
Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrotic parenchymal lung disease of unknown etiology and lack effective interventions. Using a combination of in vitro and in vivo studies, we found that overexpression of YAP1, a key effector in the Hippo pathway, promoted cell proliferation, migration, and collagen production in lung fibroblasts. Furthermore, the pro-fibrotic action of YAP1 was mediated by transcriptional activation of Twist1 through interacting with its partner TEAD. In contrast, knockdown of YAP1 inhibited extracellular matrix (ECM) deposition, which ultimately ameliorated lung fibrosis in vitro and in vivo. Additionally, we constructed a dysregulated miRNA regulatory network that affects the expression of the Hippo pathway effectors in IPF and identified miR-15a, which is significantly down-regulated in IPF patients, as one of the most essential miRNAs regulating this pathway. Moreover, knockdown of miR-15a resulted in fibroblast activation and lung fibrosis through promoting Twist expression by targeting inhibition of YAP1. In contrast, therapeutic restoration of miR-15a inhibits fibrogenesis in lung fibroblast and abrogated BLM-induced lung fibrosis in mice. These results highlight a role for miR-15a/YAP1/Twist axis in IPF that offer novel strategies for the prevention and treatment of lung fibrosis.
Similar content being viewed by others


Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrotic interstitial lung disease of unknown etiology with increasing morbidity and high mortality [1]. Until now, lung transplantation has been the most effective treatment to improve survival, and more and more studies have demonstrated the anti-fibrotic effects of nintedanib and pirfenidone in IPF [2,3,4]. Therefore, it is urgent to reveal the mechanism underlying IPF. IPF is characterized by the formation of myofibroblast foci and excessive deposition of extracellular matrix (ECM), which are mainly derived from the primary effector cells, lung fibroblasts [5]. Thus, the exploration of factors affecting the proliferation and differentiation of lung fibroblasts is useful to elucidate the molecular mechanism and to develop novel therapies for IPF.
The Hippo pathway is a highly conserved signaling pathway that participates in the regulation of organ development, tissue formation, and cancer, among others [6]. Several studies demonstrated that yes-associated protein (YAP), a key downstream effector of the Hippo pathway, promoted hepatic stellate cell activation and liver fibrosis, whereas pharmacological inhibition of YAP impeded fibrogenesis in mice [7, 8]. Liu et al. [9] found that YAP and transcriptional co-activator with PDZ-binding motif (TAZ) mediated mechanical force-induced fibroblast activation and fibrosis in IMR-90 lung fibroblasts. However, whether silencing YAP can attenuate lung fibrosis in mice is not yet clear. Furthermore, emerging evidence suggests that the Hippo pathway regulates cell proliferation and differentiation in different kinds of cells, whereas the underlying mechanisms and upstream regulator for Hippo pathway in IPF is not fully understood. Some studies have found that the YAP/TAZ-miR-130/301 circuit contributes to the development of fibrosis and pulmonary hypertension [10, 11]. An increasing number of studies, including ours, have identified the involvement of microRNAs (miRNAs) in lung fibrosis [12, 13], but it remains to be determined whether dysfunctions in the Hippo pathway during IPF are under the regulation of miRNAs and, if so, which miRNAs play the critical roles.
In the present study, our results demonstrated that YAP1 promoted fibrogenesis by transcriptional activation of Twist, whereas knockdown of YAP1 ablated lung fibrosis in vitro and in vivo. Moreover, we identified miR-15 acts as one of the most critical miRNAs regulating the YAP1/Twist in IPF. Taken together, these results indicate that YAP1 inhibition could be a novel strategy for the treatment of lung fibrosis.
Results
Overexpression of YAP1 promotes fibrogenesis in lung fibroblasts
First of all, we used gain- and loss-of-function experiments to evaluate the effect of YAP1, a key downstream effector of the Hippo pathway, in pulmonary fibrosis. The quantitative reverse transcription-PCR (qRT-PCR) assay showed significant up-regulation of YAP1 at the messenger RNA (mRNA) level (Fig. 1a). Furthermore, overexpression of YAP1 enhanced the synthesis of various fibrotic genes, such as Col 1α1, Col 3α1, and connective tissue growth factor (CTGF) (Fig. 1b–d). Simultaneously, enhanced expression of YAP1 promoted cell proliferation (Fig. 1e, f) and migration (Fig. 1g, h) and the transition of fibroblasts into myofibroblasts in cultured lung fibroblasts (Fig. 1i). Finally, forced expression of YAP1 resulted in up-regulation of fibrosis-associated proteins, including collagen 1, fibronectin 1 (Fn1), and alpha-smooth muscle actin (α-SMA) (Fig. 1j).

Overexpression of YAP1 results in fibrogenesis in lung fibroblasts. The mRNA levels of YAP1 (a), Col 1α1 (b), Col 3α1 (c), and CTGF (d) were significantly up-regulated by forced expression of YAP1; n = 6 cell batches. e, f Photomicrographs of double fluorescent staining with EDU (red) and DAPI (blue) demonstrate that YAP1 accelerated lung fibroblast proliferation; n = 5. The wound scratch assay (g, h) and immunofluorescence depicting fibroblast–myofibroblast transition (i) were used to identify the pro-fibrotic effect of YAP1; n = 5. j Fibrotic-associated protein expression was determined by western blot analysis. YAP1 increased the expression of collagen 1, Fn1, and α-SMA. The data are presented as the mean ± S.E.M.; n = 5; *P < 0.05
To further examine the pro-fibrotic effects of YAP1 in lung fibroblasts, we isolated the lung fibroblasts from adult mice and performed several parallel studies. Consistent with the results from the neonatal primary fibroblasts, we found that overexpression of YAP1 increased the mRNA level of collagen 1α1 and collagen 3α1 in adult lung fibroblasts (Fig. S1A-S1B). Meanwhile, forced expression of YAP1 promoted the proliferation of adult lung fibroblasts (Fig. S1C).
