
Abstract
F-box and WD repeat domain containing 7 (FBXW7) acts as a substrate receptor of SKP1-CUL1-F-box (SCF) E3 ubiquitin ligase and plays crucial roles in the regulation of several cellular processes, including cell growth, division, and differentiation, by targeting diverse key regulators for degradation. However, its role in regulating cellular senescence remains elusive. Here, we found that FBXW7 inactivation by siRNA-based knockdown or CRISPR/Cas9-based knockout induced significant cellular senescence in p53 wild-type cells, but not in p53 mutant or null cells, along with activation of both the p53/p21 and p16INK4a/Rb pathways. Simultaneous p53 inactivation abrogated senescence and cell growth arrest induced by FBXW7 deficiency as well as the alteration of both the p53/p21 and p16INK4a/Rb pathways. Moreover, Fbxw7 deletion accelerated replicative senescence of primary mouse embryonic fibroblasts in a p53-dependent manner. In addition, FBXW7 deletion induced the senescence-associated secretory phenotype to trigger secondary senescence. Importantly, in a radiation-induced senescence mouse model, simultaneous deletion of p53 rescued accelerated senescence and aging caused by Fbxw7 loss. Thus, our study uncovered a novel role for FBXW7 in the regulation of senescence by eliminating p53.
Similar content being viewed by others


Introduction
F-box and WD repeat domain containing 7 (FBXW7), also known as CDC4, is a substrate receptor of SKP1-CUL1-F-box (SCF) E3 ubiquitin ligase. FBXW7 directly recognizes and binds to phosphorylated substrates, such as c-MYC, c-JUN, cyclin E, Notch, and MCL1 [1, 2]. FBXW7 plays a crucial role in the regulation of several vital cellular processes, including cell growth, division, and differentiation. Most substrates regulated by FBXW7 are oncoproteins, and a high mutational rate of FBXW7 has been frequently found in many cancer types, including lung [3, 4], colorectal [5], and breast cancers [6]. Importantly, in multiple Fbxw7 mouse models, tissue-specific Fbxw7 knockout or mutation knock-in promotes tumorigenesis. For example, mice with deleted Fbxw7 in the T-cell lineage develop spontaneous thymic lymphoma [7, 8]. Therefore, FBXW7 has been designated as a well-established tumor suppressor [1, 8, 9]. However, FBXW7 was reported to confer radiation survival by targeting the tumor suppressor p53 for degradation [10,11,12] and regulate chronic myelogenous leukemia (CML) via a p53-dependent apoptosis pathway [13], which adds another layer of complexity to the role of FBXW7 in tumorigenesis. Most recently, an outstanding study showed that downregulating FBXW7 promotes chondrocyte senescence and osteoarthritis development [14]. Given that p53 plays a critical role in the induction of senescence by inducing p21 expression [15], an inhibitor of cell-cycle progression, FBXW7 may regulate senescence by controlling p53 protein levels.
Cellular senescence is a progressive and phenotypically diverse state of irreversible cell-cycle arrest that occurs in proliferating cells in response to various stresses, such as replicative exhaustion, oncogene activation, DNA damage, and cell–cell fusion [16]. Accumulating evidence has implicated senescence in an array of physiological and pathological processes, such as embryonic development, wound healing, degenerative disorders, aging, and cancer [17]. Given that senescence is an irreversible growth arrest, it generally inhibits tumor growth. However, accumulating senescent cells in tissues may also contribute to the development of tumors by modulating the tissue microenvironment, suggesting that senescence plays a dual role in the development of tumors [18]. Senescent cells exhibit a set of general hallmarks, including enlarged and flattened morphology, cell-cycle arrest, increased senescence-associated β-galactosidase (SA-β-Gal) levels, and senescence-associated secretory phenotype (SASP) [19]. In response to various stresses and stimuli, both p53 and Rb serve as hubs in the central activation pathways of senescence [20]. Activated p53 triggers the expression of p21, responsible for G1 cell-cycle transition, and further causes cell-cycle arrest, a typical hallmark of senescence. Besides p21, p53 induces E2F7 expression, which represses many E2F target genes, subsequently leading to senescence [15]. Previous studies have shown that FBXW7 interacts with and targets p53 for proteasomal degradation in both stressed and unstressed cells [10,11,12], suggesting the possibility that FBXW7 modulates senescence in a p53-dependent manner.
In this study, we report that FBXW7 inactivation readily induces cellular senescence in p53 wild-type cells, but not in p53 mutant or null cells, along with activation of both the p53/p21 and p16INK4a/Rb pathways. Simultaneous p53 knockdown or knockout not only abrogated cellular senescence induced by FBXW7 inactivation but also rescued the cell growth arrest. Moreover, in a radiation-induced senescence mouse model, simultaneous deletion of p53 rescued the increased senescence phenotype in Fbxw7+/− mice. Thus, our study uncovered a novel role for FBXW7 in the regulation of senescence by eliminating p53.
