
Abstract
The enhancer of zeste homolog 2 (EZH2) and its highly related homolog EZH1 are considered to be epigenetic silencing factors, and they play key roles in the growth and differentiation of cells as the core components of polycomb repressive complex 2 (PRC2). EZH1 and EZH2 are known to have a role in human malignancies, and alterations in these two genes have been implicated in transformation of human malignancies. Inhibition of EZH1/2 has been shown to result in tumor regression in humans and has been studied and evaluated in the preclinical setting and in multiple clinical trials at various levels. Our work thus contributes to the understanding of the relationship between regulatory molecules associated with EZH1/2 proteins and tumor progression, and may provide new insights for mechanism-based EZH1/2-targeted therapy in tumors.
Similar content being viewed by others
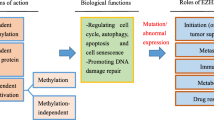

The biology of EZH1/2
The human polycomb repressive complex 2 (PRC2), a major multi-protein transcriptional repressor complex, containing four core components (Fig. 1a): a) an enhancer of zeste homolog 2 (EZH2) and its highly related homolog EZH1 (enhancer of zeste homolog 1), b) a repressor of embryonic ectodermal development (EED) [1], c) a repressor of zeste 12 (SUZ12) [2, 3], d) a retinoblastoma cells tumor-associated proteins 46 and 48 (RbAp46/48) [4]. All these major subsuites are combined with some other non-core components to modify the lysine 27 on the tail of histone H3, which mediates H3K27 monomethylation, dimethylation, and trimethylation (H3K27me1/2/3), thereby promoting chromatin compaction and achieves transcriptional repression of target genes [4, 5], which is required to control gene repression programs involved in development, regulation of tissue homeostasis or stem cell maintenance and lineage specification [6]. In drosophila, the PRC2 catalytic subunit is a single protein E(z), whereas in mammals this function is performed by two proteins, EZH1 and EZH2 [6]. That is, PRC2-EZH1 and PRC2-EZH2 play a non-negligible role in control gene silencing through the methylation of histone H3 on lys27 (H3K27me) [7].

a PRC2 complex; b-c Structure of EZH1α (up), EZH1β (down) and EZH2 (c).
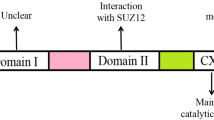
EZH1 contains EZH1α and EZH1β two isoforms, EZH1α localized in the nucleus, while EZH1β is localized in the cytoplasmic matrix of differentiated muscle cells [7]. The full length of EZH1α contains 747 amino acids, and the main functional regions from the N-terminal to the C-terminal are the WD domain (WD-40 binding domain), SANT1 (SWI3-ADA2-N-CoR-TFIIIB) domain, SANT2 domain, CXC (cysteine-rich domain) domain and SET (su(var)3–9, enhancer of zeste, trithorax) domain (Fig. 1b). The main function of the WD and SANT1 domains is to interact with EED-binding, SANT2, CXC and SET are the domains that bind to SUZ12 [7]. In addition, the CXC and SET domains are also catalytic regions. Specifically, the SET domain transfers methyl groups to H3K27, H3K27me1, and H3K27me2 in the presence of S-adenosylmethionine (SAM) to methylate them; while CXC is temporarily an unbound nucleus bodies provide binding sites [4] (Fig. 1c). Unlike EZH1α, EZH1β does not have a catalytic region (CXC and SET domain) at the C-terminus, but instead has an exclusive 554–579 amino acid sequence, NH3-KSTLLSPSSTQVVGLGVPRLFSPAP-COOH (Fig. 1b). EZH1β functions in skeletal muscle to block the assembly of EED, SUZ12, and EZH1α in the nucleus by binding to EED target genes in the cytoplasm, thereby controlling PRC2-EZH1 activity in response to atrophic oxidative stress [8] (Fig. 2a). Overall, EZH1β indirectly regulates both the assembly and function of PRC2-EZH1 in the nucleus [7]. Some studies have confirmed that chromatin compaction can also be achieved when EZH1 is not combined with PCR2, but the chromatin compaction effect of the PRC2-EZH1 complex is stronger than that of its own. EZH2 also plays an important role in this, giving the dimer forms and flexible conformation of PRC2 provide the ultimate explanation for PRC2’s ability to act on various chromatin substrates it encounters in cells [9].

a Function of EZH1 isoforms (left) and EZH2 (right); b Regulation of H3K27me3 by EZH1 and EZH2 (left); Compensatory role of EZH1 upon EZH2 inactivation (right).
EZH2 does not have isoforms like EZH1, and only a 571 amino acid N-terminal to C-terminal structure has been found. The domain of EZH2 is divided into three main parts, the first part is SDB, EBD, BAM, SAL, SRM, SANT1, the middle part is MCSS and SANT2, and the third part is also called the catalytic part is CXC and SET (Fig. 1c). One of the core components of PRC2, EED, binds to the EBD domain of EZH2, and the VEFS domain of another core member, SUZ12, reacts with SANT2 and SET, which is similar to EZH1α [4]. EZH2 was found to be mainly located in the nucleus, however, some researchers have proposed the role of EZH2 in the cytoplasm, that is, EZH2 phosphorylated at T367 (pT367-EZH2) binds to cytoplasmic proteins in triple-negative breast cancer cells and enhances invasion and metastasis [10, 11]. However, the molecular mechanism of the EZH2 shuttles between the nucleus and the cytoplasm and the proof that EZH2 exists in the cytoplasm are still inadequate.
There are similarities and structural differences between EZH1 and EZH2, which result in their similarities and functional differences. EZH1 and EZH2 are expressed in opposite ways in the course of cell development and differentiation, EZH2 predominating in proliferative cells and EZH1 in postmitotic cells. Although EZH1 exerts weak histone methyltransferase (HMTase) activity, it can replace EZH2 in the absence of which, maintaining basal H3K27me3 levels at PRC2 targets [12] and promoting chromatin compaction [7] (Fig. 2b). The compensatory phenomenon of EZH1 has been discovered and proposed by more and more scholars [13, 14], which suggests that EZH1 and EZH2 function together, and there is a specific connection between them. Mutual interference and compensatory functions of EZH1 and EZH2 were found to rearrange their own genome-scale distributions in the co-expressed EZH1/2 line [14]. Another evidence for the cooperation of EZH1 and EZH2 is their joint participation in their differentiation program in the renewal of SIX2 (SIX homeobox 2)-positive nephron progenitor cells (NPCs) [15].
Lysine modifications (H3K27me3, H3K9me2/3, H3K4me3, H3K27ac) [5] on the H3 tail by EZH1 and EZH2 in complex with PRC2 regulate chromatin to achieve gene silencing and activation, thus showing increased proliferation and invasion of cancer cells or tumors inhibitory activity. As it has been reported that EZH1/2 play critical role in tumorigenesis, continuous efforts are still to be made in the development of EZH1/2 specific and/or dual-target inhibitors with higher efficiency and lower toxicity.
In this study, we investigated the relationship of EZH1/2 proteins in tumor progression and the regulated molecules, and the summary of clinical and non-clinical research progress of targeting EZH1/2 inhibitors is to elucidate the significance of EZH1/2 as targets for tumor therapy. Overall, the development of targeted EZH1/2 inhibitors is highly promising in the treatment of tumors.
