
Abstract
Chimeric antigen receptor (CAR)-T cell therapy against T cell malignancies faces major challenges including fratricide between CAR-T cells and product contamination from the blasts. Allogeneic CAR-T cells, generated from healthy donor T cells, can provide ready-to-use, blast-free therapeutic products, but their application could be complicated by graft-versus-host disease (GvHD) and host rejection. Here we developed healthy donor-derived, CD7-targeting CAR-T cells (RD13-01) with genetic modifications to resist fratricide, GvHD and allogeneic rejection, as well as to potentiate antitumor function. A phase I clinical trial (NCT04538599) was conducted with twelve patients recruited (eleven with T cell leukemia/lymphoma, and one with CD7-expressing acute myeloid leukemia). All patients achieved pre-set end points and eleven proceeded to efficacy evaluation. No dose-limiting toxicity, GvHD, immune effector cell-associated neurotoxicity or severe cytokine release syndrome (grade ≥ 3) were observed. 28 days post infusion, 81.8% of patients (9/11) showed objective responses and the complete response rate was 63.6% (7/11, including the patient with AML). 3 of the responding patients were bridged to allogeneic hematopoietic stem cell transplantation. With a median follow-up of 10.5 months, 4 patients remained in complete remission. Cytomegalovirus (CMV) and/or Epstein-Barr virus (EBV) reactivation was observed in several patients, and one died from EBV-associated diffuse large B-cell lymphoma (DLBCL). Expansion of CD7-negative normal T cells was detected post infusion. In summary, we present the first report of a Phase I clinical trial using healthy donor-derived CD7-targeting allogeneic CAR-T cells to treat CD7+ hematological malignancies. Our results demonstrated the encouraging safety and efficacy profiles of the RD13-01 allogeneic CAR-T cells for CD7+ tumors.
Similar content being viewed by others

Introduction
T cell malignancies are highly aggressive hematological tumors which are generally associated with poor prognosis.1,2 In particular, relapsed or refractory (r/r) disease has dismal outcomes with a 5-year overall survival (OS) rate lower than 20%.1 Till now, there are no curative therapies beyond conventional chemotherapy or hematopoietic stem cell transplantation (HSCT), which can be associated with marked complications and toxicities. Therefore, there is an urgent need to develop novel therapies for this type of disease.
Immunotherapy using chimeric antigen receptor (CAR)-T cells has been under rapid development over the past decade.3,4 In multiple clinical studies using CAR-T cells targeting CD19, patients with r/r B cell leukemia and lymphoma show promising response and durable remission.5,6,7 However, the therapeutic efficacy of CAR-T cells against other types of malignancies remains under investigation. Meanwhile, the autologous nature of most CAR-T cell products has resulted in high expenses of manufacturing and inconsistent quality of the starting materials, therefore challenging the broader application of CAR-T cell therapy.8
Recently, technical advances have allowed for manufacturing allogeneic “universal” CAR-T cells, which may provide potential solutions for the issues mentioned above due to the off-the-shelf nature.8,9 Allogeneic CAR-T cells were first demonstrated for their in vivo expansion and anti-leukemic activity using an CD19-targeting CAR platform against r/r B-cell acute lymphoblastic leukemia (ALL).10 More recently, we successfully treated 6 patients with r/r B-ALL using CD19/CD22 dual-targeting allogeneic CAR-T cells that exhibited a manageable safety profile and prominent anti-leukemic function.11 These studies revealed an intriguing possibility of applying allogeneic CAR-T cell therapy to other types of malignancies. Allogeneic CAR-T therapy is particularly beneficial in treating T cell malignancies given that the autologous products can be contaminated with blasts. However, CAR-T cell fratricide, graft-versus-host disease (GvHD) and immune rejection from host T and NK cells would still become major hurdles against the effectiveness of allogeneic CAR-T therapy.12
CD7 is a transmembrane glycoprotein expressed in more than 95% of T-ALL and T-cell lymphomas, making it an attractive target in T cell malignancies.13,14 As CD7 is also expressed on most normal T cells, genetic depletion of CD7,14 or blockade of CD7 surface expression,15 are required to prevent CAR-T cell fratricide. Recent efforts have also focused on pre-selection of CD7-negative T cell side population to manufacture these CAR-T cells.16 Clinical studies using CD7-targeting CAR-T cells, derived from autologous T cells or HSCT donor T cells, have shown some anti-leukemic activity.15,16 However, other than a preliminary report on two patients,17 there has been little clinical evidence on the safety and efficacy of allogeneic CD7-CAR-T cells in a completed Phase I clinical trial. Of note, intensified lymphodepletion chemotherapy can be applied to reduce host rejection to allogeneic CAR-T cells, but will inevitably increase the incidence of infection and severe cytokine release syndrome (CRS).18 Thus, preclinical and clinical development of allogeneic CD7-CAR-T cells needs to overcome GvHD and immune rejection in addition to CAR-T cell fratricide.
Here, we present results from both preclinical and clinical assessments of RD13-01, the healthy donor-derived, CD7-targeting allogeneic CAR-T cells with specific designs to enhance persistence and potency. These CAR-T cells have been genetically depleted for CD7, T cell receptor (TCR) and human leukocyte antigen (HLA) class II, and were introduced with an NK cell inhibitor (NKi) and the common cytokine receptor γ chain (γc). RD13-01 CAR-T cells exhibited potent antitumor activity in preclinical models. A phase I clinical trial using RD13-01 was then conducted in patients with r/r CD7+ malignancies (NCT04538599), aiming to investigate the clinical safety, efficacy and pharmacokinetics of these CAR-T cells. To our knowledge, this is the first reported Phase I clinical study of CD7-targeting fully-allogeneic CAR-T cells.
Results
Generation of CD7-targeting allogeneic CAR-T cells that resist T cell- and NK cell-mediated rejection
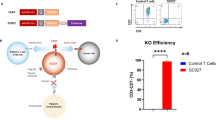
We aimed to address three major concerns regarding allogeneic CAR-T cell therapy against T cell malignancies, including fratricide, GvHD and allo-rejection. First, to protect CAR-T cells from fratricide and reduce GvHD, we generated CD7-targeting CAR-T cells (with a 4-1BB costimulatory domain) with CD7 and TCR/CD3 knockout (KO) via CRISPR/Cas9 (we named these cells CAR-dKO) (Fig. 1a, h). CAR-dKO T cells could be robustly expanded (Supplementary information, Fig. S1a), demonstrated antigen-specific tumor killing ability both in vitro and in vivo, as well as secreted inflammatory cytokines after tumor stimulation (Fig. 1b, c; Supplementary information, Fig. S1b, c).

