinberg, G. R. & Schertzer, J. D. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: implications for diabetes and cardiovascular disease. Immunol. Cell. Biol. 92, 340–345 (2014).
Sag, D., Carling, D., Stout, R. D. & Suttles, J. Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J. Immunol. 181, 8633–8641 (2008).
Park, S. Y. et al. SIRT1/adenosine monophosphate-activated protein kinase alpha signaling enhances macrophage polarization to an anti-inflammatory phenotype in rheumatoid arthritis. Front. Immunol. 8, 1135 (2017).
Zelenay, S. et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162, 1257–1270 (2015).
Rodriguez, P. C. et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J. Exp. Med. 202, 931–939 (2005).
Obermajer, N., Muthuswamy, R., Odunsi, K., Edwards, R. P. & Kalinski, P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 71, 7463–7470 (2011).
Mao, Y. et al. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res. 73, 3877–3887 (2013).
Mao, Y. et al. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin. Cancer Res. 20, 4096–4106 (2014).
Uyttenhove, C. et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 9, 1269–1274 (2003).
Hsu, Y. L. et al. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget 7, 27584–27598 (2016).
Munn, D. H. & Mellor, A. L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 34, 137–143 (2013).
Rath, M., Muller, I., Kropf, P., Closs, E. I. & Munder, M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front. Immunol. 5, 532 (2014).
Modolell, M., Corraliza, I. M., Link, F., Soler, G. & Eichmann, K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur. J. Immunol. 25, 1101–1104 (1995).
MacMicking, J., Xie, Q. W. & Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 15, 323–350 (1997).
Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2, 907–916 (2001).
Albina, J. E. & Reichner, J. S. Role of nitric oxide in mediation of macrophage cytotoxicity and apoptosis. Cancer Metastasis Rev. 17, 39–53 (1998).
Stuehr, D. J. & Nathan, C. F. Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J. Exp. Med. 169, 1543–1555 (1989).
Marigo, I. et al. T cell cancer therapy requires CD40-CD40L activation of tumor necrosis factor and inducible nitric-oxide-synthase-producing dendritic cells. Cancer Cell 30, 651 (2016).
Albina, J. E., Mills, C. D., Henry, W. L. Jr. & Caldwell, M. D. Temporal expression of different pathways of 1-arginine metabolism in healing wounds. J. Immunol. 144, 3877–3880 (1990).
Chang, C. I., Liao, J. C. & Kuo, L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res. 61, 1100–1106 (2001).
Gabrilovich, D. I., Ostrand-Rosenberg, S. & Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268 (2012).
Rodriguez, P. C. & Ochoa, A. C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol. Rev. 222, 180–191 (2008).
Rodriguez, P. C. et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 64, 5839–5849 (2004).
Zea, A. H. et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 65, 3044–3048 (2005).
Geiger, R. et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167, 829–842 (2016). e13.
Ferns, D. M. et al. Indoleamine-2,3-dioxygenase (IDO) metabolic activity is detrimental for cervical cancer patient survival. Oncoimmunology 4, e981457 (2015).
Liu, H. et al. Increased expression of IDO associates with poor postoperative clinical outcome of patients with gastric adenocarcinoma. Sci. Rep. 6, 21319 (2016).
Choe, J. Y. et al. Indoleamine 2,3-dioxygenase (IDO) is frequently expressed in stromal cells of Hodgkin lymphoma and is associated with adverse clinical features: a retrospective cohort study. BMC Cancer 14, 335 (2014).
Munn, D. H. & Mellor, A. L. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 37, 193–207 (2016).
Munn, D. H. et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22, 633–642 (2005).
Van de Velde, L. A., Gingras, S., Pelletier, S. & Murray, P. J. Issues with the specificity of immunological reagents for murine IDO1. Cell Metab. 23, 389–390 (2016).
Terness, P. et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J. Exp. Med. 196, 447–457 (2002).
Zaher, S. S., Germain, C., Fu, H., Larkin, D. F. & George, A. J. 3-hydroxykynurenine suppresses CD4+ T-cell proliferation, induces T-regulatory-cell development, and prolongs corneal allograft survival. Invest. Ophthalmol. Vis. Sci. 52, 2640–2648 (2011).
Mezrich, J. D. et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198 (2010).
Munn, D. H. et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Invest. 114, 280–290 (2004).
Sharma, M. D. et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest. 117, 2570–2582 (2007).
Holmgaard, R. B. et al. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep. 13, 412–424 (2015).
Yu, J. et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 190, 3783–3797 (2013).
Schafer, C. C. et al. Indoleamine 2,3-dioxygenase regulates anti-tumor immunity in lung cancer by metabolic reprogramming of immune cells in the tumor microenvironment. Oncotarget 7, 75407–75424 (2016).
Obermajer, N., Muthuswamy, R., Lesnock, J., Edwards, R. P. & Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 118, 5498–5505 (2011).
Jitschin, R. et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 124, 750–760 (2014).
Yu, J. et al. Noncanonical NF-kappaB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J. Immunol. 193, 2574–2586 (2014).
Smith, C. et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2, 722–735 (2012).
Liu, H. et al. GCN2-dependent metabolic stress is essential for endotoxemic cytokine induction and pathology. Mol. Cell. Biol. 34, 428–438 (2014).
Wise, D. R. & Thompson, C. B. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 35, 427–433 (2010).
Mazurek, S. & Eigenbrodt, E. The tumor metabolome. Anticancer Res. 23, 1149–1154 (2003).
Mates, J. M., Perez-Gomez, C., Nunez de Castro, I., Asenjo, M. & Marquez, J. Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int. J. Biochem. Cell. Biol. 34, 439–458 (2002).
Droge, W., Eck, H. P., Betzler, M. & Naher, H. Elevated plasma glutamate levels in colorectal carcinoma patients and in patients with acquired immunodeficiency syndrome (AIDS). Immunobiology 174, 473–479 (1987).
Gregory M. A. et al. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp. Hematol. 58, 52–58 (2017).
Emadi, A. et al. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp. Hematol. 42, 247–251 (2014).
Jacque, N. et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 126, 1346–1356 (2015).
Acknowledgements
This work was partially supported by the National Institutes of Health (NIH) R01CA184185 to P.C.R.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Mohamed, E., Al-Khami, A.A. & Rodriguez, P.C. The cellular metabolic landscape in the tumor milieu regulates the activity of myeloid infiltrates. Cell Mol Immunol 15, 421–427 (2018). https://doi.org/10.1038/s41423-018-0001-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-018-0001-7
- Springer Nature Limited
Keywords
This article is cited by
-
Unraveling the complexity of STAT3 in cancer: molecular understanding and drug discovery
Journal of Experimental & Clinical Cancer Research (2024)
-
Integrating tumor and healthy epithelium in a micro-physiology multi-compartment approach to study renal cell carcinoma pathophysiology
Scientific Reports (2024)
-
Tumor-infiltrating CD36+CD8+T cells determine exhausted tumor microenvironment and correlate with inferior response to chemotherapy in non-small cell lung cancer
BMC Cancer (2023)
-
Fatty Acid Metabolism and Cancer Immunotherapy
Current Oncology Reports (2022)
-
Interaction between HLA-G and NK cell receptor KIR2DL4 orchestrates HER2-positive breast cancer resistance to trastuzumab
Signal Transduction and Targeted Therapy (2021)