EZH1 in oncology
Abnormalities of EZH1 in tumors
It is necessary to add that, to better understand the relationship between EZH1 and tumors, we have added the EZH1 pan-cancer analysis (see Fig. 3). Surprisingly, we found that among the 39 common tumors involved in our analysis, 12 tumors (all solid tumors) including bladder urothelial carcinoma, breast invasive carcinoma, cervical squamous cell carcinoma and endocervical adenocarcinoma, glioblastoma multiforme, kidney chromophobe, kidney renal clear cell carcinoma, lung adenocarcinoma, lung squamous cell carcinoma, prostate adenocarcinoma, and uterine corpus endometrial carcinoma had low EZH1 expression compared with the normal groups, only a few with high expression of EZH1.

*P < 0.05, **P < 0.01, ***P < 0.001.
Particularly, EZH1 was observed to overexpress in mantle cell lymphoma [16], breast cancer [17], T-cell lymphoma [18] and gastric cancer [19], liver hepatocellular carcinoma, and cholangio carcinoma (Fig. 3), while mutate into follicular-patterned thyroid tumors [20]. Apart from that, the expression of EZH1 in other tumors was almost no significant difference from normal controls. In a nutshell, the expression of EZH1 in most tumors was indistinguishable from or even inferior to that in normal tissues.
The relationship with diagnosis and prognosis of EZH1
The high expression of EZH1 in some tumors was demonstrated responsible for its diagnosis and prognosis, such as breast cancer [17], gastric cancer [19], colorectal cancer [21], hepatocellular carcinoma [22], and so on. Moreover, Chakraborty’s team pointed that inactivation of the von Hippel-Lindau tumor suppressor protein (pVHL) is a hallmark lesion of the most common form of renal cell carcinoma, clear cell renal cell carcinoma (ccRCC), and pVHL-deficient ccRCC cells are highly dependent on the H3K27 methyltransferase EZH1 for survival. Therefore, targeting EZH1 may have therapeutic effects in ccRCC.
In addition to EZH1’s potential as a diagnostic marker in tumors with high EZH1 expression, the poor prognosis caused by the compensatory increase of EZH1 when EZH2 function is inactivated or depleted cannot be ignored in some tumors like mantle cell lymphoma [16]. This problem was quickly verified—a large part of the fact that EZH2 inhibitors could not proceed to the next trial phase is mainly due to its good effect in vitro but not in vivo. This is probably due to the compensatory increase in EZH1 obtained by inhibiting EZH2 by a single-target inhibitor of EZH2, which further degrades the tumour.
On the contrary, Schümann et al. pointed out that the role of EZH1 is not limited to promoting tumor progression, and its high expression may also be an indicator of good prognosis in some malignant tumors [18]. Notably, high EZH1 expression improved overall survival (OS) and progression-free survival (PFS) in T lymphoma cells [18]. In addition, the researchers discovered that EZH1 presents the epigenetic silence of the oncogenic fusion protein AML1-ETO in acute myeloid leukemia [23], which is beneficial for patient survival. And also, follicular-pattern thyroid tumors with EZH1 mutations reported in the literature are in most cases benign or they are minimally invasive or noninvasive cancers [20].
In summary, EZH1 might play opposite roles in solid cancers and blood cancers, but the situation is sometimes complicated due to the compensatory role of EZH1 in the depletion of EZH2. That is, EZH1’s special capacity to maintain the function of EZH2 when exhausted must also take precedence.
The biological roles in tumorigenesis, progression and treatment of EZH1
Over-expression of EZH1 is reported to be related to inhibition of apoptosis proteins and induction of cell cycle proteins [24]. Inhibition of the apoptotic process but promotion of cell cycle protein expression can lead to uncontrolled cell proliferation, ie., carcinogenesis, suggestive of a link between EZH1 and tumour growth. Besides, Ping et al. reported that tripartite motif-containing 21 (TRIM21) improves the treatment of apatinib in gastric cancer by inhibiting the stability of EZH1 [19]. That is, the stability of EZH1 may play a critical role in tumor development. Moreover, some microRNAs such as microRNA-370–5p [21], microRNA-20a [22], microRNA-182–5p [25], microRNA-765 [17], etc. were found to directly targeting EZH1 and participate in the regulation of biological processes such as tumor growth, invasive potential and apoptosis.
Besides, due to the mutation of EZH2 or after treatment of some EZH2 specific inhibitors in tumor cells, the compensatory effect of EZH1 will be stimulated to a large extent, which can also maintain a favorable environment for tumor development. As Aoyama et al. described, EZH1 targets bivalent genes for maintaining stem cells self-renewing in EZH2-deficient myelodysplastic syndromes [12].
Overall, the role of EZH1 in most tumors is mainly to maintain the level of H3K27me3 to ensure the proper functioning of the transcriptional silencing pattern in the case of EZH2 inactivation or depletion. Therefore, in order to maximize the effect of EZH2 small-molecule inhibitors, that is, to fully inhibit the function of EZH2 and also inhibit the compensatory effect of EZH1, it is important to use both EZH1 and EZH2 together as targets for tumor therapy.
Molecules modulated by EZH1 in tumors
Wang et al.‘s study showed that overexpression of EZH1 suppressed NF-κB signaling in aristolochic acid-injured HK-2 cells, and the molecular mechanism of which was that overexpression of EZH1 gene inhibited apoptosis of HK-2 cells, decreased ROS levels, down-regulated IL-1β, IL-6, TNF-α, Bax and Cyt C mRNA and protein expression, increased Bcl-2 expression and NFKBIA, CXCL8 and cyclin D1 [24] (Fig. 4).

Upstream and downstream proteins of EZH1 in tumors.
Besides directly modulating the target genes, EZH1 has also been found regulated by some upstream proteins. TRIM21, a protein that plays a crucial role in the regulation of many cellular events involved in tumor progression, has been reported to decrease the expression levels of the EZH1 protein by direct interaction with TRIM21 in gastric cancer [19].
EZH2 in oncology
Abnormalities of EZH2 in tumors
According to currently available studies, EZH2 and EZH1 trigger H3K27me3 to suppress the transcription of target genes. EZH2 is highly expressed in most tumors, with only a few weak expression or with somatic gain-of-function mutations (Table 1 and Fig. 5).

a Expression levels of EZH2 in TCGA tumors as shown by TIMER2. b Boxplot of EZH2 expression levels in some tumors without normal tissue data in TCGA (GTEx database). *P < 0.05, **P < 0.01, ***P < 0.001.
In an ordinary way, EZH2 is overexpressed in most solid human tumors, including cervical cancer [26], hepatocellular carcinoma [27], most brain tumors [28, 29], colorectal tumor [30], non-small cell lung cancer cells [31], thyroid cancer [32], ovarian cancer [33], breast cancer [34], etc. On the other hand, low expression and somatic gain-of-function mutations in EZH2 may be more common in hematological tumors. Researches showed that in more than 20% of diffuse large B-cell lymphomas (DLBCL) and 7% of follicular lymphomas (FL), somatic gain-of-function mutations at residue tyrosine 641 within the EZH2 SET domain (Y641N and Y641F) have been detected [26] but rarely found in solid tumors. Additionally, a lower expression level of EZH2 was detected in acute myeloid lymphoma compared to controls. (Fig. 5).
The relationship with diagnosis and prognosis of EZH2
The overexpression of both EZH1 and EZH2 is associated with most tumour aggressiveness, poor prognosis, and relapses. Owing to its enzymatic activity, EZH2 can bind to promoter of target genes to induce methylation and affect their expression. EZH2 may be regarded as an independent prognostic factor in brain tumors that its upregulation provides undesirable prognosis [28]. Similar points were heard from many studies, that is, EZH2 contributed to promoting tumor progression and affecting the overall survival (OS) and progression-free survival (PFS) of tumor patients in cervical cancer [26], triple-negative breast cancer [35], colorectal tumor [30], T-cell lymphoma [18], and cholangiocarcinoma [36]. All of these indicate that EZH2 could be considered as a diagnostic marker for most tumors.