a CAR expression and efficiency of TCR/CD3 and CD7 disruption on CAR-dKO T cells, measured by flow cytometry. b, c Tumor control ability of CAR-dKO T cells evaluated in NCG mice bearing Jurkat tumors. Mice were injected with 1 × 106 Jurkat tumor cells intravenously (i.v.) and treated with a single injection of 10 × 106 non-transduced (NT) T cells (n = 4) or CAR-dKO T cells (n = 8) 72 h after tumor inoculation. Bioluminescent images obtained at the indicated time points after tumor injection (b). Kaplan–Meier curves of mice treated with NT or CAR-dKO T cells (c), with P value calculated using a Log-rank test. d Top, composition of CD7+ and CD7− subsets in CD4+/CD8+ T cells from healthy donors (n = 5) and patients with T-ALL (n = 14). Bottom, elimination of HLA-II expression by disrupting RFX5, generating CAR-tKO T cells. e Expansion of allogeneic CD4+ T cells in the presence of CAR-dKO or CAR-tKO T cells in a mixed lymphocyte reaction (MLR) assay. f E-cadherin expression on CAR-tKO and CAR-NKi-tKO T cells. g NK cell cytotoxicity against CAR-tKO and CAR-NKi-tKO cells was detected using a luciferase-based killing assay at different NK:CAR-T ratios after 24 h of co-culture (n = 3). h Illustration of CAR-T cell development from “CAR-dKO” to “CAR-tKO” to “CAR-NKi-tKO”. Data are presented as means ± SD; All comparisons (except for Kaplan–Meier curves) were determined using student’s t-tests; *P < 0.05; **P < 0.01; ns not significant.
We then sought to improve the resistance against allo-rejection of these CAR-T cells, which is mainly mediated by the patient’s endogenous T and NK cells. Most normal T cells express CD7 (Fig. 1d), making them susceptible to CD7-CAR-T cell cytotoxicity. However, a CD7‒ side population was also detected, which can be resistant to CAR-T killing and therefore have the potential to mediate allo-rejection. In healthy donor T cells, CD7‒ T cells were primarily CD4+(Fig. 1d). Furthermore, in patients with T-ALL, the CD7‒ subset was more prominent in CD4+ (constitutes around ~20% of CD4+ cells) but not CD8+ T cells (Fig. 1d), indicating that CD4+ endogenous T cells can be the main contributor of allo-rejection against the CAR-T cells in our study. Thus, we disrupted RFX5 to ablate HLA-II in CAR-T cells in addition to CD7 and TCR/CD3 KO (we named these cells CAR-tKO) (Fig. 1d). Such genetic modification in CAR-T cells was able to diminish CD4+ T cell alloreactivity, indicated by the reduction of expansion and IFN-γ secretion of allogeneic CD4+ T cells (Fig. 1e; Supplementary information, Fig. S1d). Furthermore, allogeneic CAR-T cells can also be targeted through missing-self recognition by host NK cells, which can be negative for CD7 expression (Supplementary information, Fig. S1e). Therefore, we generated an NK cell inhibitory receptor (NKi) with the EC1-EC2 extracellular and transmembrane domains of E-cadherin (Ecad) fused to the CD28 intracellular domain. Ecad has been demonstrated to negatively regulate NK cell function via KLRG1 binding,19 and the CD28 intracellular domain was used as we found that it can enhance the inhibitory effect of NKi (Supplementary information, Fig. S1f, g). We showed that the incorporation of NKi into the CAR-T cell construct (Fig. 1f) led to reduced NK cell-mediated CAR-T cell lysis (Fig. 1g). We further confirmed the NKi function by overexpressing NKi in K562 cells and observed a reduced NK cell-mediated cytotoxicity. (Supplementary information, Fig. S1h‒j). In conclusion, we used genetic engineering to generate CD7-targeting CAR-T cells (we named these cells CAR-NKi-tKO) that resist fratricide (CD7-KO), GvHD (TCR-KO) and allo-rejections from both CD4+ T cells (HLA-II-KO) and NK cells (NKi) (Fig. 1h).
Enhancing CD7-CAR-T cell antitumor activity by introducing a γc receptor
CD7 on T cells has been shown to enhance the production of Interleukin (IL)-2,20 which is a pivotal cytokine for T cell expansion and effector function.21 Indeed, we found a decreased IL-2 production in CD19-targeting CAR-T cells after CD7-KO (Supplementary information, Fig. S2a), indicating that the function of our CAR-NKi-tKO cells might be limited due to low IL-2 signaling. Therefore, we incorporated the common cytokine receptor gamma chain (γc), a shared receptor subunit of γc cytokines,22 into the CD7-targeting CAR construct (Fig. 2a, we named these cells CAR-g-NKi) to enhance the downstream signaling. This construct was then utilized to generate CAR-T cells with tKO as described above (we named these cells CAR-g-NKi-tKO). The incorporation of γc promoted IL-2 production in CAR-T cells (Fig. 2b), together with an enhanced in vitro cytotoxicity as demonstrated by elevated cytolytic activity and perforin/granzyme A production (Fig. 2c, d). γc incorporation also increased proliferation while inhibiting apoptosis in tumor-stimulated CAR-T cells (Supplementary information, Fig. S2b). When testing antitumor function in vivo, the γc domain was found to augment CAR-T cell potency and prolong the overall survival in xenograft models (Fig. 2e). Furthermore, we found that γc-incorporated CAR-T cells achieved superior ex vivo expansion, and showed reduced levels of exhaustion markers (Fig. 2f, g), suggesting that γc enhanced the fitness of CAR-T cells. The effect of γc incorporation was independent of CAR targets, NKi expression and HLA-II-KO, as it also augmented effector potency in another CAR-T platform (Supplementary information, Fig. S2c‒i). Overall, we utilized CRISPR-mediated KOs (CD7, TCR and HLA-II), together with genetic incorporation of NKi and γc, to finalize the CAR-T cell design which will be used in clinical studies (Fig. 2h, we named these cells RD13-01).

a Schematic illustration of γc-tethered CAR design. b Secretion of IL-2 by CAR-NKi-tKO and CAR-g-NKi-tKO T cells following 8 h of stimulation using Jurkat tumor cells (n = 3). c Cytotoxicity of CAR-NKi-tKO and CAR-g-NKi-tKO T cells against Jurkat cells at indicated Effector: Target (CAR-T: Tumor) ratios after 8 h of co-culture. d Secretion of cytotoxic molecules in CD4+ and CD8+ subsets of CAR-NKi-tKO and CAR-g-NKi-tKO T cells following 8 h of stimulation using Jurkat tumor cells (n = 3). e NCG mice were injected with 1 × 106 Jurkat tumor cells (i.v.) and treated with a single injection of 10 × 106 NT, CAR-NKi-tKO or CAR-g-NKi-tKO T cells. Left, bioluminescent images obtained at the indicated time points after tumor injection; right, Kaplan–Meier curves of mice treated with NT, CAR-NKi-tKO or CAR-g-NKi-tKO T cells, with P value calculated using a Log-rank test. f Ex vivo expansion of CAR-NKi-tKO or CAR-g-NKi-tKO T cells during production. g Expression of TIM3, LAG3, and PD-1 on CAR-NKi-tKO or CAR-g-NKi-tKO T cells at indicated time points during production (n = 3). h Illustration of RD13-01 CAR-T cell degisn. Data are presented as means ± SD; All comparisons (except for Kaplan–Meier curves) were determined using student’s t-tests; *P < 0.05; **P < 0.01; ns not significant.