Results
FBXW7 silencing induces senescence in cancer cells harboring wild-type p53 along with activation of both p53/p21 and p16INK4a/Rb pathways
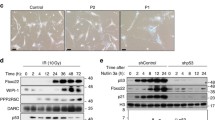
We previously found that FBXW7 binds to phosphorylated p53, leading to p53 ubiquitination and proteasomal degradation [10]. Given that p53 plays a critical role in the induction of senescence, we investigated whether FBXW7 could regulate senescence by modulating p53 levels in this study. Indeed, multiple human cancer cells harboring wild-type p53, including A549, HCT116, MCF7, SJSA, and H460 cells, displayed an enlarged and flattened cell morphology (Fig. 1A and Fig. S1), a general senescence hallmark, upon FBXW7 knockdown, indicating that senescence was induced. The percentage of SA-β-Gal was dramatically increased in FBXW7 knockdown cells (Fig. 1A and Fig. S1B). Moreover, we determined cell senescence using Sudan-Black-B (SBB) staining, an additional reliable approach to detect senescent cells [21]. Likewise, upon FBXW7 knockdown, the positive SBB staining was obviously increased, as reflected by increased intracellular blue-black granules (Fig. S1C). These data further confirmed that senescence had occurred. Interestingly, FBXW7 knockdown had no effect on cell morphology or the percentage of SA-β-Gal-positive cells in DLD-1 and MDA-MB231 cells with mutant p53 and p53-null H1299 cells, respectively (Fig. S2), suggesting that FBXW7 knockdown induces senescence in a p53-dependent manner. Senescent cell-cycle arrest is regulated by the p53/p21 and p16INK4a/Rb tumor suppressor pathways [22]. Next, we measured the levels of these crucial factors when the cell morphology was flattened after FBXW7 knockdown. Indeed, FBXW7 knockdown significantly increased the levels of p53, p21, and p16, as well as cleaved-Notch1 and c-MYC, two well-known substrates of FBXW7, while decreasing the levels of phosphorylated Rb, suggesting that both the p53/p21 and p16INK4a/Rb tumor suppressor pathways were activated (Fig. 1B).

A549, HCT116, and MCF7 cells were infected with a lentivirus expressing shFBXW7 or shGFP, selected by puromycin for seven days, and subjected to senescence-associated β-galactosidase (SA-β-Gal) staining, followed by microscopy (A left) or immunoblotting (IB) with the indicated antibodies (B). The percentage of SA-β-Gal-positive cells in the total number of cells was determined (A right). Scale bars represent 50 μm. Data are presented as the mean ± standard deviation (SD). ***p < 0.001.
Simultaneous p53 deletion abrogates the senescence induced by FBXW7 deletion
To explore the underlying mechanism of FBXW7-induced senescence, we simultaneously silenced p53, c-MYC, and Notch1 in FBXW7 knockdown A549 and MCF7 cells (Fig. S3A), as c-MYC and cleaved-Notch1, which are involved in oncogene-induced senescence [23, 24], were accumulated in FBXW7 knockdown senescent cells (Fig. 1B). We found that simultaneous p53 knockdown rescued the senescence phenotype caused by FBXW7 deficiency (Fig. S3B). Interestingly, although there are multilevel crosstalks between p53 and Notch [25], only p53, but not Notch1, knockdown reversed senescence induced by FBXW7 deficiency, suggesting induced senescence independent of Notch1. Furthermore, we generated FBXW7 and p53 deleted cells via a CRISPR/Cas9-based approach and found that FBXW7 deletion alone induced obvious senescence, while deletion of both FBXW7 and p53 suppressed the induction of senescence (Fig. 2A). Consistently, FBXW7 deletion caused an increase in the levels of p53, p21, and p16 and a decrease in Rb phosphorylation, which was rescued by simultaneous p53 deletion (Fig. 2B, lanes 3 versus 2). In addition, simultaneous p53 deletion partially rescued the cell growth arrest caused by FBXW7 deletion, which further confirmed that senescence was rescued (Fig. 2C). Collectively, these results indicate that p53 accumulation by FBXW7 inactivation plays a causal role in the promotion of senescence and cell growth arrest.

A549 and MCF7 cells with deletion of the indicated genes using CRISPR/Cas9 technology were subjected to SA-β-Gal staining, followed by microscopy (A left) or IB with the indicated antibodies (B), or cell counting kit (CCK)-8-based cell proliferation assay (C). The percentage of SA-β-Gal-positive cells in the total number of cells was determined (A right). Scale bars represent 50 μm. Data are presented as the mean ± SD. *p < 0.05, ***p < 0.001. Cell proliferation is expressed as a fold-change compared to day 1. Data are presented as the mean ± standard error of the mean (SEM) from three independent experiments. n = 3; **p < 0.01, ***p < 0.001.