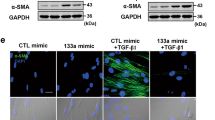
Next, we constructed YAP1-specific short hairpin RNA (shRNA) and transfected it into lung fibroblasts to examine the potential anti-fibrotic effect of YAP1 silencing during pulmonary fibrosis. As demonstrated in Fig. S2A-S2C, knockdown of YAP1 reduced the up-regulated expression of Col 1α1, Col 3α1, and CTGF driven by transforming growth factor-β1 (TGF-β1). Moreover, we found that the effect of TGF-β1 on cell proliferation, migration, and fibroblast–myofibroblast transition was nearly abated by YAP1 expression inhibition (Fig. S2D-S2F). Additionally, the similar expression of fibrotic proteins was alleviated by inhibition of YAP1 (Fig. S2G). Thus, using a combination of forced expression of YAP1 and inhibition of YAP1 in vitro, we found that overexpression of YAP1 promoted fibrogenesis, whereas inhibition of YAP1 attenuated TGF-β1-induced fibrogenesis in lung fibroblasts.
We then applied siRNA against TAZ, another transcriptional co-activator belongs to Hippo pathway, to assess the role of TAZ in lung fibrosis (Fig. S3A). As shown in Fig. S3B-S3C, silencing TAZ alleviated the TGF-β1-induced up-regulation of collagen 1α1 and collagen 3α1 (Fig. S3B-S3C). Moreover, our results showed that inhibition of TAZ could significantly mitigate the TGF-β1-driven proliferation and migration in lung fibroblasts (Fig. S3D-S3E).
YAP1 promotes lung fibrosis through transcriptional activation of Twist1
Previous studies have shown that the complex of YAP1 and TEAD translocate into the nucleus, where they associate with transcription factors and other DNA-binding proteins to modulate target gene transcription [14]. Thus, we wanted to uncover the molecular mechanisms that mediate the pro-fibrotic effects of YAP1 and to determine whether TEAD is involved in YAP1-driven lung fibrosis. To gain insight into the mechanism of YAP1, we utilized mutation of the YAP1 gene (YAP1-S94A, a loss-of-function mutant of YAP1) and the pharmacological inhibitor verteporfin, which specifically blocks the combination of YAP1 and TEAD. As demonstrated in Fig. 2a–c, overexpression of YAP1 enhanced the formation of Col 1α1, Col 3α1, and CTGF, whereas this effect was eliminated by applying verteporfin. On the other hand, interrupting the binding between YAP1 and TEAD1 by overexpressing YAP1-S94A prevented the YAP1-induced synthesis of Col 1α1, Col 3α1, and CTGF (Fig. 2a–c). Moreover, verteporfin attenuated the YAP1-induced up-regulation of fibrotic-associated proteins, and YAP1-S94A had no effect on these proteins (Fig. 2d). These data suggest that YAP1 promotes fibrogenesis by interacting with TEAD.

YAP1 promotes fibrogenesis by transcriptionally activating Twist1 expression through interaction with TEAD1. Real-time RT-PCR analysis indicated that mutation of YAP1 at the genetic level or inhibition of YAP1 by verteporfin attenuated the increased synthesis of Col 1α1 (a), Col 3α1 (b), and CTGF (c) induced by overexpression of YAP1, as well as the fibrotic protein expression (d); n = 5. e A schematic diagram of sequence complementarity between the YAP1/TEAD1 complex and Twist1. Western blot (f) and qRT-PCR (g) assays showed that blocking the combination of YAP1 and TEAD alleviated the effect of YAP1 on Twist1 expression at both the protein and mRNA levels; n = 5. h Chromatin immunoprecipitation (ChIP) assay revealed an association between YAP1/TEAD1 and Twist1 in lung fibroblasts; IgG served as a negative control; n = 3. i qRT-PCR analyses revealed that TEAD1/YAP1 was able to bind to the twist promoter region and promoted Twist1 expression in lung fibroblast;. n = 3. j Luciferase reporter constructs containing the empty vector pGL3 in the presence or absence of binding sites, as illustrated in (e), exhibited enhanced Twist1-Luc luciferase activity, whereas mut- Twist1-luc had no effect on luciferase activity; n = 5. The data are presented as the mean ± S.E.M.; *P < 0.05, **P < 0.01
Then, we applied computational analysis to explore the molecules downstream of YAP1/TEAD that might be involved in the process of lung fibrosis and found that there is a putative TEAD1-binding site within the promoter region of the Twist gene (Fig. 2e), indicating that YAP1/TEAD1 has the potential to regulate Twist expression at the transcriptional level. More importantly, forced expression of YAP1 increased the expression of Twist at both the protein and mRNA levels, whereas this effect was reversed by verteporfin, and YAP1-S94A prevented the up-regulation of Twist (Fig. 2f, g). Next, we performed a chromatin immunoprecipitation (ChIP) assay to further explore the correlation between YAP1 and Twist. As depicted in Fig. 2h, i, the ChIP and qRT-PCR assays revealed minimal binding of TEAD1 or YAP1 with the Twist promoter, which was significantly increased after transfection with TEAD1 or YAP1. Next, to examine the effect of TEAD1 or YAP1 on the promoter activity of Twist, we constructed a Twist luciferase reporter with the TEAD1-binding site (Twist-Luc) or a mutant TEAD1-binding site (mTwist-Luc). Then, we transfected lung fibroblasts with and without TEAD1 or YAP1 overexpression with the Twist-Luc and mTwist-Luc reporters. We found that overexpression of TEAD1 or YAP1 increased the luciferase activity of Twist -Luc by combining with the complementary binding site, and this activity was eliminated when we used mTwist-Luc (Fig. 2j).
