
Abstract
Clear-cell renal cell carcinoma (ccRCC) is the most common type of RCC; however, the intratumoral heterogeneity in ccRCC remains unclear. We first identified markers and biological features of each cell cluster using bioinformatics analysis based on single-cell and spatial transcriptome RNA-sequencing data. We found that gene copy number loss on chromosome 3p and amplification on chromosome 5q were common features in ccRCC cells. Meanwhile, NNMT and HILPDA, which are associated with the response to hypoxia and metabolism, are potential therapeutic targets for ccRCC. In addition, CD8+ exhausted T cells (LAG3+ HAVCR2+), CD8+ proliferated T cells (STMN+), and M2-like macrophages (CD68+ CD163+ APOC1+), which are closely associated with immunosuppression, played vital roles in ccRCC occurrence and development. These results were further verified by whole exome sequencing, cell line and xenograft experiments, and immunofluorescence staining. Finally, we divide patients with ccRCC into three subtypes using unsupervised cluster analysis. and generated a classifier to reproduce these subtypes using the eXtreme Gradient Boosting algorithm. Our classifier can help clinicians evaluate prognosis and design personalized treatment strategies for ccRCC. In summary, our work provides a new perspective for understanding tumor heterogeneity and will aid in the design of antitumor therapeutic strategies for ccRCC.

Similar content being viewed by others


Introduction
Renal cancer cells form common tumors of the urinary system; clear-cell renal cell carcinoma (ccRCC) is the most common pathological type of RCC [1]. Given the limited therapeutic efficacy of surgery, approximately 30% of patients with ccRCC exhibit distant metastasis after surgery [2]. These patients require systemic therapy, such as chemotherapy and immunotherapy. However, due to a limited understanding of the molecular biological characteristics of ccRCC, most adjuvant therapeutic drugs have failed to achieve significant benefits in treating ccRCC, with only two (renal tumor cell vaccine and sunitinib) exhibiting potential value [3]. Recently, with the deepening of our understanding of tumor immunity, immunotherapy has gained increasing attention as a potential therapeutic modality for ccRCC. However, the overall response rate of anti-PD-1 antibodies remains at 37–58% for ccRCC [4, 5]. The tumor heterogeneity associated with ccRCC pathogenesis and the tumor microenvironment (TME) comprise the main factors contributing to the poor response rate [6]. Therefore, the heterogeneity of ccRCC must be further characterized to inform the development of more effective treatment strategies.
With the development of high-throughput sequencing, tumor heterogeneity can be explored based on whole genome sequence analysis. Unlike bulk RNA-sequencing (RNA-seq), single-cell RNA-seq (scRNA-seq) analysis can be used to investigate the transcriptomic features of individual cells in the tissues of patients. Meanwhile, spatial transcriptome RNA-seq (stRNA-seq) can provide transcriptomic and positional data for intratumoral tissues. Accordingly, scRNA-seq and stRNA-seq are powerful tools for investigating cancer cells and the TME [7]. Indeed, scRNA-seq has been employed to study tumor heterogeneity in ccRCC [8,9,10], however, due to the high associated cost and strict sampling criteria, most analyses have been based on small sample sizes and single-omics technology or have lacked a strict normal control group. Therefore, larger scRNA-seq and stRNA-seq studies on numerous ccRCC samples are needed with an appropriate normal control. This will provide a more complete picture of tumor heterogeneity and the TME while limiting experimental bias caused by small sample sizes.
Obradovic et al. [11] performed scRNA-seq on 38 samples from 11 patients with ccRCC. They leveraged the VIPER algorithm to quantify single-cell protein activity based on 163,908 cells. Subsequently, Li et al. developed maps for spatial transcriptome sequencing of renal carcinoma using stRNA based on 16 samples from eight patients with ccRCC [12]. In the current study, we build on these previous reports by performing an integrated analysis using scRNA-seq and stRNA-seq data. The primary objectives of this study are to (1) identify tumor heterogeneity and subtypes in ccRCC and (2) identify new biomarkers of the different cell types and potential targets for ccRCC therapy. Collectively, the findings of this study expand the current knowledge regarding the biological characteristics of ccRCC, providing theoretical insights for designing novel therapeutic strategies for patients with ccRCC.
Materials and methods
Sample collection and whole exome sequencing analysis
Surgically resected ccRCC specimens were collected from 20 patients, including paracancerous and cancerous samples, at Shanxi Bethune Hospital with informed consent. The specimens were embedded in paraffin. DNA extraction, whole exome sequencing (WES), and data analysis were performed by Lc-Bio Technologies Co., Ltd. (Hangzhou). The single nucleotide polymorphism (SNP) analysis and CNV analysis of WES data were performed using the R package “maftools” and GISTIC 2.0 (https://cloud.genepattern.org/), respectively.
Data acquisition
The sources of all data, including scRNA-seq, bulk-seq, mRNA microarray, and DNA mutation data, as well as clinical information, are presented in Table S1 [11,12,13,14].
Data quality control and cell type annotation
We collected scRNA-seq data from eight ccRCC patients and stRNA-seq data from eight ccRCC patients (Table S2). Data quality control (QC) was performed using the R package “Seurat 4.3.0.” The QC standard was applied as follows for the RNA-seq data of each cell: >300 unique molecular identifiers (UMIs); <15% mitochondria-derived UMI counts; <10% hemoglobin-derived UMI counts. In the dimension reduction analysis, we retained the top 2000 variable genes and performed principal component analysis (PCA); the top 50 components were included in further analyses. We tested the all-resolution ratio (0–2.0) and found that a ratio of 1.1 was the most suitable for cluster analysis using Seurat’s FindClusters function (Fig. S1A). We then used the uniform manifold approximation and projection (UMAP) or t-distributed Stochastic Neighbor Embedding (tSNE) to visualize the data [15]. As the scRNA-seq data used in this study were from one batch, we did not observe a batch effect between patients (Fig. S1B, C) at the indicated resolution ratio; thus, we did not require algorithms to remove the batch effect before cluster analysis. Finally, we annotated the cell types based on markers from the CellMarker2.0 database and a previous study [8, 16]. Table S3 lists the cell type markers. To further improve the accessibility of these scRNA-seq data, we produced a visual ShinyAPP, stored at https://drive.google.com/file/d/15H0GNV6EwmvBrhiFsBhgpjfODDK6XOwc/view?usp=sharing.
Copy number variation (CNV) prediction analysis
The R package “infercnv” (version 1.10.1) was used as a reference to estimate the CNV of cancer and renal tube cells [17,18,19,20,21]. A sliding window containing 100 genes was used to calculate the average expression of these genes on each chromosome. The CNV score of each cell was defined as the mean square of the deviation in the genome (CNV score = mean[cnv-1] ^2).
Tissue array and multicolor immunofluorescence assay
The tissue chip HKid-CRCC060PG-01, comprising 30 paired tumor and para-tumor samples, was purchased from Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). The antibodies used to detect specific proteins are presented in Table S4.
Transcription factor (TF) regulation analysis
We performed pySCENIC analysis to explore the TF regulation network of each cell type [22]. Differential analysis of the area under the curve (AUC) for each module between different cell clusters was performed using the R package “SCENIC,” and an adjusted p value (adj. p val) <0.05 was considered to indicate a significant difference.
Pseudotime trajectory analysis
To examine the relationship between different cluster cells in immune cells and the sequential variation in these cells, we performed pseudotime trajectory analysis using the R package “Monocle 3” [23]. For analysis of a single-cell type, the batch effect of the different patients was removed using the “align_cds” function of the “Monocle3” R package.
Differentially expressed genes (DEGs) analysis
DEGs between different cell types or cell clusters were identified using the “FindMarkers” function of Seurat (adj. p val <0.05, |log FC| >0.5). DEGs between different ccRCC cluster samples were identified using the R package “limma” (adj. p val <0.0001).
Functional enrichment analysis
The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis of the DEGs was performed using the R package “clusterProfiler” [24]. Gene set enrichment analysis (GSEA) was performed using the R package “GSEABase.” Gene set variation analysis (GSVA) was performed using the R package “AUCell” and 50 hallmark gene sets obtained from the molecular signature database (http://www.gsea-msigdb.org/gsea/index.jsp) using default settings [22]. The cell score in spatial transcriptome analysis was calculated using the AddModuleScore function in the “Seurat” package.
Cell–cell communication analysis
The R package “CellChat” was used to evaluate cell–cell communication strength and identify receptor–ligand relationship pairs. This quantified the correlativity of the 14 different cell types identified in this study [25].