Fbxw7 deletion accelerates replicative senescence of primary mouse embryonic fibroblasts (MEFs) by inducing p53 expression
To investigate whether the FBXW7–p53 axis is involved in the senescence of normal cells, we passaged primary MEFs to induce replicative senescence. First, we generated primary Fbxw7fl/fl;p53+/+ and Fbxw7fl/fl;p53fl/fl MEFs by crossing Fbxw7fl/+;p53fl/+ mice. Consistently, Fbxw7 deletion caused significant p53 accumulation in two pairs of primary Fbxw7fl/fl;p53+/+ MEFs that had been infected with adenovirus-expressing Cre recombinase (Ad-Cre) to delete Fbxw7 (Fig. 3A). Furthermore, the protein levels of p53 were gradually increased during passaging in Fbxw7fl/fl;p53fl/fl MEFs expressing Fbxw7 and p53 as wild-type MEFs after infection with adenovirus-expressing GFP (Ad-GFP) (Fig. 3B, lanes 5 and 3 versus 1), implying a significant role of p53 in replicative senescence. We then measured the cumulative population doubling time of MEFs by serial passaging using a 3T9 protocol and found that Fbxw7fl/fl;p53+/+ MEFs underwent senescence at P4 after Fbxw7 deletion by Ad-Cre and Fbxw7fl/fl;p53+/+ and Fbxw7fl/fl;p53fl/fl MEFs underwent senescence at P6 upon Ad-GFP infection (Fig. 3C), whereas Fbxw7fl/fl;p53fl/fl MEFs were immortalized without senescence upon Fbxw7 and p53 deletion by Ad-Cre (Fig. 3B, lanes 2, 4, and 6; Fig. 3C). Meanwhile, the percentage of SA-β-Gal-positive cells was dramatically increased in Fbxw7fl/fl;p53+/+ MEFs upon Fbxw7 deletion at P6, which was completely reversed by simultaneous deletion of p53 in Fbxw7fl/fl;p53fl/fl MEFs (Fig. 3D). Taken together, these results demonstrate that Fbxw7 loss accelerates replicative senescence by inducing p53 expression.

A Two independent primary Fbxw7fl/fl;p53+/+ MEFs were infected with the adenovirus-expressing Cre recombinase (Ad-Cre) or adenovirus-expressing green fluorescent protein (Ad-GFP) for 72 h, and then subjected to IB with the indicated antibodies. B Primary Fbxw7fl/fl;p53fl/fl MEFs were infected with Ad-Cre or Ad-GFP, then passaged for indicated times, followed by IB with the indicated antibodies. C, D Primary Fbxw7fl/fl;p53+/+ and Fbxw7fl/fl;p53fl/fl MEFs were infected with Ad-Cre or Ad-GFP, followed by cell passage on a 3T9 protocol to measure the cumulative population doubling level (C), or SA-β-Gal staining at P6 (D left). The percentage of SA-β-Gal-positive cells in the total number of cells was determined (D right). Scale bar represents 50 μm. Data are presented as the mean ± SD. ***p < 0.001.
FBXW7 deletion promotes senescence and aging in a p53-dependent manner in vivo
Homozygous loss of Fbxw7 causes embryonic lethality, while mice with Fbxw7 heterozygous deletions survive, although Fbxw7 is a haplo-insufficient gene [26,27,28]. We explored the in vivo role of Fbxw7 in the regulation of senescence using Fbxw7 heterozygous loss mice (Fbxw7+/−) combined with p53+/− mice. We administered 7 Gy ionizing radiation to trigger premature aging in female mice with three genotypes (Fbxw7+/+;p53+/+, Fbxw7+/−;p53+/+, and Fbxw7+/−;p53+/−) [29]. Initially, the mice exhibited almost no systemic reactions. However, after three months, three littermates with the three genotypes showed representative hair graying, which is a typical sign of aging in animals and humans (Fig. 4A). Given that cellular senescence is usually linked to aging [30], the hair graying of Fbxw7+/−;p53+/+ mice was the most serious, indicating that the most senescence is induced upon Fbxw7 heterozygous deletion, which can be reversed by simultaneous p53 heterozygous loss. Moreover, the strongest SBB staining of liver tissues from Fbxw7+/−;p53+/+ mice further confirmed that the most senescence is induced upon Fbxw7 heterozygous deletion (Fig. 4B). In addition, both immunohistochemistry and /or immunoblotting of liver tissues collected from the littermates showed that compared to Fbxw7+/+;p53+/+ mice, the levels of p21 and p16 were significantly increased, while the phosphorylation of Rb was decreased in Fbxw7+/−;p53+/+ mice, indicating that more cells were undergoing senescence (Fig. 4C, D). Consistently, the protein levels of p53 were also increased in the liver tissues of Fbxw7 heterozygous mice (Fig. 4D, lanes 2 versus 1). Simultaneous p53 heterozygous loss also reversed the induction of p21 and p16, and the reduction in Rb phosphorylation (Fig. 4C,D, lanes 3 versus 2). Collectively, these results indicate that Fbxw7 deletion promotes senescence in a p53-dependent manner in vivo. Based on these results, we conclude that FBXW7 deficiency induces senescence both in vivo and in vitro via p53 induction.

Three littermate mice with the indicated genotypes were exposed to 7 Gy irradiation and maintained for 3 months, followed by being photographed (A). Liver tissues of mice were harvested and subjected to SBB staining (B), immunohistochemistry (IHC) (C left), or IB with the indicated antibodies (D). The area of graying hair is circled by a white line. The percentage of staining-positive cells in the total number of cells was determined from at least five random fields of liver tissues (C right). **p < 0.01; ***p < 0.001; ns not significant. Scale bars represent 50 μm.