Characteristics of RD13-01 products
We conducted a phase I clinical trial of RD13-01 in patients with r/r CD7+ hematological malignancies. The primary endpoint was safety and the secondary end points included efficacy and pharmacokinetic profiles. RD13-01 were manufactured from allogeneic third-party healthy donor peripheral blood mononuclear cells (PBMCs) in advance, and residual TCR/CD3+ T cells were removed to minimize GvHD. No karyotypic abnormalities were observed in the cellular products (Supplementary information, Fig. S3a). A fluorescence in situ hybridization (FISH) probe targeting the TRAC locus detected no rearrangements in around 500 nuclei examined (Supplementary information, Fig. S3b). Using Droplet Digital PCR (ddPCR) and qPCR, we also detected no CD7:TRAC, TRAC:CD7, RFX5:TRAC, or RFX5:CD7 chromosomal rearrangements, while CD7:RFX5 and TRAC:RFX5 rearrangements were present in around 0.5% of the harvested cells (Supplementary information, Fig. S3c‒e). Chromosome translocations were undetectable in engrafted CAR-T cells (Supplementary information, Fig. S3d). On-target indels were detected in the cellular products, and the number increased in patients after infusion, followed by a decline; off-target indels were not observed in either cellular products or post-infusion patients (Supplementary information, Fig. S3e). Single cell RNA-sequencing (scRNAseq) analyses revealed that around 60% of infused cells express genes related to T cell memory (Supplementary information, Fig. S3f‒h).
Patient characteristics and treatment protocol
Eligible patients enrolled in our clinical study had r/r hematological malignancies after 2 or more lines of therapy and had measurable disease with CD7 expression. From September 10th, 2020 to January 20th, 2021, 12 patients were enrolled, including 7 with T-ALL, 4 with T cell lymphoma and 1 with acute myeloid leukemia (AML) (Fig. 3a, Table 1; Supplementary information, Data S1). The median patient age was 34 (ranged 8‒66) years, and the median number of prior lines of therapies was 4 (ranged 2‒7). The 12 patients had been treated with at least 2 lines of chemotherapy and 3 of them had undergone HSCT previously (2 allogeneic HSCT and 1 autologous HSCT, Supplementary information, Table S1). Previous central nervous system (CNS) involvement was reported for four patients. Of the 8 patients with leukemia (including T-ALL and AML), 3 exhibited extramedullary disease. Bone marrow involvement was found in 6 out of 8 patients with leukemia (except for Patients 7 and 9), and in all 4 patients with lymphoma. Of the total 10 patients with bone marrow involvement, the median percentage of bone marrow blasts was 29% (ranged 7%–95%). We were able to analyze the tumor cells before CAR-T infusion from 10 patients via flow cytometry, all revealing ≥75% of CD7 expression on malignant cells (Table 1).

a CONSORT diagram of the clinical trial. AML, acute myeloid leukemia; CNSL, central neural system lymphoma; FCE, fludarabine cyclophosphamide and etoposide. b Diagram of clinical treatment protocol: the day of RD13-01 infusion was set as day 0; the lympho-depleting condition regimen consisted of fludarabine (30 mg/m2), cyclophosphamide (300 mg/m2) and etoposide (100 mg/day) on days ‒7 to ‒3.
12 patients received lymphodepletion chemotherapy (Fludarabine 30 mg/m2 on day ‒7 to ‒3, cyclophosphamide 300 mg/m2 on day ‒7 to ‒3, and etoposide 100 mg/day on day ‒7 to ‒3) followed by RD13-01 infusion (day 0): 3 patients received 1 × 107 cells/kg, 6 patients received 2 × 107 cells/kg, and the other 3 received 3 × 107 cells/kg (Fig. 3a, b, Table 1). The detailed information of RD13-01 products used in this study is provided in Supplementary information, Table S2.
RD13-01 safety profile
The primary end point of this study was safety; dose limiting toxicity (DLT) and incidences of treatment-emergent adverse events were examined. No DLT, GvHD, or immune effector cell-associated neurotoxicity syndrome (ICANS) occurred during the clinical trial (Table 1; Supplementary information, Tables S3 and S4). Grade 1‒2 CRS occurred in 10 patients (83%) with a median onset of 1 day after infusion (ranged 0–9) and a median duration of 6.5 days (range 4‒17), while no patients experienced severe CRS (grade ≥ 3) (Fig. 4a, Table 1; Supplementary information, Table S3). For CRS management, 4 out of the 10 patients received tocilizumab, 1 received corticosteroid, 3 received a combination of tocilizumab and corticosteroid, and 2 received non-steroidal anti-inflammatory drugs. All episodes of CRS have been controlled. In patients who developed CRS, serum levels of IFN-γ, IL-6 and IL-10 were correlated with the progression and relief of CRS symptoms (Fig. 4b). All 12 patients had grade 4 neutropenia (Supplementary information, Table S3). One out of the twelve patients (Patient 3) developed sepsis and died 24 days post infusion.

a Graph of incidences of CRS and ICANS in all patients treated. b Serum concentrations of IFN-γ, IL-6 and IL-10 in patients who developed CRS after RD13-01 infusion. Samples were taken 2 h after CAR-T infusion (“before CRS”), and at the times of initiation, peak and restoration of CRS-related symptoms. Data are presented as means ± SD. c Swimmer plot (n = 12) showing patient responses. Each bar represents an individual patient. Responses were determined on day 28 (arrowhead) and are indicated by different colors (blue, CR; green, PR; orange, PD; gray, NE). Bars with solid arrows represent patients in an ongoing follow-up. d CD7+ malignant cells in bone marrow measured using flow cytometry (left) and microscopic images (100× and 1000×, bar indicates 10 μm) of bone marrow samples (right) of Patient 5, who achieved a complete response at day 28 after RD13-01 infusion. e Computed tomography scans showed a complete response of extramedullary leukemia in Patient 9. Pre-treatment (baseline) tumor lesion was indicated by the red arrow.
EBV-associated B-cell lymphoproliferation (EBV-LPD) was identified in 1 patient (Patient 6). This 34-year-old male patient with PTCL-NOS achieved a complete response (CR) on day 28 post infusion and was diagnosed with EBV-associated DLBCL on day 55 (Supplementary information, Fig. S4a). The patient received rituximab-based therapy but eventually died of progressive disease. Furthermore, we retrospectively analyzed EBV-DNA and CMV-DNA copies in cryopreserved serum samples. Among 82 samples from 12 patients, EBV-DNA was detectable in patient 4, 6, 7, and 11, while CMV-DNA was detectable in patient 7 and 12 (Supplementary information, Fig. S4b‒e).