Now we might conclude that EZH2 is generally considered to be a tumor-promoting factor, however, some groups reported that EZH2 might also exert a tumor suppressor effect in solid tumors such as gastric cancer [37], medulloblastoma, [28], and reduce breast tumorigenesis [38]. These suggest that EZH2 exerts the opposite effect not because of the distinction between solid tumors and hematological tumors, but because of other reasons to be explored. Overall, EZH2 can be considered to be positively associated with diagnosis and prognosis in most tumors.
The above-mentioned studies suggest that the usage of EZH2’s epigenetic silencing function to suppress tumors needs to base on specific tumor characteristics. EZH2 is neither a specific silencing tumor-promoting factor nor a tumor suppressor factor, which makes the application and research of EZH2 in oncology more promising and challenging. However, the overexpression of EZH2 is associated with malignancy in most cases (Table 1, Fig. 5), such as increased tumor aggressiveness and poor clinical prognosis. Although EZH2 may have potentially committed to tumor suppression, the fact that EZH2 epigenetically silences the tumor suppressor genes is a priority that must be taken into account.
The biological roles in tumorigenesis, progression and treatment of EZH2
EZH2 signaling mainly participates in increased proliferation and invasion of cancer cells. For instance, elevated EZH2 levels often associates with cell cycle arrest inhibition [31], apoptosis and autophagy suppression [27], increased invasion and migration ability [28, 29] of tumor cells, and other biological processes.
Specifically, high expression of EZH2 in hepatocellular carcinoma [27], non-small cell lung cancer cells [31], and mantle cell lymphoma [16] was found to inhibit autophagy, cell cycle arrest and apoptosis. Moreover, EZH2 was also be reported to induce cell invasion and migration in brain tumors [28, 29], nasopharyngeal cancer [39], uveal melanoma [40], clear cell renal cell carcinoma [41], rhabdomyosarcoma [42]. In addition, modulation of cell stemness in colorectal cancer was also found [43]. Therefore, except for special tumors such as medulloblastoma, the above biological processes involved in EZH2 in tumors can be used as evaluation criteria for drug intervention.
Molecules modulated by EZH2 in tumors
Upstream regulation of EZH2
Under normal circumstances, EZH2 in cells maintains a normal level and will not over-activate transcriptional inhibition to affect the biological process of cells. However, there are indeed some signaling molecules in tumor cells that over-activate EZH2 to establish an environment conducive to the survival and development of tumor cells. (Fig. 6a). For example, EZH2 can be activated by the STAT3/HOTAIR signaling pathway to reduce pro-apoptotic proteins, thereby reducing lung cancer cell apoptosis [31]. Protein arginine methyltransferase 1 (PRMT1) asymmetrically demethylates EZH2 at R342 increases EZH2 stability and promotes epithelial-mesenchymal transition, invasion and metastasis in breast cancer cells [44]. According to Sanches’s report, EZH2 was downregulated by lysine demethylase 2B (KDM2B) and then modulated cell migration, invasion, and stemness in colorectal cancer, a chain reaction of increased AKT and PI3K resulting from their interaction was also found [43]. You et al. found that p53 inhibited the expression of O-linked N-acetylglucosamine (GlcNAc) transferase (OGT), thereby reducing the stability of EZH2 and ultimately inhibiting the development of HCC [45]. Moreover, the interaction of metabolic and epigenetic signaling pathway was reported by Yuan et al., who showed that metformin-stimulated (AMP-activated protein kinase) AMPK signaling converges at FOXO3 to stimulate SETD2 (SET domain containing 2, histone lysine methyltransferase) expression to increase H3K36me3 against EZH2-catalyzed H3K27me3, thereby reducing the stability of EZH2 and inhibiting the metastasis of prostate cancer [46].

a Upstream and downstream proteins of EZH2; b EZH2 binds to proteins in tumors.
In addition, Han’s group reported that EZH2 was upregulation by long noncoding RNA (lncRNA) prostate androgen-regulated transcript 1 (PART1)/promyelocytic leukemia zinc finger (PLZF) regulation, accompanied by enrichment of H3K27 trimethylation, thereby mediating epigenetic platelet-derived growth factor (PDGFB) silencing and downstream PI3K/Akt inhibition [37] (Fig. 6a). Moreover, Guan’s study pointed out that early growth response 1 (EGR1) activates the expression of EZH2, which in turn represses EGR1 expression through silencers downstream of the EGR1 gene, resulting in reduced breast tumorigenesis [38] (Fig. 6b).
Downstream regulation of EZH2
EZH2 has obtained much attention in recent years in the field of cancer therapy due to its aberrant expression and the ability to regulate the expression of genes by binding to their promoter and affecting methylation status. EZH2 usually transcriptional silencing its downstream target genes (Fig. 6a) including p16, p21, E-cadherin, cyclin D1, Ras, NF-κB [47], wnt [48], β-catenin [49], and GATA6 [50]. The targets directly regulated by EZH2 are also include some RNAs such as lncRNA PHACTR2-AS1 [40] and micro-181b [51]. Specifically, EZH2 promotes the methylation of H3K27 (H3K27me3) [26], the raised H3K27me3 further repressed the transcription of microRNA-139–5p to promote epithelial-mesenchymal transition (EMT) and lymph node metastasis (LNM) in pancreatic cancer [52]. EZH2 increases the transcriptional activity of miR-375 by mediating the methylation and phosphorylation of signal transducer and activator of transcription 3 (STAT3), which in turn negatively regulates forkhead box O1(FOXO1), the direct target of miR-375, and further activates the p53 signaling pathway, ultimately indicative of further progression of breast cancer [53].
Of course, not all EZH2 directly act on target genes to suppress their transcription, the extensive functional domains on EZH2 allow it to directly or indirectly bind and interact with other proteins besides PRC2 core members (Fig. 6b). EZH2 directly interacts with MYC family oncoproteins MYC and MYCN to promote neuroblastoma and small cell carcinoma tumor formation [54], EZH2 and DNA methyltransferase 3B (DNMT3B) [55] promotes malignant tumor development upon directly binding, EZH2 also combines with high mobility group A1 (HMGA1) and ubiquitin-specific peptidase 7 (USP7) to form an EZH2-HMGA1-USP7 complex, which promotes cytoplasmic chromatin fragment (CCF) formation and activates the cytoplasmic DNA sensor cyclic GMP-AMP synthase (cGAS)-STING pathway to promote breast cancer metastasis [56]. The zeste homolog 2 and androgen receptor (AR) interact to promote tumor aggressiveness [57], it can also combine with LSD1 (also known as lysine-specific demethylase 1A, KDM1A or flavin-containing amine oxidase domain-containing protein 2, AOF2) and DNMT1 as scaffolds to connect LncRNA LINP1 to inhibit KLF2 and PRSS8 (inhibits cell proliferation and boosts cell apoptosis proteins) expression and promote cervical cancer progression [58]. Interestingly, EZH2, a transcriptional repressor, was shown to convert from a gene repressor to an activator by phosphorylation of CDK1 at threonine 487 (pT487-EZH2). EZH2 binds to histone reader protein (ZMYND8) to enhance the interaction with FOXM1 and activate the transcription of matrix metalloproteinase (MMP) genes, resulting in human clear cell renal cell carcinoma (ccRCC) cell migration and invasion [41]. Similarly, EZH2 binds to the CHK1 (checkpoint kinase) promoter and activates CHK1 signaling to maintain cell stemness in epithelial ovarian cancer cells [59].