FBXW7 deletion induces senescence-associated secretory phenotype (SASP)
Senescent cells secrete various factors, including pro-inflammatory cytokines and chemokines, growth modulators, angiogenic factors, and matrix metalloproteinases, to communicate with their microenvironment and influence the surrounding cells, termed SASP or senescence messaging secretome [31, 32]. Next, we investigated whether FBXW7 deletion led to the development of SASP. First, we measured the levels of a set of factors involved in the senescence process, including interleukin (IL6), chemokines (IL8, CXCL1-3, and CCL2), other inflammatory factors (TNFα and GM-CSF), and the ligand (ICAM1). Among them, IL8, CXCL1, TNFα, and ICAM1 were the most fluctuating factors in FBXW7-deleted senescent cells (Fig. 5A). Furthermore, conditioned media from A549 sgCtrl and sgFBXW7 cells were collected to culture human fetal lung fibroblast MRC5 cells for 2 weeks. We found that MRC5 cells cultured with the conditioned medium from A549 sgFBXW7 cells displayed an enlarged and flattened morphology with increased SA-β-Gal-positive staining (Fig. 5B), indicating the occurrence of secondary senescence. These results suggest that FBXW7 deletion-induced senescence may regulate the microenvironment to influence the surrounding cells by developing SASP.

A A549 cells, with or without FBXW7 deletion, were harvested for qRT-PCR analysis for the indicated SASP factors. Data are presented as the mean ± SEM from three independent experiments. n = 3; *p < 0.05; **p < 0.01; ***p < 0.001. B The conditional medium from A549 cells, with or without FBXW7 deletion, was used to culture MRC5 cells for 2 weeks, followed by SA-β-Gal staining (top). The percentage of SA-β-Gal-positive cells in the total number of cells was determined (bottom). Scale bar represents 50 μm. Data are presented as the mean ± SD. ***p < 0.001. C A model for FBXW7 inactivation-induced senescence.
Discussion
FBXW7 acts as a substrate receptor of SCF ubiquitin ligase and plays critical roles in multiple cellular processes, such as cell proliferation and differentiation, by targeting many key regulators for degradation [2, 9, 33]. In this study, we found that FBXW7 inactivation promoted the senescence of p53 wild-type cancer and normal cells and that p53 induction played a causal role in FBXW7 deficiency-induced cellular senescence both in vitro and in vivo. The major findings of this study can be listed as follows: (1) flattened cell morphology and enhanced staining of SA-β-Gal were observed in various types of cancer and normal cells harboring wild-type p53 upon FBXW7 deficiency; (2) simultaneous p53 inactivation by siRNA-based knockdown or CRISPR/Cas9-based knockout abrogated the senescence induced by FBXW7 deficiency; (3) in an in vivo radiation-induced senescence model, heterozygous loss of Fbxw7 promoted hair graying and cell senescence in liver tissues, whereas the heterozygous loss of p53 reversed these alterations; and (4) many SASP factors secreted by FBXW7-deficient A549 cells induced senescence of MRC5 cells.
Given that FBXW7 is a well-established tumor suppressor, FBXW7 inactivation should theoretically promote cancer cell growth. Unexpectedly, in our study, FBXW7 inactivation via siRNA-based knockdown or CRISPR/Cas9-based knockout significantly induced senescence in lung cancer (A549 and H460), colon cancer (HCT116), breast cancer (MCF7), and osteosarcoma (SJSA) cells, suggesting that this is a common phenomenon (Fig. 1 and Fig. S1). Mechanistically, p53, a novel substrate of FBXW7 [10,11,12], plays a causal role in FBXW7 deficiency-induced cellular senescence both in vitro and in vivo. A recent outstanding study reported that downregulating FBXW7 promotes chondrocyte senescence and osteoarthritis development upon mechanical overloading by targeting MKK7 for degradation, which consequently stimulates the JNK signaling [14]. In addition, FBXW7 was reported to positively regulate the function of XRCC4 and EGFR by promoting XRCC4 polyubiquitination via the K63 linkage [34], and stabilizing EGFR [35], respectively. Inactivation of XRCC4 [20] or EGFR [36] induces cellular senescence. Moreover, Fbxw7 deletion in MEFs upregulates the transcription of p16INK4a and p19Arf to induce cell-cycle arrest [37]. Given simultaneous p53 deletion cannot completely abrogate the senescence and proliferation induced by FBXW7 deletion in cancer cells (Fig. 2), it is worth to further exploring other mechanisms contributing to FBXW7-regulated senescence, such as via MKK7. However, another recent study proposed that FBXW7 promotes senescence and pulmonary fibrosis, a typical age-related disease, by targeting the telomere protein TPP1 for degradation and accelerating the uncapping of telomere [38]. The seemingly contradictory roles of FBXW7 in the regulation of senescence may be attributed to the diverse substrates whose degradation is subject to FBXW7 regulation in a cell- or tissue-context-dependent manner.