Cell cultures and reagents
The human renal cell line HK2 and human renal cancer cell lines ACHN and 786 O were purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China) and cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (11875-093; Gibco) containing 10% fetal bovine serum (FBS; C04001-500; BI). The cells were grown at 37 °C and 5% CO2. The hypoxia model was produced by treating ACHN cells with 200 µmol/L CoCL2.
Cell transfection
ACHN and 786 O cells were seeded in 6-cm dishes and transfected with siRNAs once they reached 80% confluency. All investigations were performed after transfection for 48 h. The siRNA for HILPDA and NNMT were purchased from Suzhou Genepharma Co., Ltd. (Jiangsu). The siRNA sequences are provided in Table S5.
Western blotting
The protein was extracted by RIPA buffer, separated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to polyvinylidene difluoride (PVDF) membranes. After incubation with primary and secondary antibodies, the blots were exposed using an enhanced chemiluminescence kit (WBKLS0500; Merck). The antibodies used for western blotting are provided in Table S4.
RNA extraction and real-time quantitative PCR methods
Total RNA was extracted using the BioSci® Animal Tissue Cell Total RNA Extraction Kit (8034111; Dakewe), and cDNA was synthesized using the PrimeScript™ RT Reagent Kit (Perfect Real Time) (RR037A; TaKaRa). Next, qPCR was conducted using AceQ Universal SYBR qPCR Master Mix (Q511-02; Vazyme) following the manufacturer’s protocol. The specific primer sequences are provided in Table S6.
Cell viability and proliferation assays
ACHN and 786 O cells were seeded onto 96-well plates at a density of 5000 cells per well. Cell viability was measured at 0, 1, 2, 3, 4, and 5 days using a Cell Counting Kit-8 (CCK-8) (HY-K0301; MCE), according to the manufacturer’s protocol.
For colony formation assays, cells were seeded in six-well culture dishes. After being cultured for 1 week, the colonies were fixed in methanol for 30 min and stained with 0.1% crystal violet for 20 min to visualize the colonies.
5-Ethynyl-2′-deoxyuridine (EdU) staining was performed to measure cell proliferation via the EdU kit (C0075S, Beyotime Biotechnology) following the manufacturer’s protocol. Finally, cell proliferation was measured by flow cytometry.
Migration assays
Migration assays were performed using chambers (3422, Corning). ACHN and 786 O (5 × 104) cells were seeded into the upper chamber with 200 μL of serum-free RPMI-1640 medium. The bottom chamber was supplemented with 500 μL of medium containing 10% FBS. After incubation for 48 h, the cells were fixed with absolute methanol and stained with 0.1% cresyl violet for 20 min. Finally, images of these cells were acquired with an inverted microscope.
Triglyceride accumulation
Triglyceride contents of ACHN and 786 O cells were measured using a Colorimetric/Fluorometric Assay Kit (E1013-50; APPLYGEN, Beijing, China) according to the manufacturer’s instructions.
Subcutaneous xenograft experiment
All mouse experiments were conducted in accordance with protocols approved by the Capital Medical University Animal Care and Use Committee and followed guidelines for animal welfare.
Six-week-old BALB/c male nude mice were purchased from Beijing HFK Bioscience Co., Ltd. The 7860 tumor cell line was subcutaneously injected into the nude mice (5 × 106 per mouse). After 9 days, the animals were treated with an intratumoral multi‐point injection every two days with either 20 μL of PBS with complexes of 15 μg siRNA, a set of 2′‐o‐Me and 5′cholesterol‐modified HILPDA, NNMT, or negative siRNA (non‐target siRNA treatment group) with 5 μL of lipofectamine 2000. The mice were numbered by weight and randomly grouped using a random number table. Meanwhile the investigators were not blinded to the groupings.
Meanwhile, tumor volumes were measured every two days. After 21 days, the mice were euthanatized, subcutaneous tumors were excised, and images were taken. The tumor volume was calculated as length × width2/2.
Deconvolution analysis of bulk mRNA expression data
To measure the abundance of each cell type in the ccRCC tumors, we first used CIBERSORTx to analyze the merged mRNA expression data. These data included the combined TCGA-KIRC expression data, ccRCC expression data reported by Obradovic et al. [11], and E-MTAB-1980 ccRCC microarray data. A ComBat algorithm was used to remove batch differences. The signature matrix was constructed by randomly selecting 500 cells per cluster. The abundance of each cell cluster was predicted using CIBERSORTx run in Docker [26].
Building and publishing the ccRCC cluster classifier
Subtype analysis was carried out with the R package “ConsensusClusterPlus” and by dividing the meta-cohort into three subgroups based on the abundance of the prognosis-associated cell cluster. A classifier was then constructed based on eXtreme Gradient Boosting (XGBoost). First, the optimal parameters were determined using a tenfold cross-validation analysis (max depth = 4, eta = 0.5, and nrounds = 400) in the TCGA-KIRC cohort. Next, the match batch effects were minimized between different cohorts using the “ComBat” algorithm [27]. Finally, the ccRCC clusters were predicted using the XGBoost model based on the TCGA-KIRC cohort after batch correction. To facilitate further research with the model and proposed ccRCC cluster, we constructed the R package “ccRCCcluster,” available from GitHub (https://github.com/ZylRpackage2023/ccRCCcluster).
Statistical analysis
The paired Student’s t-test was used to compare paired samples, while the unpaired Student’s t-test was used to compare two groups with normal distribution and homogeneity of variance. The Mann–Whitney U-test was used to compare two groups without normal distribution. One-way analysis of variance (ANOVA) or Kruskal–Wallis variance tests were used to compare three or more groups. Fisher’s exact test was used to analyze the classified variables. Kaplan–Meier survival curves and log-rank tests were applied for survival analysis. Univariate Cox analysis was employed to calculate the hazard ratios for univariate analyses. Spearman’s correlation analysis was performed to identify correlations between two non-normally distributed variables. Statistical significance was defined as a two-tailed P value <0.05. All statistical analyses were performed using R 4.2.2.
Results
The single-cell landscape of ccRCC
Our analysis included 67,135 single cells that originated from paracancerous tissues and 119,160 tumor-derived single cells (Fig. 1B and Fig. S1D). Cluster analysis yielded 46 and 41 cell clusters, respectively, each containing >500 cells (Fig. S1E, F). The cell clusters were annotated and classified into 14 main cell types (Fig. 1A–C and Fig. S1G–I). The top five markers of the main cell types were displayed as dot plots (Fig. 1D and Table S7).

A–C UMAP plot of 20,000 single cells (randomly selected from all single cells), with each color-coded for A 14 major cell types, B sample origin (para-tumor or tumor), and C immune cell (CD45+) or non-immune cell (CD45−). D The dot plots of the top five DEGs among 14 major cell types identified in this profile. Red dots: upregulated genes (Log FC ≥0.5); Blue dots: upregulated genes (Log FC ≤−0.5). (E) The fraction of 14 major cell types originated from the eight para-tumor and eight tumor samples. F The paired comparison of 14 major cell types’ fractions between tumor and para-tumor (paired Student’s t-test, n = 16, *: <0.05; **: <0.01; ***: <0.001). G Interaction network constructed by Cellchat. H Spatial transcriptome displayed the expression level of HPD and PCK1 in tumor and para-tumor position of ccRCC samples. I The immunofluorescence assay of HDP and PCK1 in tissue chips. The scale bar represents 50 mm.
A comparison of para-tumor- and tumor-derived cells revealed that renal tubule cells existed almost exclusively in normal kidney tissue, whereas cancer cells existed only in tumor tissue. Meanwhile, a significant increase was observed in CD8+ T cells and fibroblasts in tumor tissues; in contrast, fewer CD4+ T cells were present in tumor tissues than in normal kidney tissues (Fig. 1E, F and Table S8). Cell–cell communication analysis indicated that fibroblasts communicated the most with other CD45- cell types, while macrophages primarily communicated with other CD45+ cell types in ccRCC (Fig. 1G, Fig. S1J, and Table S9). Additionally, since CD8+ T cells—increased in ccRCC—communicate readily with cancer cells, endothelial cells, and fibroblasts, we postulated that cancer cells, fibroblasts, endothelial cells, macrophages, and CD8+ T cells are vital in ccRCC occurrence and development.