RD13-01 efficacy
The objective response was evaluated on day 28 post infusion in all patients except for Patient 3 who died at day 24 due to sepsis. Of the 11 patients included in efficacy analysis, 9 (82%, with 95% CI of 52%‒95%) achieved an objective response (Fig. 4c), with 7 (64%, with 95% CI of 35%‒83%) achieving CR or CR with incomplete hematological recovery (CRi) (Table 1). Of the 8 patients with leukemia, 6 (75%, with 95% CI of 41%‒93%) achieved CR/CRi and were negative for minimal residual disease (MRD‒) (Fig. 4c). Particularly, complete responses were observed in a patient with 95% leukemia blast in the bone marrow (Patient 5, Fig. 4d), and a patient with rare extramedullary leukemia (Patient 9, Fig. 4e). All 3 evaluable patients with lymphoma achieved responses after infusion with 1 CR (Patient 6) and 2 PR (Patients 10 and 12) (Fig. 4c). Patients 8, 11, and 12 who achieved responses proceeded with an allogeneic HSCT at 3 months post CAR-T infusion. Patient 2 did not respond to CAR-T cell therapy and received salvage allogeneic HSCT 5 months post CAR-T infusion.
With a median follow-up of 10.5 months (95% CI 10.2‒10.8), 4 patients who responded to CAR-T cell therapy at day 28 (Patients 8, 9, 11, 12) remained in CR. Patient 2 remained in CR after salvage HSCT. Of the patients achieving objective responses at day 28, 3 leukemia and 1 lymphoma patients experienced disease relapse or progression at a median time of 82 days after infusion (ranged 59‒103). Bone marrow examination was performed in 3 leukemia patients after relapse, and CD7+ relapsed disease was found. (Supplementary information, Fig. S5). Patient 6 developed EBV-associated DLBCL after achieving a CR to RD13-01 and died 93 days post infusion (Supplementary information, Fig. S4a).
Kinetics of RD13-01 and endogenous T cells
We have also investigated the cellular pharmacokinetics of RD13-01 after infusion. CAR-T cells in the peripheral blood were detected by flow cytometry in both responders and non-responders, with the peak expansion 10‒14 days post infusion (Fig. 5a). In the 9 patients that achieved responses, the absolute CAR-T cell counts peaked between day 7 and 14, with a median number of 143.49 (ranged 21.66‒742.63) cells/μL. In the 2 non-responding patients, the peak CAR-T cell counts were 54.53 cells/μL and 100.85 cells/μL, respectively (Fig. 5a; Supplementary information, Fig. S6a). Using quantitative polymerase chain reaction (qPCR), CAR-T cell expansion was detected in all patients and peaked between day 7 and 28 in responders (Fig. 5b). The mean peak expansion was 1,729,247 copies/μg genomic DNA in 9 responders, while 9054 copies/μg and 1,330,908 copies/μg genomic DNA in 2 non-responders respectively (Supplementary information, Fig. S6b). The median duration of CAR-T cell persistence measured using qPCR (>500 copies/μg genomic DNA) was 28 days (ranged 10‒120 days) (Supplementary information, Table S5). For the 2 non-responders, Patient 2 had limited in vivo CAR-T expansion, whereas Patient 7 with extramedullary disease progressed rapidly when CAR expansion was observed (Fig. 5a, b). CAR-T cell peak expansion was observed after peak cytokine levels reached peak (Supplementary information, Fig. S6c), indicating that CRS is, at least partially, mediated by the endogenous immune responses. We further performed scRNAseq to clarify the exhaustion status of CAR-T cells in Patient 5 at different times after infusion. The exhaustion score was elevated on day 14 post -infusion compared with the pre-infusion products, but was then reduced on day 19 post infusion, which might be attributed to the lack of tumor cell presence (Supplementary information, Fig. S7a).

a, b Flow cytometry (a) and qPCR (b) detections of CAR-T cells in the peripheral blood of patients who achieved responses (Responders) or no response (Non-responders) at different time points post CAR-T infusion. c Stacked pie charts showing subset compositions of normal lymphocytes from responders. Plotted are median values. DNT, double negative T cell. d Fold changes of different types of endogenous lymphocytes at indicated time points compared to day 1 post-infusion. Plotted are median values for responders. e, f Surface antigen expression on normal lymphocytes (defined as the CD45-bright population) from Patient 5 at different times points. CD3 and CD7 expression on normal lymphocytes before CAR-T cell infusion (e); CD3, CD7 and CAR expression on normal lymphocytes on day 15 and 42 after infusion (f). CAR was gated from the CD3− population, and CD4/CD8 were gated from the CD3+ population. g MLR assay was performed by mixing PBMCs from Patient 5 (day 42 post infusion) with naïve T cells from the same donor origin as RD13-01 CAR-T cells, or naïve T cells from an unrelated donor (PBMC: naive T cell = 4:1), for 72 h. Percentages of patient-originated CD7−CD8+ T cells expressing various activation markers (OX40, CD137, CD25, CD40, CD69) were determined by flow cytometry to indicate allogeneic T cell activation against the CAR-T cell donor. Data are presented as means ± SD. All comparisons were calculated using Student’s t-tests. *P < 0.05; **P < 0.01; ns not significant.
In the 9 patients who achieved objective responses on day 28, a decrease of normal CD7+ T cells was observed except for Patients 6 and 8 who had a CD7+ T cell recovery on day 28 (Supplementary information, Fig. S7b). Meanwhile, the counts of CD7− T cells increased to a median of 63.93 cells/μL (ranged from 21.92 to 609.46) by day 28 (Supplementary information, Fig. S7b). Surprisingly, while CD4+CD7− T and CD7− NK cell showed moderate levels of expansion, the CD8+CD7− T cell subgroup, which was marginal before infusion, repopulated over 60% of the T cell population 14 to 21 days after CAR-T cell infusion (Fig. 5c). The CD8+CD7− T cell subset also showed the highest amplitude of expansion among the examined lymphocytes (Fig. 5d; Supplementary information, Fig. S7c), and its expansion was associated with the decreased number of CAR-T cells (Fig. 5c). These results indicated that CD8+CD7− T cells may have recognized the HLA antigen of infused CAR-T cells and played a role in allo-rejection. To test this hypothesis, we examined the allo-reactivity of T cells from Patient 5 against the naive T cells from which RD13-01 was engineered. Patient 5 achieved a complete response after RD13-01 infusion, followed with CD7−CD8+ T cell expansion, CAR-T count reduction and eventually antigen-positive tumor relapse (Fig. 5e, f; Supplementary information, Fig. S5). Post-infusion CD8+CD7− T cells from Patient 5 were found to display alloreactivity against RD13-01 donor’s naive T cells (Fig. 5g; Supplementary information, Fig. S7d), suggesting that this population may contribute to the limited CAR-T cell persistence through allo-rejection.
Discussion
Allogeneic CAR-T cell therapy is an attractive cellular immunotherapy option especially against T cell malignancies due to its ready-to-use, blast-free nature. However, rational CAR-T cell designs are necessary because of the complications associated with allogeneic CAR-T cell therapy, including GvHD and host rejection. In this study, we successfully generated healthy donor-derived, CD7-targeting CAR-T cells (RD13-01) with additional genetic modifications to overcome these challenges. The development and refinement of RD13-01 was rigorously evaluated in preclinical studies. Furthermore, we conducted the first-in-human, single-arm, dose-escalation Phase I clinical trial of allogeneic CD7-CAR-T cells, and demonstrated its safety and high response rates in patients with r/r CD7+ hematological malignancies.