To further examine whether Twist is necessary for the pro-fibrotic effects of YAP1, we transfected shRNA against Twist into lung fibroblasts to inhibit the expression of Twist. As shown in Fig. 3a, b, we found that knockdown of Twist attenuated the effect of YAP1 on collagen production. Moreover, silencing Twist alleviated YAP1-induced proliferation (Fig. 3c) and inhibited YAP1 caused cell migration in lung fibroblasts (Fig. 3d). Meanwhile, inhibition of Twist ablated the up-regulation of fibrosis-relevant proteins driven by YAP1 (Fig. 3e). These data indicated that YAP1 contributes to the development of fibrogenesis in lung fibroblasts by forming the YAP1/TEAD complex and then transcriptionally activates the expression of Twist.

Twist is necessary for the pro-fibrotic effects of YAP1. a, b Silencing Twist attenuated YAP1-induced synthesis of Col 1α1 and Col 3α1, which was measured by qRT-PCR; n = 5. c EDU staining illustrated that knockdown of Twist inhibits cell proliferation in lung fibroblasts; n = 5. d Scarification tests revealed that inhibition of YAP1 ablated the increased cell migration induced by YAP1; n = 5. e Western blot analysis showing the expression of fibrosis-relevant proteins after knockdown of Twist in the presence of YAP1 treatment; n = 5. Values are the mean ± S.E.M. of four independent experiments. *P < 0.05
Silencing YAP1 alleviated experimental pulmonary fibrosis in mice
Next, we continued to confirm the comprehensive role of YAP1 in bleomycin (BLM)-induced pulmonary fibrosis. To determine whether suppression of YAP1 could mitigate pulmonary fibrosis in mice, adenovirus-associated virus 5 (AAV5) containing a sh-YAP1 fragment was established and was designated AAV5-sh-YAP1. At 3 days after administration of BLM, AAV5-sh-YAP1 was intrabronchially injected into mice. As shown in Fig. 4a, b, our data revealed that AAV5-sh-YAP1 ameliorated the BLM-induced interstitial lung fibrosis based on Masson’s staining and HE staining. The immunohistochemistry (IHC) staining demonstrated low expression of collagen 1, Fn1, and YAP1 after treatment with AAV5-sh-YAP1 in the BLM-induced mice model (Fig. 4c), along with a dramatic reduction in the production of Col 1α1, Col 3α1 and CTGF and the levels of Fn1, α-SMA, and Twist1 at the mRNA and protein levels (Fig. 4d–g). Moreover, the AAV-mediated delivery in the current study is not specific to fibroblasts, and we thus isolated the adult lung fibroblasts from wild-type mice 3 weeks after injection of AAV5-sh-YAP1, and found that the expression of YAP1 was significantly decreased in the mice lung fibroblasts after injection of AAV5-sh-YAP1 (Fig. 4h). These data indicate that knockdown of YAP1 relieved the pulmonary injury induced by BLM, at least in part, by modulating lung fibroblasts.

Inhibition of YAP1 ameliorates pulmonary fibrosis in mice after BLM injection. a Representative images of H&E and Masson’s trichrome staining of the lungs of mice; n = 6. b Knockdown of YAP1 attenuated the hydroxyproline content in the lungs of mice treated with BLM; n = 6. c Representative immunohistochemistry (IHC) microscopy images of fibrosis-related markers in mice; n = 6. Knockdown of YAP1 by AAV5-sh-YAP1 abated the increased Col 1α1 (d), Col 3α1 (e), and CTGF (f) in BLM-treated mice; n = 6. g Inhibition of YAP1 alleviated the expression of fibrosis-related proteins in mice injected with BLM; n = 5. h The expression of YAP1 in the adult lung fibroblasts isolated from wild-type mice three weeks after injection of AAV5-sh-Scram or AAV5-sh-YAP1; n = 5. *P < 0.05
Identification of key miRNAs regulating the Hippo pathway during IPF
The core components of the Hippo pathway include YAP1, LATS1, LATS2, TEAD1, TEAD2, TEAD3, TEAD4, MOB1A, MOB1B, WWTR1, STK3, SAV1, WWC1, NF2, FRMD1, and FRMD6 [15]. Under the control of false discovery rate (FDR) < 10%, 193 miRNAs were detected as significantly differentially expressed in IPF patients compared with those in controls. According to the miRNA-target information in multiple databases (see Materials and methods), we found that 87 of 193 differentially expressed miRNAs targeted the core components of the Hippo pathway (Fig. 5a). Figure 5b shows the number of predicted regulating miRNAs for each Hippo pathway gene. In the network, miR-15a, miR-15b, and miR-497 regulated the largest number of core Hippo pathway genes (Fig. 5c), and all of these three miRNAs regulated YAP1, LATS1, LATS2, NF2, TEAD1, WWC1, SAV1, and FRMD6. Interestingly, we found the same seed sequences and eight other similar base sequences in miR-15a, miR-15b, and miR-497 (Fig. 5d). Among the three miRNAs, miR-15a was the most significantly down-regulated in IPF patients (P = 1.72 × 10−11, Fig. 5e–g). In addition, eight Hippo pathway genes, including YAP1, were potential targets for miR-15a. Meanwhile, we found that miR-15a level was significantly decreased in BLM-injected mouse lungs (Fig. 5h) as well as in TGF-β1-treated lung fibroblasts (Fig. 5i).