As a general tumor suppressor in cancer, FBXW7 is inactivated by mutations, with an overall mutation frequency of approximately 6% [39]. Upon FBXW7 inactivation, accumulated oncogenic substrates, such as c-MYC, cyclin E, and Notch, may play major roles in tumorigenesis triggered by FBXW7 inactivation. Nevertheless, cellular senescence promoted by FBXW7 inactivation may also contribute to tumorigenesis in a particular process by reprograming the microenvironment by promoting the secretion of SASP factors, which warrants further investigation. Notch1, rather than SASP alone, is required for secondary senescence [40]. Although we found that the simultaneous silencing of Notch1 could not rescue FBXW7 inactivation-induced senescence (Fig. S3B), given that Notch1 is a well-known substrate of FBXW7, it is necessary to investigate whether FBXW7 inactivation contributes to secondary senescence by inducing Notch1 expression.
Primary MEFs are canonical models to study senescence via replicative arrest through the accumulation of the INK4 family like p16INK4a [41]. p53 inactivation alone is insufficient to immortalize primary MEFs [42]. Interestingly, in our study, Fbxw7 deletion alone induced premature senescence in primary MEFs, whereas both Fbxw7 and p53 deletion immortalized primary MEFs (Fig. 3C, D). We speculate that p53 deletion coordinates with the accumulation of oncogenic substrates of FBXW7, such as c-MYC, which release primary MEFs beyond the Hayflick Limit [43], leading to immortalization.
Senescent cells accumulate in multiple tissues with age, which is one of the causative processes of aging and aging-associated diseases. In addition to aging, subsequent studies reinforced the importance of cellular senescence as a safeguard against cancer [44]. The physiological relevance of cellular senescence is not limited to tumor suppression, but also includes many biological processes, such as embryonic development [45, 46], wound healing [47, 48], tissue repair [49], organismal aging [50], and various endocrine diseases [51]. Cellular senescence can favor the damage repair of the body, but can also cause harm to the body via the microenvironment [52]. In addition, accumulating senescent cells in tissues may promote the development of tumors by modulating the microenvironment via SASP [18]. Thus, cellular senescence acts as a double-edged sword, with both beneficial and detrimental effects on physiological and pathological processes. Future investigations of the precise role of senescence may be beneficial for senolytic therapy.
In summary, our study uncovered the significant role of FBXW7 in the regulation of cellular senescence and revealed the two underlying mechanisms associated with it. First, upon FBXW7 inactivation, p53 is directly accumulated, and then, increased p53 transactivates p21, subsequently inhibiting the cyclin E/Cdk2 activity. Second, FBXW7 inactivation increases p16 expression via an unknown mechanism, which further represses the activities of cyclin D/Cdk-4,6. As a result, Rb phosphorylation is significantly inhibited and dephosphorylated Rb represses the transcription of genes required for cell-cycle progression by directly binding to the transactivation domain of E2F, finally leading to the induction of senescence. Moreover, FBXW7 inactivation-induced senescent cells secrete many SASP factors that promote secondary senescence (Fig. 5C).
Methods
Cell culture
Human cancer cells, including A549, HCT116, MCF7, SJSA, H460, H1299, DLD-1, and MDA-MB231, obtained from American Type Culture Collection (ATCC), were cultured at 37 °C in a humidified incubator with 5% CO2. A549, MCF7, SJSA, H1299, DLD-1, and MDA-MB231 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). H460 cells were maintained in RPMI-1640 medium supplemented with 10% FBS and 1% P/S. HCT116 cells were maintained in McCoy’s 5 A medium supplemented with 10% FBS and 1% P/S.
Lentiviral production and generation of stable cell lines
Short hairpins targeting FBXW7 (targeting sequence: 5′-ACA GGA CAG TGT TTA CAA A-3′), p53 (targeting sequence: 5′-CAC CAT CCA CTA CAA CTA CAT-3′), c-MYC (targeting sequence: 5′-CAG TTG AAA CAC AAA CTT GAA-3′), and Notch1 (targeting sequence: 5′-CCG GGA CAT CAC GGA TCA TAT-3′) were subcloned into pLKO.1-puro vector. Lentivirus expressing shRNA was produced by transfecting shRNA vector, psPAX2, and pMD2.G plasmids into 293 T cells. Cells were infected by lentivirus and then selected for stable expression with puromycin for seven days.
CRISPR/Cas9-based FBXW7 and p53 knockout
CRISPR/Cas9-based FBXW7 or/and p53 knockout cells were generated as described previously [53]. Briefly, single-guide RNA (sgRNA) was subcloned into the plasmid, pSpCas9(BB)-2A-Puro (PX459). Cells were simultaneously transfected with sequence-verified CRISPR plasmids expressing sgRNA targeting FBXW7 or p53, and selected with puromycin for three days. Then cells were seeded in 100 mm dish at 0.5 × 103, 1 × 103, 2 × 103 cells and cultured for 10–15 days. About 100 single clones were picked under a microscope and were further expanded. The FBXW7, p53, or FBXW7 and p53 double knockout clones were confirmed by immunoblotting and genomic DNA sequencing. The sequences of the sgRNA were used as follows: sgFBXW7: 5′-AAA GTT GGA CCA TGG TTC TG-3′; sgp53: 5′-GCA GTC ACA GCA CAT GAC GG-3′.
Generation and maintenance of MEFs
Primary MEFs were isolated from day E13.5 embryos and cultured in DMEM supplemented with 15% FBS, 0.1 mM MEM nonessential amino acids, and 1% P/S, and incubated at 37 °C in a 5% CO2 humidified incubator.