Considering that renal tubule cells exist almost exclusively in normal kidney tissue, we proposed that renal tubule cells and markers of renal tubule cells could serve as disease markers of ccRCC. Through differential expression analysis, we found that four genes (ALDOB, MT1G, HPD, and PCK1) were only expressed in renal tubule cells (Fig. S1K–N). Meanwhile, stRNA-seq data more directly showed that ALDOB, MT1G, HPD, and PCK1 were enriched in paracancer tissues (Fig. 1H and Fig. S1O). A previous study reported that ALDOB and MT1G are highly expressed in renal tissue [8]. To verify these results, we performed immunofluorescence assays for HPD and PCK1 using tissue chips. Both signals were significantly increased in para-tumor tissue compared to tumor tissue (Fig. 1I). These results indicate that the loss of HPD and PCK1 may be an indicator of ccRCC.
HPD participates in the tyrosine catabolic pathway, where it catalyzes the conversion of 4-hydroxyphenylpyruvate to homogentisate [28]. PCK1 is the main control point for regulating gluconeogenesis; the cytosolic enzyme, along with GTP, catalyzes the formation of phosphoenolpyruvate from oxaloacetate with the release of carbon dioxide and GDP [29]. Both of these metabolic enzymes (HPD and PCK1) were downregulated in ccRCC tissues, suggesting that metabolic disorders are a molecular characteristic of ccRCC.
NNMT and HILPDA are potential therapeutic targets for ccRCC
Cancer cells were the most common cells in tumor samples and determined ccRCC occurrence and progression (Table S8). The heterogeneity between different clusters of cancer cells correlated with differences between patients (Fig. 2A). To further explore tumor heterogeneity in ccRCC, sampling analysis (500 cells/cancer cell cluster) was used to predict CNV. Results showed that gene copy number loss on chromosome 3p and gene copy number amplification on chromosome 5q were common in different cancer cell clusters. To further validate these results, we calculated the CNV ratio of each gene on different chromosomes, as well as their GISTIC score. Our results agreed with those for TCGA and Shanxi Bethune Hospital cohorts, i.e., a reduction in gene copy numbers on chromosome 3p and amplification on chromosome 5q (Fig. 2B).

A The UMAP plot and The fraction of cancer cells that originated from different patients. B The CNV analysis of different cancer cell clusters in single-cell cohort, Shanxi Bethune hospital cohort, and TCGA cohorts. C The waterfall plot of mutation gene sited in Chromosome 3 in Shanxi Bethune hospital cohort. D The GSEA analysis of DEGs between cancer cells and renal tubule cells. E The GO enrichment analysis of hub genes. F Spatial transcriptome displayed the expression level of HILPDA and NNMT in tumor and para-tumor position of ccRCC samples. G Immunofluorescence staining of ccRCC tissue chips. HILPDA and NNMT were co-stained with CA9, respectively. The scale bar represents 50 mm.
Functional enrichment analysis suggested that the genes with reduced copy numbers on chromosome 3p were closely related to immune response function and may lead to tumor immune escape in ccRCC cells. Moreover, we found that the genes whose copy number was increased on chromosome 5q were associated with cell–cell adhesion and might promote oncogenesis (Table S10 and Fig. S2A, B). Cancer cell C3, from patient 7, with a high pathological and T stage, had the highest CNV score. This indicated that the calculated CNV events in tumor cells are associated with tumor development (Fig. S2C). Finally, we found that the gene site on chromosome 3 had a high mutation ratio in the Shanxi Bethune Hospital cohort (Fig. 2C). This further supported the importance of this gene site in the prevention of tumor development.
Considering that ccRCC originates from the renal tubular epithelium, we identified DEGs among cancer and renal tubular cells (Table S11). We then performed GSEA based on the Log FC rank of the DEGs. The results indicated that the NF-κB, interferon-gamma, and epithelial-mesenchymal transition pathways were activated in tumor tissues, whereas bile acid metabolism, adipogenesis, fatty acid metabolism, and xenobiotic metabolism were activated in renal tubular cells (Fig. 2D, S2D and Table S12). Owing to the heterogeneity observed between different cancer cell clusters and patients, we assessed the number of common DEGs between renal tubular cells and each cancer cell cluster. There were 83 common DEGs among nine cancer cell clusters, 56 of which were cancer cell marker genes (Fig. S2E and Table S13). GO and KEGG enrichment analyses of these 56 genes indicated enrichment in response to hypoxia and apoptotic signaling pathways in cancer cells (Fig. 2E and Table S14). Further analysis indicated that NNMT and HILPDA were only expressed in cancer cells (Fig. S2F, G). Moreover, NNMT and HILPDA showed cancer type-specific expression in samples from the TCGA database (Fig. S2H, I). However, NNMT and HILPDA expression did not demonstrate significant prognostic value in the KIRC cohort (Fig. S2J, K). Finally, stRNA-seq data showed that NNMT and HILPDA were enriched in ccRCC tissues (Fig. 2F).
To further verify the expression of NNMT and HILPDA in cancer cells, immunofluorescence assays were performed. NNMT and HILPDA were co-expressed with CA9—a conventional marker of ccRCC—, and their expression was significantly increased in tumors (Fig. 2G). Further analysis of the mRNA expression and protein levels of these genes in renal cancer cell lines (ACHN and 786 O) and a normal renal cell line (HK2) revealed that NNMT was increased in ACHN and 786 O cells, while HILPDA was only increased in 786 O cells (Fig. 3A, B). Subsequently, we knocked down NNMT and HILPDA via siRNA (Fig. S2L–S). The clone formation, EdU, and cell viability assay results indicated that NNMT and HILPDA knockdown significantly decreased the cell viability and replication capacity (Fig. 3C–J).

A The mRNA expression level of NNMT and HILPDA in HK2, ACHN, and 786 O (unpaired Student’s t-test, n = 3, *: <0.05; **: <0.01; ***: <0.001). B The protein level of NNMT and HILPDA in HK2, ACHN, and 786 O. C, D The growth curve of ACHN (C) and 786 O (D) with si-NNMT and si-HILPDA treatment measured by CCK-8 assay (unpaired Student’s t-test, n = 3, *: <0.05; **: <0.01; ***: <0.001). E The colony formation assay of ACHN and 786 O with si-NNMT and si-HILPDA treatment. F–I The EdU assay of ACHN (F, G) and 786 O (H, I) with si-NNMT and si-HILPDA treatment (unpaired Student’s t-test, n = 3, *: <0.05; **: <0.01; ***: <0.001). J The cell migration assay of ACHN and 786 O with si-NNMT and si-HILPDA treatment. K The subcutaneous xenograft experiment of 786 O with si-NNMT and si-HILPDA treatment. L The size of the subcutaneous tumor of 786 O with si-NNMT and si-HILPDA treatment (unpaired Student’s t-test, n = 3, *: <0.05; **: <0.01; ***: <0.001). M The weight of the subcutaneous tumor of 786 O with sh-NNMT and sh-HILPDA treatment (unpaired Student’s t-test, n = 3, *: <0.05; **: <0.01; ***: <0.001). NThe mRNA expression level of HIF1A, NNMT, and HILPDA in ACHN with 200 umol/L CoCL2 treatment (unpaired Student’s t-test, n = 3, *: <0.05; **: <0.01; ***: <0.001). O The protein level of NNMT and HILPDA in ACHN with 200 umol/L CoCL2 treatment. P The triglyceride level of ACHN and 786 O with si-NNMT and si-HILPDA treatment (unpaired Student’s t-test, n = 3, *: <0.05; **: <0.01; ***: <0.001).
To further explore the impact of NNMT and HILPDA on ccRCC in vivo, we created a subcutaneous xenograft mouse model injected with 786 O cells and siRNA with a 2′‐o‐Me and 5′cholesterol‐modified in the tumor. NNMT and HILPDA knockdown caused the tumor volume to be significantly reduced in the xenograft model (Fig. 3K–M). These results further highlight the potential of HILPDA and NNMT as therapeutic targets.
NNMT contributes to the metabolism of drugs and xenobiotic compounds in the liver using S-adenosylmethionine as a methyl donor [30]. HILPDA is involved in autocrine signaling, cellular responses to hypoxia, positive regulation of lipid storage (e.g., lipid droplets and secretory granules), and correct cellular localization, including cell surface localization [31]. Thus, to further explore the relationships between these loci and hypoxia and metabolism in ccRCC, we induced hypoxia in ACHN cells via CoCL2 treatment. HIF1A expression was significantly higher in ACHN cells treated with CoCL2 than in controls, indicating that the hypoxia model was successfully established (Fig. 3N). Moreover, HILPDA expression was significantly elevated in the hypoxia cell model (Fig. 3N, O). si-NNMT and si-HILPDA significantly decreased the triglyceride contents in ACHN and 786 O (Fig. 3P). These data indicated that metabolic disorder and hypoxia play distinct roles and are molecular features of ccRCC. Furthermore, NNMT and HILPDA, associated with the response to hypoxia and metabolism, are potential therapeutic targets for ccRCC.