Identification of miR-15a as a master miRNA regulating the Hippo pathway in IPF. a Core miRNA-Hippo network in IPF. Round nodes and square nodes represent miRNAs and core Hippo pathway genes, respectively. The miRNAs colored red and green represent miRNAs that are up-regulated and down-regulated in IPF, respectively. The size of each node indicates the number of predicted miRNA regulators or the number of predicted target genes. The lines between nodes indicate the miRNA-target relationship. b Histogram demonstrating the number of predicted regulating miRNAs for each Hippo pathway gene. c Histogram demonstrating the number of predicted targets for each miRNA. d Sequences of miR-15a, miR-15b, and miR-497. The seed sequences and other similar sequences are outlined in green and blue, respectively. The miR-15a (e), miR-15b (f), and miR-497 (g) were down-regulated in IPF patients based on the microarray dataset GSE32538, which contains samples from 106 IPF patient and 50 healthy controls. Down-regulation of miR-15a in BLM-treated mice (h) and in cultured lung fibroblasts in response to 10 ng/ml TGF-β1 for 48 h (i); n = 6 independent experiments. *P < 0.05
Inhibition of miR-15a promotes fibrogenesis by regulating the YAP1/Twist axis
To examine the role of miR-15a in the process of lung fibrosis, we first evaluated the regulatory effect of miR-15a on YAP1. As shown in Fig. 6a, YAP1 contains the seed sequence for miR-15a that is broadly conserved among humans, mice, and rats. We then constructed a luciferase reporter vector carrying a targeting fragment containing miR-15a-binding sites or mutant sequences (nucleotide replacement). The luciferase assay showed that miR-15a inhibited the activities of the wild-type YAP1 luciferase vector (YAP1-WT), whereas it had no effect on the vector carrying mutated binding site (YAP1-Mut) (Fig. 6b). Moreover, overexpression of miR-15a inhibited the expression of YAP1 at both the protein (Fig. 6c) and mRNA levels (Fig. 6d). These results suggested that miR-15a regulated YAP1 expression at the transcriptional level.

Knockdown of miR-15a results in fibrogenesis by regulating YAP1 in lung fibroblasts. a Sequence alignment showing miR-15a:YAP1 complementarity in mouse, rat, and human genes. The matched base pairs in the seed region are outlined in red and green. Mut mutated binding site. b Luciferase reporter activities of chimeric vectors carrying the luciferase gene and a fragment of the YAP1 3′-UTR containing wild-type or mutant miR-15a-binding sites; n = 5. c Western blot analysis of YAP1 protein levels in lung fibroblasts transfected with miR-15a; n = 5. d qRT-PCR was performed to detect the effect of miR-15a on YAP1 mRNA expression; n = 5. qRT-PCR analysis of col 1α1 (e) and col 3α1 (f) mRNA levels in lung fibroblasts after miR-15a knockdown together with YAP1/Twist inhibition; n = 5. EDU staining (g) and scratch assay (h) were employed to determine the effect of miR-15a knockdown along with YAP1/Twist silencing on cell proliferation and migration in lung fibroblasts; n = 5. i Western blot analyses were used to determine the alterations in fibrosis-related protein levels in lung fibroblasts after miR-15a knockdown together with YAP1/Twist inhibition; n = 5 independent experiments. *P < 0.05
Next, we employed a miR-15a inhibitor and YAP1- or Twist-specific shRNAs to examine the effect of miR-15a inhibition on fibrogenesis and to determine whether the YAP1/Twist axis mediates this effect. As shown in Fig. 6e, f, knockdown of miR-15a increased the expression of collagen 1α1 and collagen 3α1 at mRNA levels, whereas these effects were abolished by silencing YAP1 or Twist. To further determine the accurate pathological process contributing to the pro-fibrotic action of miR-15a inhibition, we examined the effect of miR-15a inhibition on proliferation, migration, and fibroblast-myofibroblast transition. As illustrated in Fig. 6g, h, we found that inhibition of miR-15a promoted cell proliferation and migration in cultured lung fibroblasts. Moreover, silencing YAP1 or Twist attenuated the pro-fibrotic effects of miR-15a inhibition. Meanwhile, knockdown of miR-15a resulted in the up-regulation of several fibrosis-related proteins, including collagen 1, Fn1, α-SMA, and its downstream effector YAP1 and Twist, whereas the increase in the expression of these proteins was almost abrogated after inhibition of YAP1 or Twist (Fig. 6i). These results indicated that knockdown of miR-15a leads to lung fibrogenesis by modulating the YAP1/Twist axis.
Enhanced expression of miR-15a attenuates lung fibrosis in vitro and in vivo
Next, we transfected miR-15a into lung fibroblasts to evaluate whether overexpression of miR-15a can inhibit fibrogenesis. As expected, we found that forced expression of miR-15a alleviated TGF-β1-driven synthesis of collagen 1α1 and collagen 3α1 in both neonatal primary lung fibroblasts (Fig. 7a, b) and adult lung fibroblasts (Fig. S4A-S4B). Meanwhile, enhanced expression of miR-15a inhibited cell proliferation, attenuated migration, and reduced the transition from lung fibroblasts into α-SMA-positive myofibroblasts (Fig. 7c–e, Fig. S4C). Consistent with these results, overexpression of miR-15a inhibited the up-regulation of fibrosis-relevant proteins and YAP1/Twist induced by TGF-β1 in lung fibroblasts (Fig. 7f).