In the Phase I clinical trial with RD13-01, We observed rapid tumor elimination and deep remission at all planned dose levels in patients with CD7+ leukemia and lymphoma, including 3 patients with extramedullary relapse post HSCT. Also, one patient (Patient 11) with CD7+ r/r AML received compassionate therapy of RD13-01 and achieved an MRD− CR. CD7 is expressed in ~30% of patients with AML and is associated with aggressive disease and poor prognosis.23 The antitumor effect of CD7-CAR-T therapy in AML was previously shown in a preclinical study,24 and our clinical results supported further application of allogeneic CD7-CAR-T cell therapy in patients with AML. Of note, 3 patients who achieved objective responses at day 28 received allogeneic HSCT at 3 months post CAR-T infusion, and all remained in long-term CR. No effect of RD13-01 treatment on engraftment and HSCT-associated complications was observed. Both patients on dose level 3 (Patients 11 and 12) showed early onset of CAR-T cell expansion. However, with the limited number of patients enrolled, we did not observe significant differences of CAR-T cell expansion between dose levels, which is consistent with the study of CD7-CAR-T cells generated from HSCT donors.15 Since all patients on dose level 2 achieved a response (5/6 achieved complete responses), this dose level will be adopted in future larger-scale clinical studies. As allogeneic CAR-T cells are likely to have limited persistence due to host rejection, our clinical results indicated that bridging to HSCT after an initial response to CAR-T therapy may provide benefit to disease control and long-term survival, which will require larger-scale evaluation to confirm.
Allogeneic CAR-T treatment generally requires higher doses to ensure adequate CAR-T cell number in the presence of potential host immune rejection. The dose levels in our clinical trial resembles the ones in a previously reported, smaller-scale study of allogeneic CAR-T cells,17 which are higher than the doses used for HSCT-donor-derived CAR-T cells.15 While we observed expansion of CAR-T cells in most patients, no correlation was found with clinical outcomes or different CAR-T cell donors, probably due to the limited sample size. In our study, one batch of product was used to treat 3‒5 patients, and it was intriguing that patients receiving the identical allogenic CAR-T cell product can have distinct clinical responses, suggesting that endogenous factors (i.e., tumor characteristics and recipient immune landscape) can be confounding factors in the context of a universal therapeutic agent.
The safety of RD13-01 has been demonstrated in our clinical study, and we did not observe DLT, GvHD or ICANS, which are major complications of allogeneic CAR-T therapy, in all patients treated. Further, no patient in our study experienced ≥ grade 3 CRS. A previous clinical study reported 2/2 patients with grade 3 CRS after allogeneic CD7-CAR-T cell infusion.17 We speculate that the less intense lymphodepletion we used may contribute to the lower rates of severe CRS. Notably, our lymphodepletion is still more intense than that in a clinical study on allogeneic CD19-CAR-T cells, in order to avoid the reported failure of CAR-T cell expansion.10 The genetic modifications introduced to RD13-01 may also reduce cytokine production, which still requires further investigation. One patient (Patient 6) with PTCL-NOS had a complete response to CAR-T cell therapy, but later developed EBV-LPD which was further progressed to DLBCL. EBV-LPD occurs frequently in patients with primary or secondary immunodeficiency disorders and can be a complication of chemotherapy, HSCT or solid organ transplantation.25 Our results suggest that prior EBV infection might be considered as a risk factor or exclusion criteria for CAR-T cell therapies targeting T cell malignancies.
While CD7 genetic disruption is necessary to prevent fratricide, how it affects CAR-T cell function had remained largely unknown. We have demonstrated that CD7-KO CAR-T cells can mediate short-term antitumor responses in preclinical animal models, but tumor recurrence was observed, consistent with other preclinical studies of CD7-KO CAR-T cells.14 Indeed, we found that CD7 disruption reduced IL-2 production from CAR-T cells, and therefore, a γc was tethered to the CAR construct to augment effector potency. We discovered that incorporation of γc not only boosted CAR-T cell antitumor functions, but also induced IL-2 production, indicating that γc and CD7 may regulate IL-2 production via distinct mechanisms. IL-2 is a main contributor to CAR-T cell expansion and activation,26 and a previous study utilized IL-2 receptor beta chain to potentiate CD19-CAR-T activity,27 suggesting that the design for cytokine (Signal 3) engagement can be widely applied to different platforms. In addition, the preclinical models established with Jurkat cells well showed the aggressive nature of T cell malignancies as reported by various studies,28,29,30,31 and the evaluation of CAR-T cell potency with sub-optimal doses allowed for the assessment of CAR refinement strategies such as γc incorporation. However, overexpression of the γ cytokine IL-15 was shown to induce clonal expansion in T cells,32 indicating that γc incorporation may have the risk to transform CAR-T cells. Although RD13-01 did not show prolonged persistence in patients, the potential of clonal expansion needs to be closely monitored in future clinical studies.
After CAR-T cell treatment, an expansion of normal CD7− T cells was observed, which was associated with a relatively short in vivo persistence of the CAR-T cells. As the CAR-T cells have been depleted for HLA-II to reduce alloreactivity from CD4+ T cells, the post-treatment expansion was most significant in the CD8+CD7− compartment. We further demonstrated that these expanded CD8+CD7− T cells exerted alloreactivity toward naïve T cells of the same donor from which RD13-01 CAR-T cells were engineered. Therefore, the expanded CD8+CD7− T cells could be a contributor to eradicate CAR-T cells and cause antigen-positive relapse as we observed in the patients. On the other hand, however, as previous studies have shown that T cells generated from CD7-depleted hematopoietic stem cells can maintain effector function,33 these CD7− T cells may also contribute to the low incidence of infection in our study by compensating CD7+ T lymphopenia and maintaining a T cell reservoir. Further investigations need to be focused on how to prolong CAR-T persistence without compromising the expansion of CD7− normal T cells, which will lead to the control of both tumor and infection. A potential strategy could be the expression of inhibitory ligands (i.e., PD-L1) on the allogeneic CAR-T cells which mediate resistance to the recognition and killing from recipient CD7− T cells.
Allogeneic CAR-T cells were first demonstrated for clinical antitumor effect in the CD19-targeting platform.10 Our study has revealed the potential of allogeneic CAR-T therapy in treating T cell leukemia/lymphoma and CD7+ acute myeloid leukemia besides B cell malignancies. RD13-01 is a ready-to-use allogeneic CAR-T cell product which avoids patient-derived malignant T cell contamination. It also decreases the risk of manufacture failure in urgent scenarios compared with autologous CAR-T therapy. Our study, together with previous reports on CD7-CAR-T cells generated from autologous cells or HSCT donors,15,16 supported the utilization of CD7 as a tumor-associated antigen for CAR-T cell development and further clinical evaluations. We have also revealed the risk of allo-rejection by host CD8+CD7− T cells in allogeneic CD7-CAR-T cell therapy, providing rationale for future CAR-T cell designs. Further phase II investigations are warranted to evaluate the long-term safety and efficacy in larger scales.