Clinical inhibitors targeting EZH1/2
As epigenetic silencing factors, EZH2 and EZH1 proteins play important roles in various stages of cells. Although they epigenetically silence both tumor-promoting genes and tumor suppressor genes, their high expression also contributes to different subtypes of cancers at various rates [28, 29]. Therefore, various EZH1/2 specific inhibitors have been synthesized and part of them was already studied in clinical trials. For example, CPI-0209 (Constellation Pharmaceuticals), CPI-1205 (Constellation Pharmaceuticals), Tazemetostat (EPZ-6438, Epizyme), GSK2816126 (GlaxoSmithKline), and SHR2554 (Jiangsu HengRui Medicine) were synthesized towards inhibiting EZH2. EZH1 and EZH2 dual inhibitors include Ezharmia (DS-3201b, Daiichi Sankyo), PF-06821497 (Pfizer) and HH2853 (Haihe Biopharma). Except for Tazemetostat and Ezharmia, which are approved by the Food and Drug Administration (FDA) [26], and the Ministry of Health, Labour and Welfare (MHLW), other drugs are currently under clinical evaluation (Fig. 7).

The names of compounds in clinical trials have been bolded. CID number from PubChem.
Tazemetostat (EPZ-6438) is a potent, selective, and orally available EZH2 inhibitor, its Ki and IC50 at the non-cellular level are 2.5 and 11 nM, respectively. EPZ-6438 reduces H3K27me3 levels in SMARCB1 (a tumor-suppressor gene located on chromosome 22q11.2) WT/MT (wild/mutant) cells in a concentration-dependent manner and causes strong antiproliferative effects in SMARCB1-depleted MRT cell lines with IC50 values ranging from 32 to 1000 nM [60]. There are already 26 clinical trials of EPZ-6438, it was first used in the treatment of epithelial sarcoma (NCT03874455) and is currently in clinical trials for the treatment of hematological (such as NCT05228158, NCT03010982, and NCT04703192) and solid tumors (NCT03213665, NCT03460977, and NCT03456726) in various stages. CPI-0209 (structure not published) and SHR2554 (structure not published) are currently in phase II clinical treatment for advanced solid tumors and lymphomas (NCT04104776) and phase I clinical studies for relapsed/refractory mature lymphoid neoplasms (NCT04577885). Similar to EPZ-6438, Lirametostat (CPI-1205) is also an orally bioactive EZH2 inhibitor, and its cell-free IC50 for EZH2 and EZH1 is 2 and 52 nM, respectively [61]. Additionally, CPI-1205 is currently studied in phase I to treat patients with B-cell lymphomas (NCT02395601). Because EZH2 has DNA missense mutations in many tumors [62], EZH2 inhibitor was designed to against diffuse large B-cell lymphoma cells with EZH2 gain-of-function mutations [63], the research and development of inhibitors targeting different mutation sites of EZH1/2 have gradually increased. Specifically, GSK2816126 (IC50 = 9.9 nM) [64] was first synthesized to treat germinal center B-cell-like diffuse large B-cell lymphoma-mutant and wild type, and then transformed follicular lymphoma mutant and wild type, multiple myeloma, other non-Hodgkin’s lymphomas and solid tumors in phase I clinical trials (NCT02082977). Unfortunately, this experiment has been terminated due to insufficient evidence of clinical activity at the maximum dose and schedule attained with GSK2816126 and do not justify further clinical studies.
Given that the mutual substitution and compensatory effects of EZH1 and EZH2 in tumors cannot be ignored, and also cause great resistance to cancer treatment [13, 14], the development of dual-target inhibitors of EZH1 and EZH2 is imminent. Ezharmia (DS-3201b) was designed for relapsed/refractory peripheral T-cell lymphoma and adult T-cell leukemia/lymphoma patients and is in phase II clinical studies recently (NCT04703192). Studies have shown that oral OR-S1 (DS-3201b) treatment significantly inhibited mantle cell lymphoma (MCL) tumor growth and affected B cell activation, differentiation, and cell cycle by regulating cyclin-dependent kinase inhibitor 1c (CDKN1C, also known as p57, KIP2) [65]. Moreover, its inhibitory effect on acute myeloid leukemia (AML) has also confirmed [66]. Another EZH1/2 dual inhibitor HH2853 is currently in phase I clinical trials for relapsed non-Hodgkin’s lymphomas and advanced solid tumors, while refractory non-Hodgkin lymphoma is already in phase II clinical trial recruitment process (Tables 2, 3), and HH2853 was administered orally on a continuous BID schedule for 28 consecutive days during treatment (NCT04390737). Specially, PF-06821497, also an EZH1/2 inhibitor, was developed for the treatment of relapsed/refractory small-cell lung cancer (SCLC), castration, resistant prostate cancer, and follicular lymphoma, and is currently in phase I clinical study (NCT03460977). In addition, no inhibitors against EZH1 are currently in clinical trials. The structures of the above compounds shown in Figs. 7, 8, and the clinical tests of other clinical EZH1/2 inhibitors shown in Table 2.

a EZH1 and EZH2 dual inhibitors; b EZH1 inhibitor. The names of compounds in clinical trials have been bolded. CID number from PubChem.
Non-clinical inhibitors targeting EZH1/2
Although many inhibitors targeting EZH1/2 have been designed and synthesized, there are very few drugs in clinical trials currently, and most of them are still in the R&D (research and development) or preclinical stage. For example, there are many EZH2 inhibitors were studied in academic studies as the primary drug or as a positive control, including CPI-169, CPI-360 [67], EBI-2511 [68], EI1 [69], EPZ005687 [70], EPZ011989 [71], GSK343 [72], GSK503 [73], MS1943 [74], PF-06726304 [75], and EZH1 inhibitor CPI-360 [67], in addition, EZH1/2 dual inhibitor UNC1999 [76] and JQEZ5 [77] (Table 3).
Among these inhibitors, GSK343 is a potent and selective EZH2 inhibitor with IC50 of 4 nM in cell-free assays, 60-fold selective for EZH1. Studies have confirmed that GSK343 can effectively inhibit breast cancer and prostate cancer cell proliferation. The prostate cancer cell line LNCaP is the most sensitive to GSK343, with an IC50 of 2.9 μM [72]. Different from other inhibitors, MS1943 is an orally bioavailable selective degrader of EZH2, which mediates triple-negative breast cancer (TNBC) cells cytotoxic effects through ER stress (endoplasmic reticulum stress) and UPR (unfolded protein response) induction in cells that are dependent for their growth on EZH2 [74]. In addition to these two compounds, other EZH2 inhibitors inhibited both wild-type and mutant EZH2. CPI-169 and PF-06726304 have inhibitory effects on EZH2 Y614N mutant with IC50 of 0.51 [67] and 3 nM [75], respectively. Moreover, EI1 selectively inhibited the growth of DLBCL cells carrying the Ezh2 mutation and caused cell cycle arrest and apoptosis (IC50 for EZH2 Y641F is 13 nM) [69]. EPZ011989, a small molecule inhibitor with more than 15-fold selectivity for EZH2 over EZH1, inhibits mutant and wild-type EZH2 with equal potency (Ki <3 nM), EPZ011989 reduces intracellular H3K27 methylation in human lymphoma cells WSU-DLCL2 harboring Y641F mutation with IC50 < 100 nM [71]. Furthermore, the 50% inhibitory concentration of EBI-2511 on EZH2 (A667G) is 4 nM, at the enzymatic level, and EPZ005687 targets the same EZH2 mutation site (Ki for EZH2 WT is 24 nM), which inhibits the enzymatic activity of PRC2 in a concentration-dependent manner with IC50 of 54 nM [70].