Overexpression of miR-15a attenuates TGF-β1-induced fibrogenesis in lung fibroblasts. qRT-PCR analysis of the mRNA levels of col 1α1 (a) and col 3α1 (b) in lung fibroblasts after treatment with 10 ng/ml TGF-β1 together with miR-15a overexpression; n = 5. c EDU assay was performed to determine the effect of miR-15a on TGF-β1-induced lung fibroblast proliferation; n = 5. d Scratch assay was used to examine the effect of miR-15a on TGF-β1-induced lung fibroblasts migration; n = 5. e Effect of miR-15a on TGF-β1-induced fibroblast-myofibroblast transition was determined by immunofluorescence; n = 4 independent experiments. f Western blot analysis for the determination of alterations in fibrosis-related protein levels in lung fibroblasts after treatment with TGF-β1 together with miR-15a overexpression; n = 5. *P < 0.05
Finally, we performed intratracheal injection of AAV5 carrying the miR-15a precursor sequence (AAV5-miR-15a) into mice to determine whether overexpression of miR-15a could attenuate BLM-induced experimental lung fibrosis, and we found that the expression of miR-15a was significantly up-regulated in the adult lung fibroblasts isolated from wild-type mice after injection of AAV5-miR-15a (Fig. 8a). Masson’s staining results showed that overexpression of miR-15a mitigated collagen deposition and reduced the fibrotic area in BLM-treated mice (Fig. 8b), which was also accompanied by the down-regulation of hydroxyproline content (Fig. 8c) and inhibited expression of collagen 1α1 and collagen 3α1 (Fig. 8d, e). Meanwhile, IHC showed that forced expression of miR-15a reduced the expression of Fn1 and YAP1 in BLM-treated mice (Fig. 8f). Furthermore, overexpression of miR-15a inhibited the expression of Fn1 and YAP1, as well as various fibrosis-related proteins (collagen 1 and Twist) in BLM-treated mice (Fig. 8g).

Forced expression of miR-15a alleviates BLM-induced lung fibrosis in mice. a The expression of miR-15a in the adult lung fibroblasts isolated from wild-type mice 3 weeks after injection of AAV5-miR-Ctrl or AAV5-miR-15a; n = 5. b Representative images of Masson’s trichrome-stained sections of mouse lungs. c Overexpression of miR-15a ablated the hydroxyproline content in the lungs of mice treated with BLM; n = 6. d, e Expression of col 1α1 and col 3α1 mRNA as measured by qRT-PCR in BLM-treated mice after the injection of AAV5-miR-15a; n = 6. f, g IHC and western blot analysis of the expression of FN1, YAP1, and other fibrosis-relevant proteins in BLM-treated mice after overexpression of miR-15a; n = 5. *P < 0.05
Discussion
In the present study, we characterized the pro-fibrotic property of YAP1 in a BLM-induced lung fibrosis model and elucidated the molecular events underlying the fine regulation of YAP1 in lung fibrosis. We found that overexpression of YAP1 promotes lung fibrosis by transcriptional activation of Twist expression through binding TEAD, whereas silencing YAP1 attenuated BLM-induced lung fibrosis in mice. These results indicated the critical role of YAP1 in lung fibrosis. Moreover, we constructed a dysregulated miRNA regulatory network that affects the expression of the Hippo pathway effectors in IPF and identified miR-15a as one of the most essential miRNAs regulating this pathway. Our results showed that knockdown of miR-15a resulted in fibroblast activation and lung fibrosis through promoting Twist expression by targeting inhibition of YAP1. In contrast, therapeutic restoration of miR-15a inhibited fibrogenesis in lung fibroblast and abrogated BLM-induced lung fibrosis in mice. This study indicated that manipulating the expression of YAP1 or miR-15a represents promising therapeutic strategies for lung fibrosis.
Many studies have demonstrated that the Hippo pathway participates in the process of organ fibrosis [8, 9]. Seo et al. [16] found that knockout of Salvador homolog 1 (Sav1) in tubular epithelial cells aggravated renal tubulointerstitial fibrosis in mice. Fu et al. [17] found up-regulation of Fibulin-5 in the lung tissues of patients with chronic obstructive pulmonary disease and IPF, and which demonstrated that overexpression of Fibulin-5 promoted the proliferation and migration of airway smooth muscle cells by increasing the expression of YAP and TAZ. However, the role and upstream regulators of the Hippo pathway in lung fibrosis are not clear, although several studies have found that YAP1 mediated mechanosignaling-induced lung fibroblast activation [9]. Recently, Gokey et al. [18] found the activation of YAP1 in IPF respiratory epithelium and demonstrated that the interaction between YAP1 and mTOR/p-S6 induced cell proliferation and migration and inhibited epithelial cell differentiation that may contribute to the pathogenesis of IPF. In the present study, we found up-regulation of YAP1 in both epithelial cells and fibroblasts in BLM-treated mice, and further study showed that overexpression of YAP1 promoted fibrogenesis in lung fibroblasts, accompanied by an increase in the production of fibrosis-related proteins and accelerated transition of fibroblasts into myofibroblasts, whereas silencing YAP1 exerted the opposite effects. We therefore considered that YAP1 contributed lung fibrosis by promoting fibrogenesis in lung fibroblasts and resulted in epithelial–mesenchymal transition, which warrants our further study. More importantly, we found that knockdown of YAP1 ameliorated BLM-induced pulmonary fibrosis in mice and thus demonstrated that YAP1 could be a potential target for treatment of lung fibrosis.