Materials and methods
Construct design and RD13-01 production
RD13-01 retroviral construct contains a CAR and a NKi ligand moiety. The CAR is comprised of a CD7-binding scFv, a 4-1BB costimulatory domain, a CD3ζ signaling domain, and a γc intracellular domain. The NKi ligand contains an EC1-EC2 extracellular domain of E-cadherin fused to a CD28 costimulatory domain. The CAR and the NKi ligand are linked by a F2A self-cleaving peptide. The retrovirus was produced in 293GP cells, cryopreserved at −80 °C, and thawed immediately before transduction. Cas9 plasmid was linearized and purified via FastPure® Gel DNA Extraction Mini Kit (Vazyme). Cas9 mRNA was synthesized by in vitro transcription with linearized Cas9 plasmid using MEGAscript™ T7 Transcription Kit (Invitrogen), and then purified via Monarch RNA Cleanup Kit (New England Biolabs). All sgRNAs were generated on Dr. Oligo 192 synthesizer using standard solid phase method. The crude product was further purified by RP-HPLC. Both termini were modified with 2′-O-methyl 3′phosphorothioate modification. Both Mass-spectrum and Next-generation sequencing were used to confirm the sequence correctness. The single guide RNA (sgRNA) was dissolved in RNase-free water and the concentration was measured using a UV microplate reader. The genomic sgRNA target sequences (with the protospacer adjacent motif in bold) are as follows: TRAC: 5′-AGAGTCTCTCAGCTGGTACACGG-3′; CD7: 5′-GTAGACATTGACCTCCGTGATGG-3′; RFX5: 5′-GGGGTTGCGGATCCACCTATAGG-3′.
Donors for CAR-T cell manufacturing were selected according to the criteria of blood donation with ages ranging from 18 to 40. The MHC-I molecules of the donors were unmatched with those of the patients. For CAR-g-NKi-tKO CAR-T cell production, CD3+ T cells were isolated and incubated with anti-CD3/CD28 beads (Thermo Fisher Scientific, 40203D) at a 2:1 bead/cell ratio and cultured in X-VIVO™ 15 medium (Lonza, 04-744Q) supplemented with 2.5% human serum albumin (HSA) and 200 IU/mL IL-2. On day 2, the Dynabeads were removed, then the T cells were washed twice with OPTI-MEM and resuspended in OPTI-MEM at a final concentration of 1–3 × 108 cells/mL. 0.4 mL of resuspended T cells were mixed with 120 µg of Cas9 mRNA and electroporated using a BTX Agile Pulse apparatus (500 V, 1 ms). After that, the T cells were immediately cultured at 37 °C with 5% CO2. On day 3, 40 μg mixture of total sgRNAs targeting TRAC, CD7, and RFX5 at an equal molecular ratio was electroporated as described above. On day 4, the T cells were centrifuged at 2000× g for 2 h at 32 °C with CAR-encoded retrovirus in retronectin-coated plates. The CAR-T cells were expanded at 37 °C for an extra 6 days.
In vitro cytotoxicity assay
Jurkat-Luci tumor cells were generated and used in a luciferase-based cytotoxic T lymphocyte (CTL) assay. Briefly, a firefly luciferase-encoding lentivirus was transduced into Jurkat tumor cells. The luciferase-expressing cells were resuspended at 1 × 105 cells/mL in X-VIVO 15 medium and incubated with CAR-T cells at different E:T ratios for 8 h at 37 °C. Subsequently, 100 μL of the co-culture was transferred to a 96-well white luminometer plate, and 100 μL of substrate (Vazyme, Cat#DD1201-01) was added. The luminescence was immediately determined by Infinite 200 PRO (TECAN). The results were reported as the percentage of killing based on the luciferase activity in the wells with tumor cell monoculture [% killing = 100 – ((relative light units (RLU) from well with effector and target cell coculture)/(RLU from well with target cells) × 100)].
Cytokine production assay
A total of 1 × 105 CAR-T cells/well were stimulated with Jurkat or Nalm6 cells at a 1:1 ratio in a 96-well tissue culture plate for 8 h. Supernatants were collected and cytokines were measured using an enzyme-linked immunosorbent assay (ELISA) kit and antibodies against human IL-2 (R&D, Cat#DY202), and human IFN-γ (R&D, Cat#DY285B).
Flow cytometry
All cells were harvested and resuspended in phosphate-buffered saline, and stained with the following antibodies: anti-human-CD3-FITC (BD Biosciences Cat#555916, RRID:AB_396217), anti-CD7-PE (BioLegend Cat#343106, RRID:AB_1732011), anti-HLA-II-APC (BioLegend Cat#361714, RRID:AB_2750316), anti-PD1-BV421 (BioLegend Cat#329920, RRID:AB_10960742), anti-TIM3-FITC (BioLegend Cat#345022, RRID:AB_2563937), anti-LAG3-PE (BioLegend Cat#369306, RRID:AB_2629592), anti-CD45RO-APC (BioLegend Cat#304210, RRID:AB_314426), anti-CD4-FITC (BD Biosciences Cat#555346, RRID:AB_395751), anti-CD45-PerCP (BioLegend Cat#304026, RRID:AB_893337), anti-OX40-BV510 (BioLegend Cat# 350026, RRID:AB_2616912), anti-CD137-APC (BioLegend Cat# 309810, RRID:AB_830672), anti-CD69-APC/Cyanine7 (BioLegend Cat#310914, RRID:AB_314849), anti-CD40-APC (BioLegend Cat#334310, RRID:AB_2260153), anti-CD25-BV605 (BioLegend Cat#302632, RRID:AB_11218989), anti-CD8-APC (BD Biosciences Cat#555369, RRID:AB_398595), anti-Granzyme A-PE (BioLegend Cat# 507206, RRID:AB_315472), and anti-Perforin-PE (BioLegend Cat#308106, RRID:AB_314704), at concentrations according to the manufacturer’s instructions. CAR expression was detected by staining with a His-Tagged human CD7 (Arco Biosystems, Cat#CD7-H25H7), followed by staining with a APC-anti-His-Tag antibody (BioLegend Cat#362605, RRID:AB_2715818). Flow cytometry was performed on a BD Celesta, and the data were analyzed using the FlowJo 10 software (FlowJo, RRID:SCR_008520).
Animal model
6- to 8-week-old NOD-Prkdcem26Cd52Il2rgem26Cd22/NjuCrl (NCG) (GemPharmatech, RRID:SCR_017239) immuno-deficient mice were engrafted with Jurkat-GFP-Luci cells or Nalm6-GFP-Luci cells through i.v. injection, and 1 × 107 of CD7-CAR-T cells or 1 × 106 of CD19-CAR-T cells were i.v. injected 4 days after tumor inoculation. Tumor burdens were monitored using an in vivo imaging system (Caliper Life Sciences) at the indicated time points. Live imaging software was used to visualize and calculate total luminescence. To analyze the tumor cells from the peripheral blood of the mice, 100 μL of blood sample was collected by orbital puncture. After red blood cell lysis, the cells were incubated for subsequent flow cytometric analysis. Mice were euthanized when they developed signs of excessive tumor burden or when the weight loss exceeded 20% of baseline, and survival was analyzed accordingly. All animal experiments were conducted in compliance with laboratory animal welfare and ethics committee.