We then carried out a SCENIC analysis to reveal the TF regulatory network in ccRCC cancer cells. PRKAA2, which encodes AMPK, is upregulated in cancer cells, and AMPK is an important energy-sensing enzyme that monitors cellular energy status [32]. JUN and FOSB are AP-1 TF subunits, and AP-1 TF is closely related to hypoxia-related and NF-κB pathways [33]. IRX3 affects lipid metabolism in liver cells by regulating mitochondrial function [34]. The TFs upregulated in ccRCC may be potential drug targets for antitumor therapy (Figure S2T).
APOC1 is a biomarker of M2-like polarization in tumor-associated macrophages (TAMs)
A total of 34,299 macrophages were clustered into five macrophage sub-clusters (C1–C5) (Fig. 4A). Macrophage C1 was enriched while macrophage C4 was decreased in tumor tissue (Fig. 4B). We then calculated the Macrophage C1 scores based on the stRNA-seq data, and found them to be significantly increased in tumor tissue (Fig. 4C). Therefore, we posit that the macrophage C1 group is TAMs. Differential expression analysis revealed that macrophage C1 possessed the CD68+CD163+APOC1+ phenotype (Figs. S3A–F, Fig. 4D, and Table S15). VEGFA and VEGFB, which can promote tumor growth, were also expressed by cells in macrophage C1 (Fig. S3G, H). In contrast, macrophage C4 exhibited high expression of the gene encoding CLEC10A (CD301), an M2-type macrophage marker that may be associated with M2 macrophage polarity [35] (Fig. S3A). Macrophage C5 highly expressed genes encoding SERPINB2, THBS1, PTX3, and CXCL8, which are associated with macrophage infiltration and M1-like macrophage polarization [36,37,38] (Fig. S3A).

A The UMAP plot of five cell clusters of macrophages. B The paired comparison of five cell clusters’ fraction of macrophages between tumor and para-tumor (paired Student’s t-test, n = 16, *: <0.05; **: <0.01; ***: <0.001). C Spatial transcriptome displayed the Macrophage C1 cell cluster was enriched in tumor position. D Spatial transcriptome displayed the expression level of APOC1 in tumor and para-tumor position in the ccRCC sample. E Heatmap of the already known marker of TAMs. The mean expression level of each cell cluster was transformed into a row Z-score. F Immunofluorescence staining of CD163 and APOC1 in ccRCC tissue chip. CD163 + APOC1+ macrophages only emerged in tumor tissues. The scale bar represents 50 mm. G Heatmap of immune checkpoint expression on macrophages. The row Z-score was implicated to represent the expression level. H The GSEA analysis revealed the activated pathway enriched in macrophage C1. I The violin plots of the AUCell scores (M1 score and M2 score) for each cell cluster of macrophages. J Differentiation trajectory of macrophage cells in ccRCC, with each color-coded for clusters (left) and pseudotime (right).
To confirm that macrophage C1 comprises TAMs, we assessed TAM markers and found that they were all highly expressed in macrophage C1 (Fig. 4E). Immunofluorescence staining confirmed the presence of macrophage C1 (CD163+APOC1+) in ccRCC tissues (Fig. 4F). We then examined the distribution of immune checkpoints in the TME of ccRCC patients, more specifically, the checkpoints with high antigen expression in macrophages C1 and C4 (Fig. 4G). Recent studies have reported M2-like TAMs in ccRCC. APOC1, associated with cellular triglyceride metabolism, has been identified as an M2-like TAM marker and is associated with cholesterol metabolism in macrophages [39,40,41,42,43]. Using GSEA, we found that macrophage C1 was enriched in the “WT VS PPARG KO MACROPHAGE ROSIGLITAZONE STIM UP” gene set (Fig. 4G and Table S16). Given that the PPARγ pathways were activated in M2-like macrophages and macrophage C1 highly expressed CD163, we further examined the expression of other M2-like macrophage markers [44]. We found that CSF1R, MRC1, and PDCD1LG2 were highly expressed in macrophage C1, whereas CLEC10A was highly expressed in macrophage C4 (Fig. S3I). To further assess the M1 and M2 polarization of macrophages, we collected macrophage gene expression data and generated M1 and M2 gene sets through DEG analysis (Table S17). GSVA indicated that macrophage C5 had a high M1 score, while macrophage C1 and C4 had higher M2 scores (Fig. 4I). Therefore, we posit that macrophage C1 is an M2-like TAM.
Since macrophage C4 also had the molecular features of M2-like macrophages, we further explored the relationship between macrophage C1 and C4 using Monocle 3. The results indicated that macrophage C1 differentiated from macrophage C4 in ccRCC tissues (Fig. 4J). This may explain why macrophage C4 levels were decreased in the tumor tissue.
To further identify the TF-regulated network in TAMs, we performed SCENIC analysis and found that the AP-1 TFs JUN and FOSB were upregulated in macrophage C1 (Fig. S3J). Simultaneously, DDIT3, a C/EBP family protein, was upregulated in macrophage C1. AP-1 and C/EBP can promote the M2-like polarization of macrophages [45,46,47]. Finally, NR1H3 (LXRa), which was increased in M2 macrophages, was also upregulated in macrophage C1 [48] (Fig. S3J). This further confirms that macrophage C1 comprises TAMs with M2-like polarization characteristics in ccRCC.
The cell differentiation trajectory of tumor-driven CD8+ T cells in ccRCC is activated–exhausted–proliferated
In this study, we detected 13,956 CD8 + T cells, including 11,919 in tumor tissue and 2037 in normal tissue (Fig. 5A). As shown in the UMAP figure, after recluster analysis of CD8+ T C1 cells, CD8+ T cells were assigned to four clusters (Fig. 5A). CD8+ T C2 cells (IL7R+KLRB1+) were enriched in para-tumor and tumor tissues, whereas those from C1_1 (LAG3+ HAVCR2+), C1_2 (GZMM+ ICOS+), and C3 (STMN1+) were significantly enriched in tumor tissues (Fig. 5B, Fig. S4A, and Table S18). Additionally, stRNA-seq data directly indicated that the CD8+ T cell C1_1 score, C1_2 score, and C3 score were significantly increased in tumor tissues (Fig. 5C–E). Therefore, these clusters were designated tumor-driven CD8+ T cells.

A The UMAP plot of four cell clusters of CD8 + T cells after re-clustering. B The paired comparison of four cell clusters’ fraction of CD8 + T cells between tumor and para-tumor (paired Student’s t-test, n = 16, *: <0.05; **: <0.01; ***: <0.001). C–E Spatial transcriptome displayed the CD8 + T C1_1 cell cluster (C), CD8 + T C1_2 cell cluster (D), and CD8 + T C3 cell cluster (E) were enriched in tumor position. F The GSEA analysis of DEGs between CD8 + T C1_1 and CD8 + T C2 cell cluster. G Heatmap of immune checkpoints upregulated or downregulated in four cell clusters of CD8 + T cells. A row Z-score was used to represent the expression level. H The bubble diagram of the Tex markers’ expression for each cell cluster of CD8 + T cells. I Heatmap of AUCell score of 50 hallmark pathways in each cell cluster of CD8 + T cells. J The UMAP plot of four cell clusters of CD8 + T cells to display the phase of each cell. K The histogram illustrating the different cell cycles in each cell type of CD8 + T cells. L The ridge plot to display G2M.Score of each cell type of CD8 + T cells. M Spatial transcriptome displayed the expression level of STMN1 in tumor and para-tumor position in the ccRCC sample. N Immunofluorescence staining of CD8, PDCD1, and STMN1 in ccRCC tissue chip. CD8 + PDCD1 + VCAM1 + T cell only emerged in tumor tissues. The scale bar represents 50 mm. O Differentiation trajectory of CD8 + T cells in ccRCC, with each color-coded for clusters (top) and pseudotime (bottom). P Heatmap of AUC scores of selected regulons altered in CD8 + T cells. AUC scores were measured by SCENIC per cell.