Accumulating evidences demonstrated that TEAD is one of the most canonical transcriptional factors mediating the pathophysiological effects of YAP1 [14, 19, 20]. The present study found that enhanced expression of YAP1 promoted lung fibrogenesis via transcriptionally modulating the activity of TEAD, whereas mutation of binding sites between YAP1 and TEAD abolished the pro-fibrotic effect of YAP1. Meanwhile, blocked combination of YAP1 and TEAD by Verteporfin also mitigated the modification of YAP1 in the process of lung fibrosis. A large number of studies indicated Twist contributing to the pathological process of lung fibrosis [21,22,23]. Pozharskaya et al. [23] revealed that the expression of Twist was significantly elevated in type II epithelial cells and fibroblasts in lungs of IPF patients. Moreover, Yang et al. [24] demonstrated that overexpression of inhibitor of DNA-binding 2 attenuates pulmonary fibrosis through regulation of c-Abl and Twist. In addition to promoting epithelial–mesenchymal transition, Twist has been found to activate fibroblasts and lead to systemic sclerosis [25]. However, a recent study from Tan et al. [22] found that loss of Twist in the mesenchymal compartment in mice leads to increased expression of CXCL12, which promoted accumulation of T cells in the lung and resulted in pulmonary fibrosis. Our study showed that silencing Twist in lung fibroblast abolished the pro-fibrotic role of AMO-miR-15a. Thus, although Twist plays various functions in the process of IPF, the detailed and precise effect of Twist needs to be further investigated. In this study, we found that YAP1 contributed to lung fibrosis by transcriptionally activating the Twist expression via binding to TEAD. Taken together, our results suggest the novel mechanisms of Hippo signals in IPF that may represent a potential therapy for pulmonary fibrosis.
Some studies have reported that miRNAs contribute to the development of IPF and may act as targets for the treatment of this disease. Dakhlallah et al. [26] found that enhanced miR-17~92 cluster expression attenuated lung fibrosis. Xiao et al. [27] showed that overexpression of miR-29 mitigated lung fibrosis in mice. Our previous studies showed that miR-26a prevents lung fibrosis by affecting both lung fibroblasts and epithelial cells [13, 28]. In this study, we found the down-regulated miR-15a, miR-15b and miR-497 that regulated the largest number of core Hippo pathway genes. Among the three miRNAs, miR-15a was the most significantly down-regulated in IPF patients. A growing body of evidence demonstrated that miR-15a inhibited cell proliferation and fibrotic relevant diseases [29, 30]. Tijsen et al. [30] found that inhibition of miR-15a aggravated overload-induced cardiac fibrosis in mice. Rawal et al. [31] found that suppression of miR-15a/b promoted fibrotic remodeling in Type 2 diabetic mouse heart, whereas forced expression of miR-15a/b inhibited the activation of diabetic human cardiac fibroblasts. However, the role and mechanism of miR-15 in lung fibrosis is not clear. Our study found that overexpression of miR-15a alleviated fibroblast activation and lung fibrosis in vitro and in vivo. In contrast, knockdown of miR-15a promoted fibrogenesis in lung fibroblasts, whereas that was nearly alleviated by YAP1 or Twist inhibition. Thus, our study demonstrated that miR-15a inhibited lung fibrosis, at least in part, by regulating the YAP1/Twist axis. Interestingly, miR-15a, miR-15b, and miR-497 contain the same seed sequences, and it is thus warranted to determine whether miR-15b and miR-497 also play anti-fibrotic effects in IPF. In addition, we suppose whether we could find critical regulators that promoted the expression of these three miRNAs simultaneously to exert a better effect against lung fibrosis than any of them.
Although dozens of miRNAs have been reported to inhibit pulmonary fibrosis in mice, there is little clinical or pre-clinical study about the application of miRNAs in the treatment of lung fibrosis. As we know, IPF is caused by the combined effects of deregulation of multiple miRNAs and genes rather than individual genes or miRNAs [32]. In fact, by analyzing the microarray dataset GSE32538, we previously found that more than 80% of the dysregulated miRNAs were down-regulated in IPF [12]. Thus, we speculate that exploring and modulating the factors causing the global down-regulation of miRNAs in IPF may provide insights into this disease. In this study, we screened for dysregulated miRNAs that regulate the Hippo pathway during IPF and identified miR-15a as one of the most key miRNAs. In addition, some studies have reported that YAP1 controls miRNA biogenesis and contributes to human cancers [33, 34]. Thus, further studies are warranted to investigate whether YAP1 is responsible for the global down-regulation of miRNAs in IPF.
Taken together, the present study demonstrated that YAP1 promoted lung fibrogenesis through transcriptionally activating Twist via binding TEAD that conferred by miR-15a loss. In addition, our results showed that YAP1 silencing or miR-15a supplement could be novel strategies for the treatment of lung fibrosis.
Materials and methods
Animal model and treatment
C57BL/6 mice with a mean weight of 20 g were obtained from Changsheng Biotechnology (Liaoning, China) and maintained according to the regulation approved by the Institutional Animal Ethics Committee of Harbin Medical University. Pulmonary fibrosis was induced with a single intratracheal instillation of BLM dissolved in saline as previously described [13]. Detailed descriptions of animal model and treatment are described in the supplement.