These EZH2 inhibitors may not be put into clinical use because their inhibitory effect on EZH2 is much greater than that of EZH1, which ignores the compensatory effect of EZH1 when EZH2 is depleted, which may lead to an unsatisfactory therapeutic effect on tumors. Nevertheless, there are only a few single-target EZH1 and EZH1/2 dual-target inhibitors currently. We only found one EZH1 inhibitor, CPI-360, which is a SAM-competitive EZH1 inhibitor with an IC50 of 102.3 nM. Published data show that CPI-360 effectively reduces overall H3K27me3 and H3K27me2 levels in KARPAS-422 cells with EC50 of 56 and 65 nM, respectively. CPI-360 also affected the activity of the Y641N mutant in EZH2-containing KARPAS-422 cells. In addition, CPI-360 gradually blocked KARPAS-422 cells in the G1 cell cycle phase, followed by induction of apoptosis [67]. An EZH1/2 dual-target inhibitor, UNC1999, was later found to be slowly absorbed in rat plasma, reaching a maximum concentration of 118.8 ± 12.0 ng/ml 1.5 h after oral administration [78]. UNC1999 is an orally active selective inhibitor of EZH2 and EZH1 (non-cellular IC50 is 2 and 45 nM, respectively) and a potent inducer of autophagy. Studies have shown that UNC1999 has strong inhibitory activity against EZH2 Y641N and EZH2 Y641F mutants in vitro, and shows a concentration-dependent inhibitory effect on cell proliferation in DLBCL cell lines containing EZH2 Y641N mutations [76]. Different from other types of inhibitors, JQZE5 is a reversible dual-target inhibitor of EZH1/2 [77], but so far no supporting research data has been made public. The structures of the above compounds shown in Figs. 7, 8.
Although many EZH1/2 inhibitors have entered the clinical trial stage, only one EZH2 inhibitor Tazemetostat (EPZ-6438) and EZH1/2 dual-target inhibitor Ezharmia (DS-3201b) has approved. Therefore, the development and research of EZH1/2 inhibitors still need a lot of work, and the focus of future research should be on inhibitors with dual EZH1/2 targets.
EZH1/2 inhibitors in combination with other drugs
Due to the limitations of monotherapy, many clinical trials currently conducted using combinations of two or more inhibitors (Table 4).
Epizyme, the company that developed tazemetostat, has conducted 12 clinical trials of tazemetostat in combination with various other drugs. Except for a few of these projects in completed (NCT03028103, NCT02220842, NCT02220842) and terminated (NCT04590820) states, most of them are either recruiting or not yet recruiting (NCT05152459). Besides, EZH1/2 dual inhibitor DS-3201b in combination with first-line drug irinotecan for advanced colorectal cancer is currently recruiting for a phase I/II clinical trial in patients with small cell lung cancer (NCT03879798). Moreover, SHR2554 is in phase II trials in combination with 14 other drugs in breast tumors (NCT04355858) and combination with SHR1791 in advanced solid tumors and B-cell lymphomas (NCT04407741). Other strategies being focusing on EZH1/2 inhibitors in combination with other drugs shown in Table 4.
Combination with non-EZH1/2 target molecule inhibitors
One of the ongoing projects of Epizyme is the combination of tazemetostat and umbralisib (PI3Kδ and CK1-ε dual inhibitors) for the therapy of recurrent/refractory follicular lymphoma (NCT05152459). Moreover, in patients with relapsed/refractory (R/R) follicular lymphoma (FL), tazemetostat demonstrated single-agent antitumor activity, compared with the phosphoinositide 3-kinase (PI3K) inhibitors (idelalisib, copanlisib, umbralisib, and duvelisib), which indicated for phase III/IV and late-stage (3L/4L+) treatment of R/R FL, showed lower risk, while achieving similar efficacy outcomes [79]. The above suggests that the appropriate combination of EZH1/2 and PI3K inhibitors can reduce the dose to reduce the risk, on the other hand, it might increase the anticancer efficacy.
The EZH1/2 inhibitor UNC1999 was shown to enhance the antitumor effect of sorafenib (VEGFRs and Raf inhibitor) in hepatocellular carcinoma (HCC), that is, the combination of the two exhibited synergistic antitumor effects in vitro and in vivo [80]. Mechanistically, the increased levels of H3K27me3 induced by sorafenib led to HCC resistance, and the use of UNC1999 inhibited EZH2 and EZH1 to block this resistance. In conclusion, sorafenib plus EZH1/2 inhibitors may constitute a new therapeutic approach for HCC. Similarly, depletion of EZH2 with EZH2 inhibitors increases the strong sensitivity of non-small cell lung cancer cells to cisplatin [81]. Besides, proteasome inhibitors combined with UNC1999 showed a strong synergistic effect in prostate cancer cells [82]. All of these indicate that non-EZH1/EZH2 target inhibitors can be used in combination with EZH1/2 inhibitors as an effective tumor treatment strategy.
Combination with monoclonal antibodies
Notably, the work of Wang’s team revealed the relationship between EZH2 and the tumor microenvironment. Studies have shown that tumor-infiltrating Tregs (TI-Tregs) are dependent on EZH2 in tumors, which can be exploited to pharmacologically and genetically disrupt EZH2 activity in Tregs, thereby remodeling the tumor microenvironment and enhancing the recruitment and function of CD8 (+) and CD4 (+) effector T cells to eliminate tumors [83]. In vivo, depletion of EZH2 acts synergistically on melanoma suppression during anti-CTLA-4 or IL-2 immunotherapy of melanoma in mice [84]. Moreover, the combination of tazemetostat, atezolizumab, and obinutuzumab is being used for the therapy of lymphoma in phase I clinical trial (NCT02220842) conducted by Epizemy. This combination therapy enhances the recruitment of innate and adaptive immunity and is effective against CD20+ B-cell malignancies. Especially, atezolizumab and obinutuzumab are monoclonal antibodies targeting PD-L1 and anti-CD20, respectively.
What’s more, EPZ-6438 was even used to conquer lymphoma, DLBCL, and follicular lymphoma with other five drugs: rituximab (anti CD20), cyclophosphamide, vincristine (chemotherapy drug for leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, et al.), doxorubicin (topoisomerase-II inhibitor, commonly used as a tumor chemotherapeutic agent), and prednisolone (a synthetic drug similar to cortisone, used to relieve rheumatic and allergic conditions and to treat leukemia) (NCT02889523). This combination helps to regulate the immune microenvironment. On the one hand, it is beneficial to overcome malignant tumors such as lymphoma and leukemia, and on the other hand, it is also helpful for the control of autoimmune diseases.
Another EZH2 inhibitor CPI-1205 is combined with ipilimumab (anti CTLA-4) in phase I clinical treatment of advanced solid tumors (NCT03525795). It also companies with enzalutamide and abiraterone/prednisone in phase I/II state to intervene metastatic castration resistant prostate cancer (NCT03480646). CTLA-4 (cytotoxic T lymphocyte-associated antigen-4) affects the body’s immune system, impairing its ability to kill cancer cells, its monoclonal antibody can inhibit its activity, and combined with EZH2 inhibitors can effectively target cancer cells. In the long term, the subtle role of EZH2 in the tumor microenvironment can also be considered in the study of novel EZH1/2 inhibitors.