MEF cell passage on a 3T9 protocol
Primary MEFs (P1) were infected with adenovirus (Ad-Cre) for 72 h. A total of 9 × 105 cells were seeded in a 60-mm dish, cultured for 72 h, and then counted at each passage until there are not enough cells to subculture. PDL (population doubling level) was calculated using a standard formula: cumulative PDL = initial PDL + 3.32 × [log (current cell yield) − log (cell plated)].
Immunoblotting and antibodies
Cells were lysed in lysis buffer [50 mM Tris–Cl (pH 7.5), 150 mM NaCl, 1 mM DTT, 1 mM EDTA, 0.5% sodium deoxycholate, 50 mM NaF, 1 mM Na3VO4, 1% NP-40, 0.1% SDS] with protease inhibitors and phosphatase inhibitors, followed by SDS-PAGE gel electrophoresis and antibody incubation [54]. The target proteins were visualized by chemiluminescence. Antibodies used are listed as follows: FBXW7 (A301-720A, Bethyl Laboratories), p53 (OP43 from Calbiochem for human sample, 2524 and 32532 from Cell Signaling Technology for mouse sample), p21 (2947 from Cell Signaling Technology for human sample, ab188224 from abcam for mouse sample), p16 (92803 from Cell Signaling Technology for human sample, ab211542 from abcam for mouse sample), p-Rb (8516, Cell Signaling Technology), cleaved-Notch1 (4147, Cell Signaling Technology), c-MYC (5605, Cell Signaling Technology), ACTIN (A5441, Sigma), and GAPDH (2118, Cell Signaling Technology).
SA-β-galactosidase staining for senescence
The staining of senescence-associated β-galactosidase was conducted according to the manufacturer’s instructions (C0602, Beyotime). Briefly, cultured cells were fixed by β-galactosidase staining fixative at room temperature for 15 min, and then incubated in the staining working solution for ~12–24 h at 37 °C. The stained cells were observed and photographed under a light microscope.
Sudan-Black-B (SBB) staining
Cells adhered to the glass slides were immersed in Sudan-black stain for 2 min, washed with 75% ethanol to remove excess stain, and then immersed in distilled water for 1 min, and counterstained with nuclear fast red stain for 1 min. For paraffin sections of tissues, after deparaffinization and rehydration, the sections were stained according to the procedure for cell staining. The slides were sealed with glycerin gelatin and then observed and photographed in time.
CCK8 assay
Cells were seeded in 96-well plates in triplicate at 3000 cells per well. Cell proliferation was evaluated by a CCK8 assay according to the manufacturer’s instructions (HY-K0301, MedChemExpress), and results were expressed as the fold-change compared with the control.
Mouse studies
Fbxw7+/− mice were generated by crossing Fbxw7fl/fl (Jackson laboratory, Stock No: 017563) mice with Vasa-Cre (Jackson laboratory, Stock No: 018980) transgenic mice. Mice were genotyped using the following primers to detect floxed Fbxw7 (497 bp), deleted Fbxw7 (662 bp), and WT (315 bp): Fbxw7-cKO-F: 5′-ATT GAT ACA AAC TGG AGA CGA GG-3′; Fbxw7-cKO-R: 5′-ATA GTA ATC CTC CTG CCT TG GC-3′; Fbxw7-KO-F: 5′-GGC TTA GCA TAT CAG CTA TGG-3′. p53+/− mice were obtained and genotyped as previously described [55]. For the animal studies, all procedures were approved by Zhejiang University Laboratory Animal Center. Animal care was provided in accordance with the principles and procedures outlined in Chinese National Research Guide for the Care and Use of Laboratory Animals.
Mice radiation exposure
Female mice aged 6–8 weeks were exposed to 7 Gy of radiation (X-RAD 160; PXi) and maintained for 3 months, followed by being photographed and sacrificed for liver tissue collection.
Immunohistochemical staining
For immunohistochemistry (IHC), 5 μm thick sections of mouse liver tissues were stained with the antibodies, as previously described [56]. The following antibodies were used: p21 (ab188224, abcam), p16 (ab211542, abcam), and p-RB (Ser807/811) (8516, Cell Signaling Technology).