Cell culture
Primary lung fibroblasts were isolated from lungs of 3-day-old C57BL/6 mice. Detailed descriptions of cell culture and treatment are described in the supplement.
Western blot
Western blot was performed as previously described [35]. Detailed descriptions of western blot assays are described in the supplement.
Quantitative RT-PCR (qRT-PCR)
Total RNA from collected cells or tissues was extracted using TRIzol reagent as previously described [36]. The relative miRNA and long non-coding RNA expression levels were calculated based on Ct values and were respectively normalized to U6 or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels in each sample. Detailed descriptions of qRT-PCR assays are described in the supplement.
Luciferase reporter assays
Using PCR, we amplified the 3’-untranslated region (UTR) of YAP1 containing the predicated target sites for miR-15a, as previously described [37]. The PCR fragment was inserted into the multiple cloning sites in the pMIR-REPORT luciferase miRNA expression vector (Ambion, USA). HEK293T cells (1 × 105/well) were co-transfected with 0.1 μg of the luciferase YAP1 chimeric vector and miR-15a mimics by Lipofectamine 2000 (Invitrogen, USA). We collected the cell lysate after 24 h of transfection and measured luciferase activities with a Dual-Luciferase Reporter Assay kit (Promega, USA).
Bioinformatics analysis
The miRNA expression dataset GSE32538, which consists of 109 IPF patient samples and 50 healthy control samples, was downloaded from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) [38]. Quantile normalization and log2 transformation were applied to the raw data. Values of replicate probes corresponding to an miRNA were averaged to represent the expression value of that miRNA. Two-sample t-tests were carried out to select the differentially expressed miRNAs. P values were adjusted by the Benjamini and Hochberg correction procedure to account for multiple tests with a FDR < 10%. Considering that miRNA targets predicted by multiple algorithms might be more reliable, we used the miRNA-target interactions of Homo sapiens appearing in at least two of nine datasets (TargetScan, miRanda, PicTar, miRBase, DIANA-microT, PITA, RNAhybrid, miRTarBase, and miRecords). The miRNA-target network was constructed using Cytoscape web tool (http://js.cytoscape.org). All analyses were performed in R 3.2.3 (https://www.r-project.org/).
Statistical analysis
Data were presented as the mean ± S.E.M. of at least three independent experiments. One-way analysis of variance followed by Dunnett’s test was used for multiple comparisons. In addition, two-tailed P < 0.05 was considered statistically significant difference. Statistical analyses were carried out using GraphPad Prism 5.0 and SPSS 14.0.
References
Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med. 2018;24:39–49.
Raghu G. Idiopathic pulmonary fibrosis: lessons from clinical trials over the past 25 years. Eur Respir J. 2017;50:pii: 1701209
van Manen MJG, Birring SS, Vancheri C, Vindigni V, Renzoni E, Russell AM, et al. Effect of pirfenidone on cough in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2017;50:pii: 1701157.
Richeldi L, Kreuter M, Selman M, Crestani B, Kirsten AM, Wuyts WA, et al. Long-term treatment of patients with idiopathic pulmonary fibrosis with nintedanib: results from the TOMORROW trial and its open-label extension. Thorax. 2018;73:581–583.
Li Y, Liang J, Yang T, Monterrosa Mena J, Huan C, Xie T, et al. Hyaluronan synthase 2 regulates fibroblast senescence in pulmonary fibrosis. Matrix Biol. 2016;55:35–48.
Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–28.
Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, et al. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology. 2018;154:1465–79 e1413.
Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF, et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015;63:679–88.
Liu F, Lagares D, Choi KM, Stopfer L, Marinkovic A, Vrbanac V, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308:L344–357.
Bertero T, Cottrill KA, Annis S, Bhat B, Gochuico BR, Osorio JC, et al. A YAP/TAZ-miR-130/301 molecular circuit exerts systems-level control of fibrosis in a network of human diseases and physiologic conditions. Sci Rep. 2015;5:18277.
Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, et al. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep. 2015;13:1016–32.
Li H, Zhao X, Shan H, Liang H. MicroRNAs in idiopathic pulmonary fibrosis: involvement in pathogenesis and potential use in diagnosis and therapeutics. Acta Pharm Sin B. 2016;6:531–9.
Liang H, Xu C, Pan Z, Zhang Y, Xu Z, Chen Y, et al. T`he antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol Ther. 2014;22:1122–33.
Lin KC, Moroishi T, Meng Z, Jeong HS, Plouffe SW, Sekido Y, et al. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat Cell Biol. 2017;19:996–1002.
Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–312.
Seo E, Kim WY, Hur J, Kim H, Nam SA, Choi A, et al. The Hippo-Salvador signaling pathway regulates renal tubulointerstitial fibrosis. Sci Rep. 2016;6:31931.
Fu J, Zheng M, Zhang X, Zhang Y, Chen Y, Li H, et al. Fibulin-5 promotes airway smooth muscle cell proliferation and migration via modulating Hippo-YAP/TAZ pathway. Biochem Biophys Res Commun. 2017;493:985–91.
Gokey JJ, Sridharan A, Xu Y, Green J, Carraro G, Stripp BR, et al. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight. 2018;3:pii: 98738.
Saito K, Kawasoe R, Sasaki H, Kawaguchi A, Miyata T. Neural progenitor cells undergoing Yap/Tead-mediated enhanced self-renewal form heterotopias more easily in the diencephalon than in the telencephalon. Neurochem Res. 2018;43:171–80.