Combination with metabolic enzyme inhibitors or inducers
Given that studies have confirmed the close relationship between EZH2 and metabolism, relative clinical trials are already underway. For example, tazemetostat combined with CYP3A4 inhibitor (itraconazole) or inducer (rifampicin) to modulate metabolic pathways for the treatment of diffuse large B cell lymphoma, primary mediastinal lymphoma, mantle cell lymphoma, advanced solid tumor, and marginal zone lymphoma, is currently in the completion stage of clinical phase I trials (NCT03028103). Additionally, EPZ6438 also used in four drugs combination manner including fluconazole (antifungal and yeast infection drugs), omeprazole (digestive system drugs), and repaglinide (antihyperglycemic class of drugs) for the treatment of diffuse large B cell lymphoma, primary mediastinal lymphoma, mantle cell lymphoma, advanced solid tumor, and marginal zone lymphoma (NCT03028103). The above suggests that the combined use of EZH2 inhibitors and metabolism-related inhibitors or inducers can also become an effective means of treating clinically complex tumors.
Novel use of old drugs in blocking EZH1/2
In addition to combining with new targeted therapy drugs, repurposing old drugs is also a time-saving and efficient strategy. Gambogenic acid was originally identified from Gamboge as an inhibitor of the FGFR signaling pathway in erlotinib-resistant non-small cell lung cancer (NSCLC) with antitumor effects [85]. It was also later shown to be a potent EZH2 inhibitor that specifically covalently binds cys668 within the EZH2-SET domain and leads to EZH2 ubiquitination [86]. There are many similar drugs that can target and modulate epigenetic factors, and then adjusting the treatment strategy may make the drugs more fully utilized. That is to say, the use of less drug dosage or type plays a more precise and stronger anti-cancer effect.
Conclusions and future perspective
Overexpression or gain-of-function mutations within the catalytic SET domains of EZH1/2 has been reported to disrupt normal PRC2 function. Couple with the mutual compensatory and replacement effects of EZH1 and EZH2, resulting in abnormal tumor cell growth and increased adhesion and invasiveness, which is not conducive to patient survival. Therefore, EZH1/2 are important for targeted therapy. However, clinical efficacy using only EZH2-specific inhibition showed only modest effects on cancer. Therefore, further efforts should focus on combination therapy or finding dual inhibitors for effective therapy. Currently, dual inhibition of EZH1 and EZH2, as well as targeted and non-targeted combinations, are under clinical research with promising results.
Furthermore, although the selectivity profile of many EZH1/2 inhibitors is well understood based on our current knowledge of the working mode of the EZH2 protein, the rational design of dual EZH1/2 inhibitors with high selectivity and bioavailability remains a challenge. Future work will certainly yield further insights into the current bottleneck. Last but not least, the development of new drugs requires a lot of resources and usually consumes a lot of time, which creates a lot of resistance for patients with malignant tumors who urgently need clinical solutions. Therefore, while considering new drug development and research, we should also focus on clinical drugs that already used in the past, explore new ways of using such drugs, and consider combining them with clinical EZH1/2 drugs to fight tumors. Our general purpose is to use the least amount of medicine (including but not limited to the amount and type of medicine used) to achieve the maximum effect.
References
Lee CH, Yu JR, Kumar S, Jin Y, LeRoy G, Bhanu N, et al. Allosteric activation dictates PRC2 activity independent of its recruitment to chromatin. Mol Cell. 2018;70:422–34.e6.
Chen S, Jiao L, Shubbar M, Yang X, Liu X. Unique Structural platforms of SUZ12 dictate distinct classes of PRC2 for chromatin binding. Mol Cell. 2018;69:840–52.e5–.
Chammas P, Mocavini I, Di Croce L. Engaging chromatin: PRC2 structure meets function. Br J cancer. 2020;122:315–28.
Zhao Y, Guan YY, Zhao F, Yu T, Zhang SJ, Zhang YZ, et al. Recent strategies targeting embryonic ectoderm development (EED) for cancer therapy: allosteric inhibitors, PPI inhibitors, and PROTACs. Eur J Medicinal Chem. 2022;231:114144.
Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, Chin CR, et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature. 2021;589:299–305.
Völkel P, Bary A, Raby L, Chapart A, Dupret B, Le Bourhis X, et al. Ezh1 arises from Ezh2 gene duplication but its function is not required for zebrafish development. Sci Rep. 2019;9:4319.
Bodega B, Marasca F, Ranzani V, Cherubini A, Della Valle F, Neguembor MV, et al. A cytosolic Ezh1 isoform modulates a PRC2-Ezh1 epigenetic adaptive response in postmitotic cells. Nat Struct Mol Biol. 2017;24:444–52.
Liu P, Shuaib M, Zhang H, Nadeef S, Orlando V. Ubiquitin ligases HUWE1 and NEDD4 cooperatively control signal-dependent PRC2-Ezh1α/β-mediated adaptive stress response pathway in skeletal muscle cells. Epigenetics Chromatin. 2019;12:78.
Grau D, Zhang Y, Lee CH, Valencia-Sánchez M, Zhang J, Wang M, et al. Structures of monomeric and dimeric PRC2:EZH1 reveal flexible modules involved in chromatin compaction. Nat Commun. 2021;12:714.
McMullen ER, Skala SL, Gonzalez ME, Djomehri S, Chandrashekar DS, Varambally S, et al. Subcellular localization of EZH2 phosphorylated at T367 stratifies metaplastic breast carcinoma subtypes. Breast Cancer. 2021;28:496–505.
Anwar T, Arellano-Garcia C, Ropa J, Chen YC, Kim HS, Yoon E, et al. p38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat Commun. 2018;9:2801.
Aoyama K, Oshima M, Koide S, Suzuki E, Mochizuki-Kashio M, Kato Y, et al. EZH1 targets bivalent genes to maintain self-renewing stem cells in EZH2-insufficient myelodysplastic syndrome. iScience. 2018;9:161–74.
Wassef M, Luscan A, Aflaki S, Zielinski D, Jansen P, Baymaz HI, et al. EZH1/2 function mostly within canonical PRC2 and exhibit proliferation-dependent redundancy that shapes mutational signatures in cancer. Proc Natl Acad Sci. 2019;116:6075–80.
Yamagishi M, Hori M, Fujikawa D, Ohsugi T, Honma D, Adachi N, et al. Targeting excessive EZH1 and EZH2 activities for abnormal histone methylation and transcription network in malignant lymphomas. Cell Rep. 2019;29:e7.
Liu H, Hilliard S, Kelly E, Chen CH, Saifudeen Z, El-Dahr SS. The polycomb proteins EZH1 and EZH2 co-regulate chromatin accessibility and nephron progenitor cell lifespan in mice. J Biol Chem. 2020;295:11542–58.
Li W, Bi C, Han Y, Tian T, Wang X, Bao H, et al. Targeting EZH1/2 induces cell cycle arrest and inhibits cell proliferation through reactivation of p57(CDKN1C) and TP53INP1 in mantle cell lymphoma. Cancer Biol Med. 2019;16:530–41.
Zeng Z, Yang Y, Wu H. MicroRNA-765 alleviates the malignant progression of breast cancer via interacting with EZH1. Am J Transl Res. 2019;11:4500–7.
Schümann FL, Groß E, Bauer M, Rohde C, Sandmann S, Terziev D, et al. Divergent effects of EZH1 and EZH2 protein expression on the prognosis of patients with T-cell lymphomas. Biomedicines. 2021;9.
Ping M, Wang S, Guo Y, Jia J. TRIM21 improves apatinib treatment in gastric cancer through suppressing EZH1 stability. Biochem Biophys Res Commun. 2022;586:177–84.