The GSEA analysis was performed to explore the biological characteristics of C1_1 CD8+ T cells based on DEGs between the C1_1 and C2 CD8+ T cells. The pathway associated with PD1, a well-known TEX (T cell exhausted) marker, was significantly upregulated in C1_1 CD8+ T cells (Fig. 5F). Meanwhile, previous studies have shown that LAG3 and HAVCR2 also are TEX markers in ccRCC [8]. Therefore, we posit that the C1_1 CD8+ T cells, which highly expressed LAG3 and HAVCR2, were CD8+ exhausted T cells (Fig. S4C).
To further identify the immunological features of the CD8+ T cells, we assessed the expression of immune checkpoints in these cells (Fig. 5G). Most of the inhibitory checkpoints, including HAVCR2, LAG3, CD27, and TNFRSF9, were significantly upregulated in the C1_1 and C3 CD8+ T cells, while ICOS, a T cell activation marker, was significantly upregulated in the C1_2 CD8+ T cells [49]. These results suggested that the C1_2 CD8+ T cells were activated while C3 CD8+ T cells exhibited TEX characteristics. To verify this, we examined the expression of TEX markers using a bubble diagram. Compared with C2 cell clusters, the expression of most TEX markers (PDCD1, TOX, CTLA4, CXCL13, and TIGIT) was increased in C1_1 and C3 CD8+ T cells, whereas CD69 was decreased in C1 and C3 CD8+ T cells (Fig. 5H). Noticeably, the C3 CD8+ T cells had the highest expression of CXCL13. stRNA-seq analysis also indicated that these TEX markers were upregulated in ccRCC (Fig. S4B). These results further confirmed that C3 had TEX characteristics.
To further identify the molecular features of C3 CD8+ T cells, we performed a GSEA analysis. We found that “G2M checkpoint” and “E2F targets” pathways were significantly activated in C3 CD8+ T cells (Fig. 5I). Associated with the function of STMN1 in the cell cycle, we proposed that C3 CD8+ T cells were characterized by abnormal cell cycling. Indeed, cell cycle analysis further indicated that >60% of the C3 CD8+ T cells were in the G2M phase and had a higher G2M score than the C1 and C2 cells (Fig. 5J–L). Accordingly, we designated the C3 CD8+ T cells CD8+ proliferated T cells. Moreover, considering that the scRNA-seq data and stRNA-seq data suggested that STMN1 is only expressed in C3 CD8+ T cells and is highly expressed in ccRCC tissue, we postulate that STMN1 is a C3 CD8+ T cell marker (Fig. 5M, Fig. S4D, E). Finally, immunofluorescence staining of CD8, STMN1, and PDCD1 confirmed the presence of C3 CD8+ T cells (STMN1+) in ccRCC tissues, with TEX characteristics (Fig. 5N).
To further establish the relationship between CD8+ T cell clusters, we performed pseudotime trajectory analysis using Monocle 3. The trajectory was visualized as a UMAP plot (Fig. 5O). Interestingly, cluster C3, which was associated with TEX, was located at the end of the differentiation trajectory. Therefore, we proposed the following reaction process after CD8+ T cells encounter tumor cells: activated (C1_2: GZMM+ICOS+) → exhausted (C1_1: LAG3+HAVCR2+) → proliferating (C3: STMN1).
Finally, we performed SCENIC analysis (Fig. 5P) and found that the EOMES and STAT3 pathways were upregulated in the C1_1 CD8+ T cells. In previous studies, loss of STAT3 significantly reduced the proportion and number of tumor-specific exhausted T cells and impaired granzyme B production [50]. Meanwhile, EOMES has also been reported as a TEX [51]. In a recent study, Wu et al. reported that BHLHE40 was the key TF in TEX using CRISPR-Screen and in vitro modeling of CD8+ T cell exhaustion. Finally, we found that E2F1 expression was increased in C3 CD8+ T cells. E2F family proteins regulate the cell cycle and are associated with cell proliferation. Hence, we posit that E2F1 has an important role in maintaining C3 CD8+ T cells in the G2M phase.
Collectively, these results indicate that C1_1 CD8+ T cells are exhausted while C3 CD8+ T cells are proliferating. Accordingly, these TFs might represent effective drug targets for tumor therapy.
Fibroblasts are enriched on the tumor border and may promote angiogenesis in ccRCC
In total, 15,328 fibroblasts were identified (Fig. 6A) and were enriched in tumor tissues. These fibroblasts were divided into six clusters, with each cluster originating from different patients, similar to cancer cells, indicating tumor heterogeneity (Fig. 6A, B). Considering that C1–3 and C5 were primarily tumor tissue cells, we designated them tumor-associated fibroblasts (TAFs). To further identify the locations in the tumors, we calculated the TAF score based on stRNA-seq data and found the TAFs to be located on the border of the tumor. Therefore, we posit that TAFs have a vital role in tumor growth regulation (Fig. 6C and Fig. S5A). We then compared the differently activated pathways in normal and tumor fibroblasts and identified “UV−response−up,” “inflammatory−response,” “hypoxia,” “hedgehog−signaling,” “notch−signaling,” “interferon−alpha−response,” and “angiogenesis” pathways as significantly activated in tumor fibroblasts (Fig. 6D). This corresponded with the results from the cancer cells, further confirming that fibroblasts exhibit tumor heterogeneity.

A The UMAP plot of six fibroblast clusters from eight patients. B The cycle plot indicated different fibroblast clusters from different patients. C Spatial transcriptome displayed the TAF cell cluster was enriched in tumor border. D Heatmap of AUCell score of 50 hallmark pathways in fibroblast from tumor and para-tumor. E Spatial transcriptome displayed the expression level of PLXDC1 (TEM7) in tumor and para-tumor position in the ccRCC sample. F Immunofluorescence staining of PLXDC1(TEM7) and ACTA2 (α-SMA) in tissue chip. ACTA2 + TEM7+ fibroblasts only occur in ccRCC tissues, while they are almost absent in normal kidneys. The scale bar represents 50 mm. G The UMAP plot of four endothelial cell clusters from tumor and para-tumor tissue. H The paired comparison of four cell clusters’ fraction of endothelial cells between tumor and para-tumor (paired Student’s t-test, n = 16, *: <0.05; **: <0.01; ***: <0.001). I Spatial transcriptome displayed the endothelial C2 cell cluster was enriched in the tumor border. J Immunofluorescence staining of PECAM1 (CD31) and ESM1 in tissue chip. The scale bar represents 50 mm. K Spatial transcriptome displayed the expression level of ESM1 in tumor and para-tumor position in the ccRCC sample. L Heatmap of AUCell score of 50 hallmark pathways in endothelial cells from tumor and para-tumor.
Considering that hypoxia and angiogenesis pathways are closely related to angiogenesis, we posit that TAFs can promote angiogenesis in ccRCC. Comparative DEG analysis between normal and tumor fibroblasts for each patient revealed 13 intersecting DEGs (Fig. S5B and Table S19). The expression of NDUFA4L2 was significantly upregulated in cancer cells and expressed in tumor fibroblasts (Fig. S5C). Meanwhile, PLXDC1 was only expressed in tumor fibroblasts (Fig. S5C); previous studies showed that PLXDC1 is associated with angiogenesis [52]. PLXDC1 and ACTA2 are upregulated in ccRCC and positively correlated with each other in the TCGA-KIRC cohort (Fig. 6E and Fig. S5D–F). Therefore, we hypothesized that TAFs can promote angiogenesis in tumor tissues. To test this hypothesis, we performed immunofluorescence staining on tissue chips and showed that PLXDC1(TEM7)+ ACTA2 (α-SMA)+ fibroblasts were highly enriched in tumor tissues (Fig. 6F and Fig. S5G).
Enhanced angiogenesis is a molecular characteristic of tumor-derived endothelial cells located at the tumor border
A total of 15,088 endothelial cells derived from tumor or non-malignant tissues were detected and categorized into four clusters. C2 (PECAM1+ESM1+) was enriched at the tumor border and had the highest FLT1 (VEGFR1) expression, indicating that endothelial C2 cells exhibited the highest level of angiogenesis (Fig. 6G–I and Fig. S5H–I, and Table S20). We then performed an immunofluorescence assay and stRNA-seq analysis to identify tumor-specific markers for endothelial C2 cells (Fig. 6J, K and Fig. S5J). The results identified PECAM1+ESM1+ cells in tumor tissues. In previous studies, PECAM1 was verified as a signature of endothelial cells [53], while here it was found to be significantly upregulated in ccRCC (Figure S5K, L). ESM1 is involved in angiogenesis and promotes angiogenic sprouting; herein, ESM1 was specifically expressed in ccRCC and strongly correlated with PECAM1 expression (Fig. S5M, N) [54]. These results further support the notion that tumor-derived endothelial cells exhibit higher angiogenesis than other endothelial cells. GSVA pathway analysis showed that angiogenesis and oxidative−phosphorylation pathways were enriched in C2 cells(Fig. 6L). This may be a response to hypoxia in tumors and provides further evidence of angiogenesis of tumor-derived endothelial cells in ccRCC.