Quantitative RT-PCR
Quantitative RT-PCR analysis was performed as described previously [57]. Briefly, total RNA was isolated from cells using TRIzol reagent (15596018, Invitrogen). cDNA was generated from RNA using the Prime-Script RT reagent kit (RR037A, Takara), following the manufacturer’s instructions. Quantitative real-time PCR (qRT-PCR) was performed using SYBR Premix Ex Taq (RR420A, TaKaRa) on an Applied Biosystems StepOnePlusTM Real-Time PCR instrument. Relative expression levels of indicated genes were determined using the comparative Ct (2ΔΔCt) method with GAPDH as an endogenous normalization control. The following primers were used for qRT-PCR analysis: IL6 forward: 5′-ACT CAC CTC TTC AGA ACG AAT TG-3′, reverse: 5′-CCA TCT TTG GAA GGT TCA GGT TG-3′; IL8 forward: 5′-TTT TGC CAA GGA GTG CTA AAG A-3′, reverse: 5′-AAC CCT CTG CAC CCA GTT TTC-3′; CXCL1 forward: 5′-TCA TTG TGA AGG CAG GGG AA-3′, reverse: 5′- AAT TAA GCC CCT TTG TTC TAA GCC -3′; CXCL2 forward: 5′-AGC TCT CCT CCT CGC ACA-3′, reverse: 5′- GAG TGT GGC TAT GAC TTC GGT-3′; CXCL3 forward: 5′-CGC CCA AAC CGA AGT CAT AG-3′, reverse: 5′-GCT CCC CTT GTT CAG TAT CTT TT-3′; CCL2 forward: 5′- CAG CCA GAT GCA ATC AAT GCC-3′, reverse: 5′-TGG AAT CCT GAA CCC ACT TCT-3′; TNFα forward: 5′-CCT CTC TCT AAT CAG CCC TCT G-3′, reverse: 5′-GAG GAC CTG GGA GTA GAT GAG-3′; GM-CSF forward: 5′- GCT GCT GAG ATG AAT GAA ACA GTA-3′, reverse: 5′-AGC AGT CAA AGG GGA TGA CAA-3′; ICAMI forward: 5′-ATG CCC AGA CAT CTG TGT CC-3′, reverse: 5′-GGG GTC TCT ATG CCC AAC AA-3′; GAPDH forward: 5′-AGG GCA TCC TGG GCT ACA C-3′, reverse: 5’-GCC AAA TTC GTT GTC ATA CCA G-3′.
Statistical analysis
Statistical analyses were performed by two-tailed Student’s t-test, using Statistical Program for Social Sciences software 20.0. Data are expressed as mean ± standard error of the mean (S.E.M) or standard deviation (SD). The statistical significance was set as p < 0.05.
Data availability
The authors declare that all data supporting the findings of this study are available with the article or from the corresponding author upon reasonable request.
References
Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17:115.
Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93.
Yokobori T, Yokoyama Y, Mogi A, Endoh H, Altan B, Kosaka T, et al. FBXW7 mediates chemotherapeutic sensitivity and prognosis in NSCLCs. Mol Cancer Res. 2014;12:32–7.
Xiao G, Li Y, Wang M, Li X, Qin S, Sun X, et al. FBXW7 suppresses epithelial-mesenchymal transition and chemo-resistance of non-small-cell lung cancer cells by targeting snai1 for ubiquitin-dependent degradation. Cell Prolif. 2018;51:e12473.
He D, Ma Z, Fang C, Ding J, Yang W, Chen P, et al. Pseudophosphatase STYX promotes tumor growth and metastasis by inhibiting FBXW7 function in colorectal cancer. Cancer Lett. 2019;454:53–65.
Meyer AE, Furumo Q, Stelloh C, Minella AC, Rao S. Loss of Fbxw7 triggers mammary tumorigenesis associated with E2F/c-Myc activation and Trp53 mutation. Neoplasia. 2020;22:644–58.
Onoyama I, Tsunematsu R, Matsumoto A, Kimura T, de Alboran IM, Nakayama K, et al. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J Exp Med. 2007;204:2875–88.
Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014;14:233–47.
Yumimoto K, Nakayama KI. Recent insight into the role of FBXW7 as a tumor suppressor. Semin Cancer Biol. 2020;67:1–15.
Cui D, Xiong X, Shu J, Dai X, Sun Y, Zhao Y. FBXW7 confers radiation survival by targeting p53 for degradation. Cell Rep. 2020;30:497–509e494.
Galindo-Moreno M, Giraldez S, Limon-Mortes MC, Belmonte-Fernandez A, Reed SI, Saez C, et al. SCF(FBXW7)-mediated degradation of p53 promotes cell recovery after UV-induced DNA damage. FASEB J. 2019;33:11420–30.
Tripathi V, Kaur E, Kharat SS, Hussain M, Damodaran AP, Kulshrestha S, et al. Abrogation of FBW7alpha-dependent p53 degradation enhances p53’s function as a tumor suppressor. J Biol Chem. 2019;294:13224–32.
Reavie L, Buckley SM, Loizou E, Takeishi S, Aranda-Orgilles B, Ndiaye-Lobry D, et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell. 2013;23:362–75.
Zhang H, Shao Y, Yao Z, Liu L, Zhang H, Yin J, et al. Mechanical overloading promotes chondrocyte senescence and osteoarthritis development through downregulating FBXW7. Ann Rheum Dis. 2022;81:676–86.
Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129–43.
Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705.
van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–46.
Schosserer M, Grillari J, Breitenbach M. The dual role of cellular senescence in developing tumors and their response to cancer therapy. Front Oncol. 2017;7:278.
González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021;288:56–80.
Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–76.
Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. 2019;179:813–27.
Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40.
Campaner S, Doni M, Hydbring P, Verrecchia A, Bianchi L, Sardella D, et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat Cell Biol. 2010;12:54–59.
Kagawa S, Natsuizaka M, Whelan KA, Facompre N, Naganuma S, Ohashi S, et al. Cellular senescence checkpoint function determines differential Notch1-dependent oncogenic and tumor-suppressor activities. Oncogene. 2015;34:2347–59.
Dotto GP. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat Rev Cancer. 2009;9:587–95.
Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol. 2004;14:1852–7.