Jung CK, Kim Y, Jeon S, Jo K, Lee S, Bae JS. Clinical utility of EZH1 mutations in the diagnosis of follicular-patterned thyroid tumors. Hum Pathol. 2018;81:9–17.
Zhang Y, Li L, Lu KX, Yu LB, Meng J, Liu CY. LncRNA SNHG3 is responsible for the deterioration of colorectal carcinoma through regulating the miR-370-5p/EZH1 axis. Eur Rev Med Pharm Sci. 2021;25:6131–7.
Zhang Q, Deng X, Tang X, You Y, Mei M, Liu D, et al. MicroRNA-20a suppresses tumor proliferation and metastasis in hepatocellular carcinoma by directly targeting EZH1. Front Oncol. 2021;11:737986.
Dou L, Yan F, Pang J, Zheng D, Li D, Gao L, et al. Protein lysine 43 methylation by EZH1 promotes AML1-ETO transcriptional repression in leukemia. Nat Commun. 2019;10:5051.
Wang L, Liu N, Xue X, Zhou S. The effect of overexpression of the enhancer of zeste homolog 1 (EZH1) gene on aristolochic acid-induced injury in HK-2 human kidney proximal tubule cells in vitro. Med Sci Monit. 2019;25:801–10.
Li J, Fan X, Wang Q, Gong Y, Guo L. Long noncoding RNA PRNCR1 reduces renal epithelial cell apoptosis in cisplatin-induced AKI by regulating miR-182-5p/EZH1. Kidney Blood Press Res. 2021;46:162–72.
Zheng J, Chen L. Non-coding RNAs-EZH2 regulatory mechanisms in cervical cancer: The current state of knowledge. Biomed pharmacother. 2022;146:112123.
Zhang H, Liang H, Wu S, Zhang Y, Yu Z. MicroRNA-638 induces apoptosis and autophagy in human liver cancer cells by targeting enhancer of zeste homolog 2 (EZH2). Environ Toxicol Pharm. 2021;82:103559.
Paskeh MDA, Mehrabi A, Gholami MH, Zabolian A, Ranjbar E, Saleki H, et al. EZH2 as a new therapeutic target in brain tumors: molecular landscape, therapeutic targeting and future prospects. Biomed Pharmacother. 2022;146:112532.
Zhang S, Liao W, Wu Q, Huang X, Pan Z, Chen W, et al. LINC00152 upregulates ZEB1 expression and enhances epithelial-mesenchymal transition and oxaliplatin resistance in esophageal cancer by interacting with EZH2. Cancer Cell Int. 2020;20:569.
Shao X, Zhao T, Xi L, Zhang Y, He J, Zeng J, et al. LINC00565 promotes the progression of colorectal cancer by upregulating EZH2. Oncol Lett. 2021;21:53.
Li HS, Xu Y. Inhibition of EZH2 via the STAT3/HOTAIR signalling axis contributes to cell cycle arrest and apoptosis induced by polyphyllin I in human non-small cell lung cancer cells. Steroids 2020;164:108729.
Sawicka-Gutaj N, Shawkat S, Andrusiewicz M, Ziółkowska P, Czarnywojtek A, Gut P, et al. EZH2 and SMYD3 expression in papillary thyroid cancer. Oncol Lett. 2021;21:342.
Bai Z, Wu Y, Bai S, Yan Y, Kang H, Ma W, et al. Long non-coding RNA SNGH7 Is activated by SP1 and exerts oncogenic properties by interacting with EZH2 in ovarian cancer. J Cell Mol Med. 2020;24:7479–89.
Zheng XJ, Li W, Yi J, Liu JY, Ren LW, Zhu XM, et al. EZH2 regulates expression of FOXC1 by mediating H3K27me3 in breast cancers. Acta Pharm Sin. 2021;42:1171–9.
Gao B, Liu X, Li Z, Zhao L, Pan Y. Overexpression of EZH2/NSD2 histone methyltransferase axis predicts poor prognosis and accelerates tumor progression in triple-negative breast cancer. Front Oncol. 2020;10:600514.
Sasaki M, Sato Y. An immunohistochemical panel of insulin-like growth factor II mRNA-binding protein 3 (IMP3), enhancer of zeste homolog 2 (EZH2), and p53 is useful for a diagnosis in bile duct biopsy. Virchows Arch 2021;479:697–703.
Patil S, Steuber B, Kopp W, Kari V, Urbach L, Wang X, et al. EZH2 regulates pancreatic cancer subtype identity and tumor progression via transcriptional repression of GATA6. Cancer Res. 2020;80:4620–32.
Xu L, Meng X, Xu N, Fu W, Tan H, Zhang L, et al. Gambogenic acid inhibits fibroblast growth factor receptor signaling pathway in erlotinib-resistant non-small-cell lung cancer and suppresses patient-derived xenograft growth. Cell Death Dis. 2018;9:262–76.
Fan DC, Zhao YR, Qi H, Hou JX, Zhang TH. MiRNA-506 presents multiple tumor suppressor activities by targeting EZH2 in nasopharyngeal carcinoma. Auris Nasus Larynx. 2020;47:632–42.
Wen Y, Hou Y, Yi X, Sun S, Guo J, He X, et al. EZH2 activates CHK1 signaling to promote ovarian cancer chemoresistance by maintaining the properties of cancer stem cells. Theranostics 2021;11:1795–813.
Zhou J, Liu M, Sun H, Feng Y, Xu L, Chan AWH, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut 2018;67:931–44.
Song JY, Perry AM, Herrera AF, Chen L, Skrabek P, Nasr MR, et al. Double-hit signature with TP53 abnormalities predicts poor survival in patients with germinal center type diffuse large B-cell lymphoma treated with R-CHOP. Clin Cancer Res. 2021;27:1671–80.
Ma J, Zhang J, Weng YC, Wang JC. EZH2-mediated microRNA-139-5p regulates epithelial-mesenchymal transition and lymph node metastasis of pancreatic cancer. Mol Cells. 2018;41:868–80.
Chu W, Zhang X, Qi L, Fu Y, Wang P, Zhao W, et al. The EZH2-PHACTR2-AS1-Ribosome axis induces genomic instability and promotes growth and metastasis in breast cancer. Cancer Res. 2020;80:2737–50.
Wang X, Cao W, Zhang J, Yan M, Xu Q, Wu X, et al. A covalently bound inhibitor triggers EZH2 degradation through CHIP-mediated ubiquitination. Embo j. 2017;36:1243–60.
Gui T, Liu M, Yao B, Jiang H, Yang D, Li Q, et al. TCF3 is epigenetically silenced by EZH2 and DNMT3B and functions as a tumor suppressor in endometrial cancer. Cell Death Differ. 2021;28:3316–28.
Chakraborty AA, Nakamura E, Qi J, Creech A, Jaffe JD, Paulk J, et al. HIF activation causes synthetic lethality between the VHL tumor suppressor and the EZH1 histone methyltransferase. Sci Transl Med. 2017;9:eaal5272.
Tang B, Sun R, Wang D, Sheng H, Wei T, Wang L, et al. ZMYND8 preferentially binds phosphorylated EZH2 to promote a PRC2-dependent to -independent function switch in hypoxia-inducible factor-activated cancer. Proc Natl Acad Sci USA. 2021;118:e2019052118.
Schmidt A, Behrendt L, Eybe J, Warmann SW, Schleicher S, Fuchs J, et al. The effect of direct and indirect EZH2 inhibition in rhabdomyosarcoma cell lines. Cancers 2021;14:41–56.