Mixed lymphocyte reaction (MLR) assay for CD4+ T cell alloreactivity
T cells from an allogeneic donor were isolated and activated using anti-CD3/CD28 Dynabeads (Gibco). These cells were further used as stimulators after HLA-II upregulation and subsequent mitomycin-c treatment. To generate primed alloreactive recipient T cells, pretreated donor T cells were mixed with fresh PBMCs from the CAR-T cell donor, at a 1:2 ratio in X-VIVO 15 media supplemented with 10% FBS and 300 IU/mL IL-2. IL-2 was withdrawn from day 4. Ten days later, cells were harvested, labeled with CSFE (Thermo Fisher Scientific, C34570), and co-cultured with CAR-T cells (with dKO or tKO) for 6 h. Following coculture, the cells were incubated with anti-CD3 and anti-CD4 antibodies and permeabilized for 10 min using BD FACS Permeabilizing Solution 2, followed by incubation with anti-IFN-γ antibody and analysis using flow cytometry.
NK cell alloreactivity measurement
K562-luciferase cells were transduced with lentivirus expressing a chimeric receptor that comprises an EC1-EC2 extracellular domain, an E-cadherin transmembrane domain, and the CD28 intracellular domain (K562-NKi). Three days after infection, the engineered K562 cells or CAR-T cells with the integrated NKi were subjected to coculture with NK92 cells. KLRG1 expression was introduced in NK-92 cells through KLRG1 mRNA electroporation. Target cells (K562 or CAR-T cells) were cocultured with NK-92 cells at different E:T ratios at 37 °C. Luminescence was determined immediately after 4 h of coculture to evaluate the NK cell cytotoxicity. To test the cytokine release from NK cells, 1 × 105 NK-92 cells/well were cocultured with K562-NKi cells at a 1:1 ratio in a 96-well tissue culture plate for 12 h. Cytokines produced by NK92 cells were measured using a human IFN-γ ELISA kit.
Design and oversight of clinical trial
The first-in-human, single-arm, dose-escalation phase I clinical trial of RD13-01 therapy for patients with r/r CD7+ hematological malignancies was conducted at the Bone Marrow Transplantation Center, the First Affiliated Hospital, Zhejiang University School of Medicine. The study protocol was approved by the First Affiliated Hospital, School of Medicine, Zhejiang University Institutional Review Board and registered in ClinicalTrials.gov (NCT04538599). The 13 participants or their guardians provided written informed consent in accordance with the Declaration of Helsinki. Patient 11 (diagnosed with AML) also consented for compassionate use. One patient was excluded because of CNS involvement.
A “3 + 3” dose-escalation design was applied in this study. The primary objective of the study was to assess the safety and tolerability of RD13-01, while the secondary objective was to assess the preliminary anti-tumor activity and characterize the pharmacokinetics of RD13-01. Patients were given a lymphodepletion chemotherapy with fludarabine, cyclophosphamide, and etoposide before RD13-01 infusion. All 12 enrolled patients received one infusion of RD13-01 between September 10th 2020 and January 20th 2021, except for the first enrolled patient who received split-dosing infusion for safety concerns. Patient 2 received a second dose of RD13-01 on day 22 due to suboptimal expansion and unsatisfactory clinical response.
The Safety Review Committee (SRC), established by the Principal Investigator, is to ensure appropriate safeguards are in place for the safety of participating subjects, the scientific rigor and integrity while the trial is ongoing. During dose escalation phase, SRC will evaluate the safety and tolerability, and pharmacokinetics of RD13-01 to decide whether or not dose escalation will continue. The SRC consisted of Principal Investigators or delegates, Medical Monitors from pharmaceutical industry or delegates, and Contract Research Organization (CRO) Medical Monitor or delegates. The Study team members may also be invited to the SRC as appropriate. If necessary, further consultation of internal or external experts are performed by the SRC. Based upon safety, pharmacokinetics and other relevant data, the SRC will decide on the recommended phase 2 dose and Maximal Tolerable Dose.
Assessment of toxic effects and response
CRS and ICANS were graded according to the ASTCT consensus.34 Other toxicities during and after therapy were assessed according to the National Institutes of Health Common Terminology Criteria for Adverse Events Version 5.0 (http://ctep.cancer.gov/). Therapy responses were assessed using flow cytometry as well as morphological and imaging analyses according to National Comprehensive Cancer Network Guidelines (NCCN, RRID:SCR_012959).
Assessment of CAR-T cell persistence and expansion
Following RD13-01 infusion, serial peripheral blood samples were collected in K2EDTA BD vacutainer tubes (BD Biosciences). The persistence of RD13-01 in fresh peripheral blood was determined using flow cytometry and qPCR. For flow cytometry analyses, CD45+CD7− lymphocytes were first gated from PBMCs, and CAR+ cells were subsequently gated. For qPCR analyses, genomic DNA was extracted using AxyPrep Blood Genomic DNA Miniprep Kit (Axygen) from cryopreserved peripheral blood and bone marrow samples, and qPCR was performed in triplicates using AceQ qPCR Probe Master Mix (Vazyme Biotech) and a CFX ConnectTM real-time PCR system (Bio-Rad). Copy numbers per microgram of genomic DNA were calculated from a standard curve of 10-fold serial dilutions of purified CAR plasmid containing 2 × 101‒2 × 106 copies/μL.
Statistical analysis
The cutoff date of clinical evaluation was November 30, 2021. Variables were tabulated and summarized with descriptive statistics. For the time-to-event analyses, the Kaplan–Meier method was used to describe the progression-free survival and overall survival. Serum concentrations of cytokines in clinical samples were compared using the Mann–Whitney method whereas other comparisons were performed using the Student’s t-test. Analysis of differences in exhaustion score between different groups was determined by two-sided Wilcoxon rank-sum test. All P values presented are two-tailed. P values lower than 0.05 were considered statistically significant. Statistical analyses were performed using GraphPad Prism 7 software (GraphPad Prism, RRID:SCR_002798).
Data availability
Any requests for raw and analyzed data will be reviewed by the First Affiliated Hospital, School of Medicine, Zhejiang University Institutional Review Board. Patient-related data that are not included in the paper were generated as part of a clinical trial and are subject to patient confidentiality. Any data and materials (e.g., tissue samples or imaging data) that can be shared will need approval from the First Affiliated Hospital, School of Medicine, Zhejiang University Institutional Review Board and a Material Transfer Agreement in place. All data shared will be de-identified. ScRNA-seq sets are available through the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accessions GSE192453. All codes or mathematical algorithms supporting the results of this study are available upon reasonable request.
References
Marks, D. I. & Rowntree, C. Management of adults with T-cell lymphoblastic leukemia. Blood. 129, 1134–1142 (2017).