Finally, we performed SCENIC analysis to explore the TF-regulated network in endothelial cells (Fig. S5O). The pathways associated with FOXF1, HOXD9, and HOXD8 were significantly upregulated in endothelial C2 cells. FOXF1 is a downstream target of VEGF that promotes angiogenesis [55,56,57], while HOXD8 and HOXD9 are expressed in vascular endothelial cells [58, 59]. These TFs could serve as potential drug targets for ccRCC treatment.
Three TME subtypes associated with ccRCC Prognosis
To further investigate the relationship between the identified cell clusters and patient outcomes in the clinical cohorts, we predicted the abundance of each cell cluster using CIBERSORTx with mRNA expression data (Fig. S6A and Table S21). Due to the significant heterogeneity of cancer cells and fibroblasts, we split cancer cells into cancer cells and fibroblasts in C1–3 and TAFs in C5. We found that nine cell clusters were associated with poor overall survival (OS; p < 0.05), including macrophage C1, C1_1 CD8+ T cells, and C3 CD8+ T cells, which were tumor-associated cell clusters (Fig. 7A–D, Fig. S6B). The abundance of macrophage C1, C1_1 CD8+ T cells, and C3 CD8+ T cells increased in the higher-grade and higher-stage ccRCC samples (Fig. 7E, F). This demonstrates that M2-like TAMs, CD8+ exhausted T cells, and CD8+ proliferated T cells promote ccRCC development. Therefore, we further explored cell communication between cancer cells, macrophage C1, C1_1 CD8+T cells, and C3 CD8+T cells, revealing that MIF–CD74 might be important in cancer cell chemotaxis and recruitment (Fig. 7G).

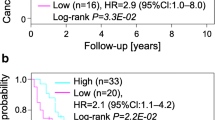
A Association between the relative abundance of cell clusters calculated by CIBERSORTx and overall survival. Clusters associated with bad outcomes (p < 0.05) are colored in red, while clusters associated with good outcomes (p < 0.05) are colored in blue. B–D Kaplan–Meier survival curve for patients with high or low abundance of macrophage C1 (B), CD8 + T cell C1_1 (C), and CD8 + T cell C3 (D) in TCGA-KIRC cohort. A log-rank p value <0.05 was considered statistically significant. E, F Line charts show macrophage C1, CD8 + T cell C1_1, and CD8 + T cell C3 enriched in high-grade (E) and late-stage (F) ccRCC tissues (one-way ANOVA test, *: <0.05; **: <0.01; ***: <0.001). G The cellchat analysis of cell communication between cancer cells, Macrophage C1, CD8 + T cell C1_1, and CD8 + T cell C3. H The heatmap of the abundance of cell clusters identified in this study in meta-cohort. I Kaplan–Meier survival curve of three patient subgroups in TCGA cohort. J Kaplan–Meier survival curve of three patient subgroups in E-MTAB-1980 cohort. K Kaplan–Meier survival curve of three patient subgroups in Obradovic et al’s cohort.
To further identify the immune characteristics of the TME in ccRCC, we performed unsupervised cluster analysis and divided the 811 ccRCC patients from the meta-cohort into three subtypes (Fig. 7H and Fig. S6C–G). Patients in the C3 subtype exhibited a shorter OS than those in the C2 and C1 subtypes (Fig. 7I–K). Interestingly, C1_1 CD8+ T cell, C3 CD8+ T cell, and macrophage C1 levels increased in patients in the C1 subtype (Fig. S6H). These results further support the vital role of TEX, CD8+ proliferating T cells, and the polarization of M2 macrophages in the TME of ccRCC, suggesting that we should design immunotherapy strategies targeting these cells in ccRCC.
Development of a classifier to predict the ccRCC subtype using bulk mRNA expression data
To render our ccRCC clusters reproducible and generalizable, we developed a classifier to predict ccRCC clusters in new samples. First, based on the mRNA expression data, we performed pairwise differential analysis across the ccRCC clusters in the training cohort (TCGA). In total, 859 overlapping DEGs were prioritized as ccRCC cluster-related genes for further analysis (Fig. S7A, Fig. 8A, and Table S22). Metascape analysis revealed that these genes were closely associated with tube morphogenesis (Fig. S7B and Table S23). Next, the XGBoost algorithm was used to create a classifier to predict ccRCC clusters. We validated the accuracy of our classifier using the “ccRCCcluster” R package applied to the TCGA (training) cohort, E-MTAB-1980 cohort, and the Obradovic et al. cohort, achieving accuracies of 1.000, 0.901, and 0.9045, respectively (Fig. 8B, Fig. S8, and Table S24). These results demonstrate the robustness of our classifier.

A The heatmap of ccRCC subtype-related gene expression level in meta-cohort. B The cycle plot and Fisher’s test of three subtypes samples in the validation cohort.
Discussion
Due to the limited therapeutic benefit provided by surgical intervention, the mortality rate of patients with metastatic ccRCC remains high [1]. Meanwhile, drugs used for treating ccRCC, such as TKI and anti-PD-1 antibodies, also exhibit a limited therapeutic effect [3]. Therefore, the mechanisms underlying drug resistance in ccRCC require further exploration. Tumor heterogeneity, including cancer and tumor-infiltrating cells, is the primary cause of drug resistance in most cancers [60]. However, research on this phenomenon in patients with ccRCC is lacking. Considering that scRNA-seq can measure the RNA features of single cells in tumors, we further investigated tumor heterogeneity using this technique. Our study explored the novel cell subgroups, altered pathways, and regulatory networks driven by TFs in ccRCC. We also identified new cell clusters and corresponding cell markers using immunofluorescence and stRNA-seq technology in ccRCC. Using a deconvolution algorithm and bulk ccRCC RNA-seq data, we calculated the abundance of each cell cluster and identified the ccRCC subtype. Finally, we created a classifier to predict the ccRCC subtypes. These results expand our understanding of intratumoral heterogeneity, provide treatment targets, and will aid clinicians in designing personal therapeutic strategies for ccRCC.
When exploring intratumoral heterogeneity in ccRCC, we found that cancer cells derived from different patients exhibited significant tumor heterogeneity. This phenomenon may explain why most antitumor therapies are not effective for all ccRCC patients. Therefore, we explored the biological features shared by all cancer cells in a ccRCC sample and found that gene copy number loss on chromosome 3p and gene copy number amplification on chromosome 5q were common features. We then performed WES of our ccRCC samples to directly validate this phenomenon. Gene loss on chromosome 3 is associated with the tumor immune response. Functional enrichment analysis revealed that hypoxia and metabolic disorders are molecular features of ccRCC cells. An immunofluorescence assay and subcutaneous xenograft model revealed that NNMT, a protein associated with cell metabolism, and HILPDA, a protein associated with hypoxia, were significantly upregulated in cancer cells and potential therapeutic targets for ccRCC. Exploiting these characteristics of cancer cells can lead to the design of effective treatment strategies for ccRCC.
We also confirmed that the TME is highly complex and closely related to intratumoral heterogeneity in ccRCC. As a result, several key cell types were studied. First, we found that CD68+CD163+APOC1+ macrophages undergoing M2-like polarization were significantly increased in tumors and associated with poor outcomes in patients with ccRCC. A similar phenomenon has been reported previously [40]. We also identified another M2-like macrophage subset (CD68+CD163+FCER1A+) in ccRCC. Although this type of macrophage is rare in ccRCC, it decreased in tumors and may have an antitumor role; however, this finding is not consistent with the previous theory of M2-promoting cancer. These findings require further investigation and validation.