Jandke A, Da Costa C, Sancho R, Nye E, Spencer-Dene B, Behrens A. The F-box protein Fbw7 is required for cerebellar development. Dev Biol. 2011;358:201–12.
Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–9.
Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612.
Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–35.
Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118.
Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94.
Lau AW, Fukushima H, Wei W. The Fbw7 and betaTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Front Biosci (Landmark Ed). 2012;17:2197–212.
Zhang Q, Karnak D, Tan M, Lawrence TS, Morgan MA, Sun Y. FBXW7 facilitates nonhomologous end-joining via K63-linked polyubiquitylation of XRCC4. Mol Cell. 2016;61:419–33.
Rocher-Ros V, Marco S, Mao JH, Gines S, Metzger D, Chambon P, et al. Presenilin modulates EGFR signaling and cell transformation by regulating the ubiquitin ligase Fbw7. Oncogene. 2010;29:2950–61.
Alexander PB, Yuan L, Yang P, Sun T, Chen R, Xiang H, et al. EGF promotes mammalian cell growth by suppressing cellular senescence. Cell Res. 2015;25:135–8.
Masuda K, Ishikawa Y, Onoyama I, Unno M, de Alboran IM, Nakayama KI, et al. Complex regulation of cell-cycle inhibitors by Fbxw7 in mouse embryonic fibroblasts. Oncogene. 2010;29:1798–809.
Wang L, Chen R, Li G, Wang Z, Liu J, Liang Y, et al. FBW7 mediates senescence and pulmonary fibrosis through telomere uncapping. Cell Metab. 2020;32:860–77 e869.
Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–12.
Teo YV, Rattanavirotkul N, Olova N, Salzano A, Quintanilla A, Tarrats N. et al. Notch signaling mediates secondary senescence. Cell Rep. 2019;27:997–1007.e1005.
Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–11.
Conzen SD, Cole CN. The three transforming regions of SV40 T antigen are required for immortalization of primary mouse embryo fibroblasts. Oncogene. 1995;11:2295–302.
Shay JW, Wright WE. Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol. 2000;1:72–76.
Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature. 2019;574:268–72.
Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–18.
Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–30.
Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–85.
Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–33.
Merkt W, Zhou Y, Han H, Lagares D. Myofibroblast fate plasticity in tissue repair and fibrosis: Deactivation, apoptosis, senescence and reprogramming. Wound Repair Regen. 2021;29:678–91.
Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederlander NJ, Jeganathan K, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10:825–36.
Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol. 2020;16:263–75.
van Deursen JM. Senolytic therapies for healthy longevity. Science. 2019;364:636–7.
Gong L, Shu J, Chen X, Pan H, Chen G, Bi Y, et al. DEPTOR inhibits lung tumorigenesis by inactivating the EGFR-mTOR signals. Cancer Lett. 2021;519:263–76.
Cui D, Dai X, Shu J, Ma Y, Wei D, Xiong X, et al. The cross talk of two family members of beta-TrCP in the regulation of cell autophagy and growth. Cell Death Differ. 2020;27:1119–33.
Xiong X, Zhao Y, Tang F, Wei D, Thomas D, Wang X, et al. Ribosomal protein S27-like is a physiological regulator of p53 that suppresses genomic instability and tumorigenesis. elife. 2014;3:e02236.
Chen X, Xiong X, Cui D, Yang F, Wei D, Li H, et al. DEPTOR is an in vivo tumor suppressor that inhibits prostate tumorigenesis via the inactivation of mTORC1/2 signals. Oncogene. 2020;39:1557–71.
Bi Y, Gong L, Liu P, Xiong X, Zhao Y. Nuclear ErbB2 represses DEPTOR transcription to inhibit autophagy in breast cancer cells. Cell Death Dis. 2021;12:397.
Acknowledgements
We thank Chao Bi from the core facility platform of Zhejiang University School of Medicine for her technical support. We also thank the Laboratory Animal Center of Zhejiang University for the technical support.
Funding
This work was supported by the National Natural Science Foundation of China (92053117, 81972591, and 82188102 to YZ, 81974429 and 82172898 to XX, 82002924 to DC), and the Natural Science Foundation of Zhejiang Province (LY22H160029 to DC, LZ22H160003 to YZ).
Author information
Authors and Affiliations
Contributions
LG and DC designed and performed the experiments, analyzed the data, and drafted the manuscript. DL designed and performed the experiments, and analyzed the data. XS and HP designed and performed the experiments. XX analyzed and interpreted the data, and revised the manuscript. YZ designed the study, analyzed and interpreted the data, and revised and finalized the manuscript. All authors have reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Dr Angelo Peschiaroli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Gong, L., Cui, D., Liu, D. et al. FBXW7 inactivation induces cellular senescence via accumulation of p53. Cell Death Dis 13, 788 (2022). https://doi.org/10.1038/s41419-022-05229-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05229-2
- Springer Nature Limited
This article is cited by
-
Circulating Osteopontin Predicts Clinical and Radiological Response in First-Line Treatment of Advanced Non-Small Cell Lung Cancer
Lung (2024)
-
FBXW7 in breast cancer: mechanism of action and therapeutic potential
Journal of Experimental & Clinical Cancer Research (2023)