Rodriguez-Zuniga, M. J. M., Cortez-Franco, F. & Qujiano-Gomero, E. Adult T-cell leukemia/lymphoma. Review of the literature. Actas Dermosifiliogr (Engl Ed) 109, 399–407 (2018).
Brown, C. E. & Mackall, C. L. CAR T cell therapy: inroads to response and resistance. Nat. Rev. Immunol. 19, 73–74 (2019).
MacKay, M. et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat. Biotechnol. 38, 233–244 (2020).
Neelapu, S. S. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Maude, S. L. et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448 (2018).
Abramson, J. S. et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396, 839–852 (2020).
Depil, S., Duchateau, P., Grupp, S. A., Mufti, G. & Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 19, 185–199 (2020).
Mo, F., Mamonkin, M., Brenner, M. K. & Heslop, H. E. Taking T-cell oncotherapy off-the-shelf. Trends Immunol. 42, 261–272 (2021).
Benjamin, R. et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet 396, 1885–1894 (2020).
Hu, Y. et al. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin. Cancer Res. 27, 2764–2772 (2021).
Fleischer, L. C., Spencer, H. T. & Raikar, S. S. Targeting T cell malignancies using CAR-based immunotherapy: challenges and potential solutions. J. Hematol. Oncol. 12, 141 (2019).
Campana, D. et al. Stages of T-cell receptor protein expression in T-cell acute lymphoblastic leukemia. Blood 77, 1546–1554 (1991).
Gomes-Silva, D. et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 130, 285–296 (2017).
Pan, J. et al. Donor-derived CD7 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia: first-in-Human, phase I trial. J. Clin. Oncol. 39, 3340–3351 (2021).
Lu, P. et al. Naturally selected CD7 CAR-T therapy without genetic manipulations for T-ALL/LBL: first-in-human phase I clinical trial. Blood 140, 321–334 (2022).
Li, S. et al. Eradication of T-ALL cells by CD7-targeted universal CAR-T cells and initial test of ruxolitinib-based CRS management. Clin. Cancer Res. 27, 1242–1246 (2021).
Chou, C. K. & Turtle, C. J. Assessment and management of cytokine release syndrome and neurotoxicity following CD19 CAR-T cell therapy. Expert Opin. Biol. Ther. 20, 653–664 (2020).
Rosshart, S. et al. Interaction of KLRG1 with E-cadherin: new functional and structural insights. Eur. J. Immunol. 38, 3354–3364 (2008).
Jung, L. K., Roy, A. K. & Chakkalath, H. R. CD7 augments T cell proliferation via the interleukin-2 autocrine pathway. Cell. Immunol. 141, 189–199 (1992).
Yee, C. et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl. Acad. Sci USA 99, 16168–16173 (2002).
Nata, T. et al. Targeting the binding interface on a shared receptor subunit of a cytokine family enables the inhibition of multiple member cytokines with selectable target spectrum. J. Biol. Chem. 290, 22338–22351 (2015).
Chang, H., Yeung, J., Brandwein, J. & Yi, Q. L. CD7 expression predicts poor disease free survival and post-remission survival in patients with acute myeloid leukemia and normal karyotype. Leuk. Res. 31, 157–162 (2007).
Gomes-Silva, D. et al. CD7 CAR T cells for the therapy of acute myeloid leukemia. Mol. Ther. 27, 272–280 (2019).
Heslop, H. E. How I treat EBV lymphoproliferation. Blood 114, 4002–4008 (2009).
Fraietta, J. A. et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 24, 563–571 (2018).
Kagoya, Y. et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 24, 352–359 (2018).
Xu, Y. et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 12, 49 (2019).
Mamonkin, M. et al. Reversible transgene expression reduces fratricide and permits 4-1BB costimulation of CAR T cells directed to T-cell malignancies. Cancer Immunol. Res. 6, 47–58 (2018).
Maciocia, P. M. et al. Targeting the T cell receptor beta-chain constant region for immunotherapy of T cell malignancies. Nat. Med. 23, 1416–1423 (2017).
Mamonkin, M., Rouce, R. H., Tashiro, H. & Brenner, M. K. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood 126, 983–992 (2015).
Hsu, C. et al. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood 109, 5168–5177 (2007).
Kim, M. Y. et al. CD7-deleted hematopoietic stem cells can restore immunity after CAR T cell therapy. JCI Insight 6, e149819 (2021).
Greco, A. et al. Altered urinary excretion of human kininase activity in hypertension. Adv. Exp. Med. Biol. 120B, 645–649 (1979).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81730008, 81770201, 81870153), Key Project of Science and Technology Department of Zhejiang Province (2019C03016, 2018C03016-2, 2021C03010). We thank Dr. Christine Brown for her intellectual feedback and suggestions. We thank Bioheng Biotech for study collaboration and funding support for the clinical trial. We thank Sharon Lin and Mang Cang for editing the paper.
Author information
Authors and Affiliations
Contributions
Y.H., Y.Z., J.R., and H.H. designed the overall study. Y.H., Y.Z., D.W., J.R., and H.H. wrote the manuscript. Y.H., M.Z., H.Zhao, W.G., and H.H. designed the clinical trial. J.R., Y.Z., G.C., T.G., X.J., X.H., Y.W., H.Zhang and Q.M. performed the preclinical experiments. M.G., S.Y., X.Zheng, Xiaolong L., X.Zhang, M.C., and K.W. were responsible for manufacturing and quality control of CAR-T cells. Y.H., M.Z., W.G., L.H., H.Zhao, G.W., Xiuju L., and J.C. performed the clinical trial. Y.H., Y.Z., J.R., W.G., Q.C., C.Z., and H.Zhao. analyzed the data. H.H., J.R., D.W., and Y.H. interpreted the results and supervised the study.
Corresponding authors
Ethics declarations
Competing interests
Y.Z., W.G., G.C., L.H., T.G., X.J., X.Zheng, S.Y., Xiaolong L., X.Zhang, M.C., Xiuju L., M.G., K.W., X.H., Y.W., and J.R. are employees of Nanjing Bioheng Biotech Co. All other authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, Y., Zhou, Y., Zhang, M. et al. Genetically modified CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy for relapsed/refractory CD7-positive hematological malignancies: a phase I clinical study. Cell Res 32, 995–1007 (2022). https://doi.org/10.1038/s41422-022-00721-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-022-00721-y
- Springer Nature Singapore Pte Ltd.
This article is cited by
-
Infusion and delivery strategies to maximize the efficacy of CAR-T cell immunotherapy for cancers
Experimental Hematology & Oncology (2024)
-
Advances in CAR-T-cell therapy in T-cell malignancies
Journal of Hematology & Oncology (2024)
-
CRISPR/Cas-based CAR-T cells: production and application
Biomarker Research (2024)
-
Genetically engineered hypoimmunogenic cell therapy
Nature Reviews Bioengineering (2024)
-
Fratricide-resistant CD7-CAR T cells in T-ALL
Nature Medicine (2024)