Second, we found that C1_1 (LAG3+HAVCR2+) and C3 (STMN1+) CD8+ T cells were highly enriched in ccRCC samples and associated with worse clinical outcomes. Further functional enrichment analysis verified that C1_1 CD8+ T cells were exhausted, which agrees with the finding of a past study [61]. We also found that C3 CD8+ T cells remain in the G2M phase causing us to designate this cluster as proliferating CD8+ T cells. Interestingly, the cells in this cluster had TEX characteristics and were closely associated with poor patient prognosis. Although proliferated T cells had been reported in other types of cancer, they have not been commonly reported in ccRCC [62]. Associated with CXCL13 expression, we postulate that the proliferated CD8+ T cells play an important role in tumor progression. Our trajectory analysis revealed an activated–exhausted–proliferated process for the CD8+ T cells, with C3 CD8+ T cells at the end of the trajectory. Noticeably, these proliferated CD8+T cells primarily remained within the G2M phase, suggesting that they exhibit a slower multiplication rate. This may involve CD8+ T cells being exhausted to a certain extent. This theory may explain why C3 CD8+ T cells appear at the end of the trajectory. Additionally, we discovered novel TF alterations that may underlie TEX. These findings deepen our understanding of tumor immune exhaustion and immune escape in patients with ccRCC.
Finally, we found that MIF–CD74 activity is vital in the chemotaxis and recruitment of TAMs and CD8+ exhausted T cells. Therefore, the MIF–CD74 axis may be a potential target in ccRCC therapy. Compared with the heterogeneity of cancer cells, the heterogeneity of immune cells described above was lower between different ccRCC patients. This may inform antitumor strategies based on tumor immune cells that have greater efficiency than those based on cancer cells.
Various tumor stromal cells promote tumor development and have become drug targets for antitumor therapy [63]. Tumor stromal cells in ccRCC also display significant heterogeneity. First, we discovered that most fibroblasts were present in the tumor cells and localized at the tumor borders. This suggest that these fibroblasts serve as “soil” to promote tumor development. Meanwhile, these fibroblast cells displayed significant heterogeneity among different ccRCC patients. Therefore, antitumor treatments should be formulated based on the common characteristics of all fibroblasts. We also found that enhanced angiogenesis is a biological characteristic of vascular endothelial cells located at the tumor border. This further supports the notion that TAFs promote tumor progression by enhancing endothelial cell angiogenesis within the tumor border. Immunofluorescence staining further confirmed the upregulation of angiogenic markers in these cells. Hence, anti-angiogenesis targeted therapy could be an effective treatment strategy for ccRCC.
Based on gene expression patterns and unsupervised clustering, numerous ccRCC subtypes were identified. Therefore, these subtypes are difficult to reproduce in new samples, and their biological significance is often intricate. Our study divided ccRCC samples from multiple cohorts into three clusters based on the abundance of different cell types and clusters. Cluster 3 had the highest abundance of M2-like macrophages (macrophage C1), CD8+ exhausted T cells (T cell C1_1), and CD8+ proliferated T cells (T cell C3), and correlated with worse clinical outcomes. Finally, we built a classifier using XGBoost and gene expression data to predict ccRCC subtypes in additional clinical samples.
Compared to previous studies, our study examined a larger number of samples with scRNA-seq, stRNA-seq, and bulk RNA-seq and employed a more sophisticated bioinformatic analysis. Additionally, we performed WES, cell line assays, murine tumor xenograft model experiments, and immunofluorescence analysis using tissue chips to verify our results. Our classifier enables reproducible tumor classification. However, many of our conclusions require further verification in in vitro and in vivo studies.
Conclusion
Analyses of the single-cell landscape and new cell clusters obtained in our study advance the current understanding of ccRCC tumor heterogeneity and pathogenesis. The molecular mechanisms revealed can inform the development of an antitumor strategy for treating ccRCC. Furthermore, the identified cell type-specific markers can improve tumor diagnosis and potentially serve as drug targets for cell therapy. Finally, the classifier we developed can accurately predict ccRCC subtypes, guide prognosis prediction, and aid in the design of a personalized treatment strategy for ccRCC. In summary, our work provides a new perspective for understanding tumor heterogeneity and will guide the design of antitumor therapeutic strategies for ccRCC.
Data availability
The public datasets used and/or analyzed during the current study are available from The Mendeley Data (https://data.mendeley.com/datasets/nc9bc8dn4m/1 and https://doi.org/10.17632/g67bkbnhhg.1), Gene Expression Omnibus (GSE129253), The mRNA microarray data (E-MTAB-1980: https://www.ebi.ac.uk/arrayexpress/) and University of California Santa Cruz (UCSC) Xena browser (https://xenabrowser.net) database. The WES data produced in our study was uploaded in supplementary materials (Tables S25 and S26). The raw WES data can be obtained by asking the authors. The code of “ccRCCcluster” was published on GitHub (https://github.com/ZylRpackage2023/ccRCCcluster). The shinyAPP to Visualize scRNA-seq data stored in https://drive.google.com/file/d/15H0GNV6EwmvBrhiFsBhgpjfODDK6XOwc/view?usp=sharing.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Xu P, Zhang S, Cheng J, Cao B, Huang J, Li Y, et al. Prognostic value of tumour contour irregularity on surgical strategies for T1bN0M0 renal cell carcinoma: a multi-institutional study. Eur J Radiol. 2023;159:110665.
Porta C, Cosmai L, Leibovich BC, Powles T, Gallieni M, Bex A. The adjuvant treatment of kidney cancer: a multidisciplinary outlook. Nat Rev Nephrol. 2019;15:423–33.
Grimm M-O, Bex A, De Santis M, Ljungberg B, Catto JWF, Rouprêt M, et al. Safe use of immune checkpoint inhibitors in the multidisciplinary management of urological cancer: the European Association of Urology Position in 2019. Eur Urol. 2019;76:368–80.
Kotecha RR, Motzer RJ, Voss MH. Towards individualized therapy for metastatic renal cell carcinoma. Nat Rev Clin Oncol. 2019;16:621–33.
Golkaram M, Kuo F, Gupta S, Carlo MI, Salmans ML, Vijayaraghavan R, et al. Spatiotemporal evolution of the clear cell renal cell carcinoma microenvironment links intra-tumoral heterogeneity to immune escape. Genome Med. 2022;14:143.
Vegliante R, Pastushenko I, Blanpain C. Deciphering functional tumor states at single-cell resolution. EMBO J. 2022;41:e109221.
Hu J, Chen Z, Bao L, Zhou L, Hou Y, Liu L, et al. Single-cell transcriptome analysis reveals intratumoral heterogeneity in ccRCC, which results in different clinical outcomes. Mol Ther. 2020;28:1658–72.
Borcherding N, Vishwakarma A, Voigt AP, Bellizzi A, Kaplan J, Nepple K, et al. Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun Biol. 2021;4:122.
Zhang Y, Narayanan SP, Mannan R, Raskind G, Wang X, Vats P, et al. Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc Natl Acad Sci USA. 2021;118:e2103240118.
Obradovic A, Chowdhury N, Haake SM, Ager C, Wang V, Vlahos L, et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell. 2021;184:2988–3005.e16.
Li R, Ferdinand JR, Loudon KW, Bowyer GS, Laidlaw S, Muyas F, et al. Mapping single-cell transcriptomes in the intra-tumoral and associated territories of kidney cancer. Cancer Cell. 2022;40:1583–99.e10.
Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860–7.
Hörhold F, Eisel D, Oswald M, Kolte A, Röll D, Osen W, et al. Reprogramming of macrophages employing gene regulatory and metabolic network models. PLoS Comput Biol. 2020;16:e1007657.
Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. https://doi.org/10.1038/nbt.4314 (2018).
Hu C, Li T, Xu Y, Zhang X, Li F, Bai J, et al. CellMarker 2.0: an updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2022;51:D870–6.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401.
Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–96.
Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309.
Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355:eaai8478.
Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171:1611–24.e24.
Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–6.
Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14:979–82. https://doi.org/10.1038/nmeth.4402.
Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021;12:1088.
Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37:773–82.
Jt L, We J, Hs P, Ae J, Jd S. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–3.
Rüetschi U, Rymo L, Lindstedt S. Human 4-hydroxyphenylpyruvate dioxygenase gene (HPD). Genomics. 1997;44:292–9.
Xu D, Wang Z, Xia Y, Shao F, Xia W, Wei Y, et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. 2020;580:530–5.
Wang W, Yang C, Wang T, Deng H. Complex roles of nicotinamide N-methyltransferase in cancer progression. Cell Death Dis. 2022;13:267.
de la Rosa Rodriguez MA, Deng L, Gemmink A, van Weeghel M, Aoun ML, Warnecke C, et al. Hypoxia-inducible lipid droplet-associated induces DGAT1 and promotes lipid storage in hepatocytes. Mol Metab. 2021;47:101168.