Mesrouze Y, Bokhovchuk F, Meyerhofer M, Fontana P, Zimmermann C, Martin T, et al. Dissection of the interaction between the intrinsically disordered YAP protein and the transcription factor TEAD. eLife. 2017;6:pii: e25068.
Zhao X, Sun J, Chen Y, Su W, Shan H, Li Y, et al. lncRNA PFAR promotes lung fibroblast activation and fibrosis by targeting miR-138 to regulate the YAP1-Twist axis. Mol Ther. 2018;26:2206–17.
Tan J, Tedrow JR, Nouraie M, Dutta JA, Miller DT, Li X, et al. Loss of Twist1 in the mesenchymal compartment promotes increased fibrosis in experimental lung injury by enhanced expression of CXCL12. J Immunol. 2017;198:2269–85.
Pozharskaya V, Torres-Gonzalez E, Rojas M, Gal A, Amin M, Dollard S, et al. Twist: a regulator of epithelial-mesenchymal transition in lung fibrosis. PLoS ONE. 2009;4:e7559.
Yang J, Velikoff M, Agarwal M, Disayabutr S, Wolters PJ, Kim KK. Overexpression of inhibitor of DNA-binding 2 attenuates pulmonary fibrosis through regulation of c-Abl and Twist. Am J Pathol. 2015;185:1001–11.
Palumbo-Zerr K, Soare A, Zerr P, Liebl A, Mancuso R, Tomcik M, et al. Composition of TWIST1 dimers regulates fibroblast activation and tissue fibrosis. Ann Rheum Dis. 2017;76:244–51.
Dakhlallah D, Batte K, Wang Y, Cantemir-Stone CZ, Yan P, Nuovo G, et al. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;187:397–405.
Xiao J, Meng XM, Huang XR, Chung AC, Feng YL, Hui DS, et al. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol Ther. 2012;20:1251–60.
Liang H, Gu Y, Li T, Zhang Y, Huangfu L, Hu M, et al. Integrated analyses identify the involvement of microRNA-26a in epithelial-mesenchymal transition during idiopathic pulmonary fibrosis. Cell Death Dis. 2014;5:e1238.
Lovat F, Fassan M, Gasparini P, Rizzotto L, Cascione L, Pizzi M, et al. miR-15b/16-2 deletion promotes B-cell malignancies. Proc Natl Acad Sci USA. 2015;112:11636–41.
Tijsen AJ, van der Made I, van den Hoogenhof MM, Wijnen WJ, van Deel ED, de Groot NE, et al. The microRNA-15 family inhibits the TGFbeta-pathway in the heart. Cardiovasc Res. 2014;104:61–71.
Rawal S, Munasinghe PE, Nagesh PT, Lew JKS, Jones GT, Williams MJA, et al. Down-regulation of miR-15a/b accelerates fibrotic remodelling in the Type 2 diabetic human and mouse heart. Clin Sci (Lond). 2017;131:847–63.
Liang H, Liu S, Chen Y, Bai X, Liu L, Dong Y, et al. miR-26a suppresses EMT by disrupting the Lin28B/let-7d axis: potential cross-talks among miRNAs in IPF. J Mol Med (Berl). 2016;94:655–65.
Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD, et al. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell. 2014;156:893–906.
Chaulk SG, Lattanzi VJ, Hiemer SE, Fahlman RP, Varelas X. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J Biol Chem. 2014;289:1886–91.
Liang H, Zhao X, Wang C, Sun J, Chen Y, Wang G, et al. Systematic analyses reveal long non-coding RNA (PTAF)-mediated promotion of EMT and invasion-metastasis in serous ovarian cancer. Mol Cancer. 2018;17:96.
Liang H, Pan Z, Zhao X, Liu L, Sun J, Su X, et al. LncRNA PFL contributes to cardiac fibrosis by acting as a competing endogenous RNA of let-7d. Theranostics. 2018;8:1180–94.
Cai B, Ma W, Ding F, Zhang L, Huang Q, Wang X, et al. The long noncoding RNA CAREL controls cardiac regeneration. J Am Coll Cardiol. 2018;72:534–50.
Yang IV, Coldren CD, Leach SM, Seibold MA, Murphy E, Lin J, et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax. 2013;68:1114–21.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (31671187, 81770284); the Major Program of National Natural Science Foundation of China (81530010); the Wuliande Foundation of Harbin Medical University (WLD-QN1707); and the University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province (UNPYSCT-2016197).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by R.A. Knight
Supplementary information
Rights and permissions
About this article

Cite this article
Chen, Y., Zhao, X., Sun, J. et al. YAP1/Twist promotes fibroblast activation and lung fibrosis that conferred by miR-15a loss in IPF. Cell Death Differ 26, 1832–1844 (2019). https://doi.org/10.1038/s41418-018-0250-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0250-0
- Springer Nature Limited
This article is cited by
-
The possible correlation between miR-762, Hippo signaling pathway, TWIST1, and SMAD3 in lung cancer and chronic inflammatory diseases
Scientific Reports (2024)
-
YAP1 inhibits the senescence of alveolar epithelial cells by targeting Prdx3 to alleviate pulmonary fibrosis
Experimental & Molecular Medicine (2024)
-
Preventive effects of Ramelteon on bleomycin-induced pulmonary fibrosis in mice
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Roles of Twist1 in lipid and glucose metabolism
Cell Communication and Signaling (2023)
-
Divergent roles of the Hippo pathway in the pathogenesis of idiopathic pulmonary fibrosis: tissue homeostasis and fibrosis
Inflammation and Regeneration (2023)