Winder WW. Energy-sensing and signaling by AMP-activated protein kinase in skeletal muscle. J Appl Physiol. 2001;91:1017–28.
Zhao S, Li B, Zhao R, Pan Z, Zhang S, Qiu W, et al. Hypoxia-induced circADAMTS6 in a TDP43-dependent manner accelerates glioblastoma progression via ANXA2/ NF-κB pathway. Oncogene. 2023;42:138–53.
Ma Y, Chen G, Yi J, Li Q, Tan Z, Fan W, et al. IRX3 plays an important role in the pathogenesis of metabolic-associated fatty liver disease by regulating hepatic lipid metabolism. Front Endocrinol. 2022;13:895593.
Eggink LL, Roby KF, Cote R, Kenneth Hoober J. An innovative immunotherapeutic strategy for ovarian cancer: CLEC10A and glycomimetic peptides. J Immunother Cancer. 2018;6:28.
Sen P, Helmke A, Liao CM, Sörensen-Zender I, Rong S, Bräsen J-H, et al. SerpinB2 regulates immune response in kidney injury and aging. J Am Soc Nephrol. 2020;31:983–95.
You Y, Tian Z, Du Z, Wu K, Xu G, Dai M, et al. M1-like tumor-associated macrophages cascade a mesenchymal/stem-like phenotype of oral squamous cell carcinoma via the IL6/Stat3/THBS1 feedback loop. J Exp Clin Cancer Res. 2022;41:10.
Khosravi M, Najafi M, Amirfarhangi A, Karimi M, Fattahi F, Shabani M. The increase of pFAK and THBS1 protein and gene expression levels in vascular smooth muscle cells by histamine-treated M1 macrophages. Iran J Allergy Asthma Immunol. 2019;18:72–9.
Clarke R, Von Ende A, Schmidt LE, Yin X, Hill M, Hughes AD, et al. Apolipoprotein proteomics for residual lipid-related risk in coronary heart disease. Circ Res. 2023;132:452–64.
Jia Y, Zhang B, Zhang C, Kwong DL-W, Chang Z, Li S, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in esophageal squamous cell carcinoma. Adv Sci. 2023;10:e2204565.
Ren L, Yi J, Yang Y, Li W, Zheng X, Liu J, et al. Systematic pan-cancer analysis identifies APOC1 as an immunological biomarker which regulates macrophage polarization and promotes tumor metastasis. Pharm Res. 2022;183:106376.
Hao X, Zheng Z, Liu H, Zhang Y, Kang J, Kong X, et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 2022;56:102463.
Low-Kam C, Rhainds D, Lo KS, Barhdadi A, Boulé M, Alem S, et al. Variants at the APOE /C1/C2/C4 locus modulate cholesterol efflux capacity independently of high-density lipoprotein cholesterol. J Am Heart Assoc. 2018;7:e009545.
Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–43.
Yang Y, Qin J, Lan L, Li N, Wang C, He P, et al. M-CSF cooperating with NFκB induces macrophage transformation from M1 to M2 by upregulating c-Jun. Cancer Biol Ther. 2014;15:99–107.
Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang M, et al. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther. 2016;24:915–25.
Wang Y, Zhu J, Zhang L, Zhang Z, He L, Mou Y, et al. Role of C/EBP homologous protein and endoplasmic reticulum stress in asthma exacerbation by regulating the IL-4/signal transducer and activator of transcription 6/transcription factor EC/IL-4 receptor α positive feedback loop in M2 macrophages. J Allergy Clin Immunol. 2017;140:1550–61.e8.
Bories G, Colin S, Vanhoutte J, Derudas B, Copin C, Fanchon M, et al. Liver X receptor activation stimulates iron export in human alternative macrophages. Circ Res. 2013;113:1196–205.
Xiao Z, Mayer AT, Nobashi TW, Gambhir SS. ICOS is an indicator of T-cell-mediated response to cancer immunotherapy. Cancer Res. 2020;80:3023–32.
Sun Q, Zhao X, Li R, Liu D, Pan B, Xie B, et al. STAT3 regulates CD8+ T cell differentiation and functions in cancer and acute infection. J Exp Med. 2023;220:e20220686.
Noviello M, Manfredi F, Ruggiero E, Perini T, Oliveira G, Cortesi F, et al. Bone marrow central memory and memory stem T-cell exhaustion in AML patients relapsing after HSCT. Nat Commun. 2019;10:1065.
Kim GH, Won JE, Byeon Y, Kim MG, Wi TI, Lee JM, et al. Selective delivery of PLXDC1 small interfering RNA to endothelial cells for anti-angiogenesis tumor therapy using CD44-targeted chitosan nanoparticles for epithelial ovarian cancer. Drug Deliv. 2018;25:1394–402.
Zhou Z, Christofidou-Solomidou M, Garlanda C, DeLisser HM. Antibody against murine PECAM-1 inhibits tumor angiogenesis in mice. Angiogenesis. 1999;3:181–8.
Fukumoto M, Kondo K, Uni K, Ishiguro T, Hayashi M, Ueda S, et al. Tip-cell behavior is regulated by transcription factor FoxO1 under hypoxic conditions in developing mouse retinas. Angiogenesis. 2018;21:203–14.
Wang G, Wen B, Deng Z, Zhang Y, Kolesnichenko OA, Ustiyan V, et al. Endothelial progenitor cells stimulate neonatal lung angiogenesis through FOXF1-mediated activation of BMP9/ACVRL1 signaling. Nat Commun. 2022;13:2080.
Wang Y-Q, Xu Z-M, Wang X-L, Zheng J-K, Du Q, Yang J-X, et al. LncRNA FOXC2-AS1 regulated proliferation and apoptosis of vascular smooth muscle cell through targeting miR-1253/FOXF1 axis in atherosclerosis. Eur Rev Med Pharm Sci. 2020;24:3302–14.
Bolte C, Ustiyan V, Ren X, Dunn AW, Pradhan A, Wang G, et al. Nanoparticle delivery of proangiogenic transcription factors into the neonatal circulation inhibits alveolar simplification caused by hyperoxia. Am J Respir Crit Care Med. 2020;202:100–11.
Toshner M, Dunmore BJ, McKinney EF, Southwood M, Caruso P, Upton PD, et al. Transcript analysis reveals a specific HOX signature associated with positional identity of human endothelial cells. PLoS ONE. 2014;9:e91334.
Iordache F, Constantinescu A, Andrei E, Maniu H. Histone acetylation regulates the expression of HoxD9 transcription factor in endothelial progenitor cells. Rom J Morphol Embryol. 2015;56:107–13.
Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–68.
Drake CG, Stein MN. The immunobiology of kidney cancer. J Clin Oncol. JCO2018792648 (2018).
Wang F, Long J, Li L, Wu Z-X, Da T-T, Wang X-Q, et al. Single-cell and spatial transcriptome analysis reveals the cellular heterogeneity of liver metastatic colorectal cancer. Sci Adv. 2023;9:eadf5464.
Shi Q, Xu R, Song G, Lu H, Xue D, He X, et al. GATA3 suppresses human fibroblasts-induced metastasis of clear cell renal cell carcinoma via an anti-IL6/STAT3 mechanism. Cancer Gene Ther. 2020;27:726–38.
Acknowledgements
We would like to thank all the investigators for participating in the present study. This work was supported by the Ministry of Science and Technology of People’s Republic of China (2023YFC2306003), the National Natural Science Foundation of China (No. 81972652 and No. 32270635) and the National Natural Science Foundation of Beijing Municipality (No. 7232082).
Author information
Authors and Affiliations
Contributions
YZ, XH, and MY designed and conducted the study. YZ, XH, and MY drafted the manuscript. XW, YY, and LZ revised the manuscript. YZ, XH, and MY performed the data analysis. YZ, XH, MY, MZ, and LZ conduct experimental operations. YZ, XH, and MY contributed equally to this work. XW, YY, and LZ contributed equally to this work. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The studies involving human participants were reviewed and approved by the ethics committee of Shanghai OUTDO Biotech Corporation Limited and Shanxi Bethune Hospital. The patients/participants provided their written informed consent to participate in this study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Y., Huang, X., Yu, M. et al. The integrate profiling of single-cell and spatial transcriptome RNA-seq reveals tumor heterogeneity, therapeutic targets, and prognostic subtypes in ccRCC. Cancer Gene Ther 31, 917–932 (2024). https://doi.org/10.1038/s41417-024-00755-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-024-00755-x
- Springer Nature America, Inc.