De Martino M, Nicolau-Neto P, Ribeiro Pinto LF, Traverse-Glehen A, Bachy E, Gigantino V, et al. HMGA1 induces EZH2 overexpression in human B-cell lymphomas. Am J cancer Res. 2021;11:2174–87.
Han H, Wang S, Meng J, Lyu G, Ding G, Hu Y, et al. Long noncoding RNA PART1 restrains aggressive gastric cancer through the epigenetic silencing of PDGFB via the PLZF-mediated recruitment of EZH2. Oncogene 2020;39:6513–28.
Guan X, Deng H, Choi UL, Li Z, Yang Y, Zeng J, et al. EZH2 overexpression dampens tumor-suppressive signals via an EGR1 silencer to drive breast tumorigenesis. Oncogene 2020;39:7127–41.
Sanches JGP, Song B, Zhang Q, Cui X, Yabasin IB, Ntim M, et al. The role of KDM2B and EZH2 in regulating the stemness in colorectal cancer through the PI3K/AKT pathway. Front Oncol. 2021;11:637298–313.
Li Z, Wang D, Lu J, Huang B, Wang Y, Dong M, et al. Methylation of EZH2 by PRMT1 regulates its stability and promotes breast cancer metastasis. Cell Death Differ. 2020;27:3226–42.
You Z, Peng D, Cao Y, Zhu Y, Yin J, Zhang G, et al. P53 suppresses the progression of hepatocellular carcinoma via miR-15a by decreasing OGT expression and EZH2 stabilization. J Cell Mol Med. 2021;25:9168–82.
Yuan H, Han Y, Wang X, Li N, Liu Q, Yin Y, et al. SETD2 restricts prostate cancer metastasis by integrating EZH2 and AMPK signaling pathways. Cancer Cell. 2020;38:350–65.e7.
Wang L, Jin Q, Lee JE, Su IH, Ge K. Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc Natl Acad Sci USA. 2010;107:7317–22.
Jin B, Zhang P, Zou H, Ye H, Wang Y, Zhang J, et al. Verification of EZH2 as a druggable target in metastatic uveal melanoma. Mol Cancer. 2020;19:52–67.
Tao T, Chen M, Jiang R, Guan H, Huang Y, Su H, et al. Involvement of EZH2 in aerobic glycolysis of prostate cancer through miR-181b/HK2 axis. Oncol Rep. 2017;37:1430–6.
Guan X, Shi A, Zou Y, Sun M, Zhan Y, Dong Y, et al. EZH2-mediated microRNA-375 upregulation promotes progression of breast cancer via the inhibition of FOXO1 and the p53 signaling pathway. Front Genet. 2021;12:633756–70.
Wang L, Chen C, Song Z, Wang H, Ye M, Wang D, et al. EZH2 depletion potentiates MYC degradation inhibiting neuroblastoma and small cell carcinoma tumor formation. Nat Commun. 2022;13:12–28.
Duan D, Shang M, Han Y, Liu J, Liu J, Kong SH, et al. EZH2-CCF-cGAS axis promotes breast cancer metastasis. Int J Mol Sci. 2022;23:1788–804.
Khan H, Jia W, Yu Z, Zaib T, Feng J, Jiang Y, et al. Emodin succinyl ester inhibits malignant proliferation and migration of hepatocellular carcinoma by suppressing the interaction of AR and EZH2. Biomedicine Pharmacother. 2020;128:110244–52.
Wu L, Gong Y, Yan T, Zhang H. LINP1 promotes the progression of cervical cancer by scaffolding EZH2, LSD1, and DNMT1 to inhibit the expression of KLF2 and PRSS8. Biochem Cell Biol. 2020;98:591–9.
Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110:7922–7.
Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M, et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-cell lymphomas. J medicinal Chem. 2016;59:9928–41.
Honma D, Kanno O, Watanabe J, Kinoshita J, Hirasawa M, Nosaka E, et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017;108:2069–78.
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012;492:108–12.
Kagiyama Y, Fujita S, Shima Y, Yamagata K, Katsumoto T, Nakagawa M, et al. CDKN1C-mediated growth inhibition by an EZH1/2 dual inhibitor overcomes resistance of mantle cell lymphoma to ibrutinib. Cancer Sci. 2021;112:2314–24.
Fujita S, Honma D, Adachi N, Araki K, Takamatsu E, Katsumoto T, et al. Dual inhibition of EZH1/2 breaks the quiescence of leukemia stem cells in acute myeloid leukemia. Leukemia 2018;32:855–64.
Bradley WD, Arora S, Busby J, Balasubramanian S, Gehling VS, Nasveschuk CG, et al. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem Biol. 2014;21:1463–75.
Lu B, Shen X, Zhang L, Liu D, Zhang C, Cao J, et al. Discovery of EBI-2511: a highly potent and orally active EZH2 inhibitor for the treatment of non-hodgkin’s lymphoma. ACS Med Chem Lett. 2018;9:98–102.
Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of EZH2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA. 2012;109:21360–5.
Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6.
Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, et al. EPZ011989, a potent, orally-available EZH2 inhibitor with robust in vivo activity. ACS Med Chem Lett. 2015;6:491–5.
Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–6.
Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–92.
Ma A, Stratikopoulos E, Park KS, Wei J, Martin TC, Yang X, et al. Discovery of a first-in-class EZH2 selective degrader. Nat Chem Biol. 2020;16:214–22.
Kung PP, Rui E, Bergqvist S, Bingham P, Braganza J, Collins M, et al. Design and synthesis of pyridone-containing 3,4-dihydroisoquinoline-1(2H)-ones as a novel class of enhancer of zeste homolog 2 (EZH2) inhibitors. J medicinal Chem. 2016;59:8306–25.
Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8:1324–34.
Kulkarni RA, Bak DW, Wei D, Bergholtz SE, Briney CA, Shrimp JH, et al. A chemoproteomic portrait of the oncometabolite fumarate. Nat Chem Biol. 2019;15:391–400.
Zhou C, He A, Kang Q, Li C, Liu T, Cai W, et al. Development of a UPLC-MS/MS method for determination of a dual EZH1/2 inhibitor UNC1999 in rat plasma. Bioanalysis 2022;14:67–74.
Proudman D, Nellesen D, Gupta D, Adib D, Yang J, Mamlouk K. A matching-adjusted indirect comparison of single-arm trials in patients with relapsed or refractory follicular lymphoma who received at least two prior systemic treatments: tazemetostat was associated with a lower risk for safety outcomes versus the PI3-kinase inhibitors idelalisib, duvelisib, copanlisib, and umbralisib. Adv Ther. 2022;39:1678–96.
Kusakabe Y, Chiba T, Oshima M, Koide S, Rizq O, Aoyama K, et al. EZH1/2 inhibition augments the anti-tumor effects of sorafenib in hepatocellular carcinoma. Sci Rep. 2021;11:21396–410.
Koyen AE, Madden MZ, Park D, Minten EV, Kapoor-Vazirani P, Werner E, et al. EZH2 has a non-catalytic and PRC2-independent role in stabilizing DDB2 to promote nucleotide excision repair. Oncogene 2020;39:4798–813.
Rizq O, Mimura N, Oshima M, Saraya A, Koide S, Kato Y, et al. Dual inhibition of EZH2 and EZH1 sensitizes PRC2-dependent tumors to proteasome inhibition. Clin Cancer Res. 2017;23:4817–30.
Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 2018;23:3262–74.
Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, et al. The histone methyltransferase EZH2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 2017;20:854–67.
Acknowledgements
This study was supported by research funding from National Natural Science Foundation of China (No. 82073895, 82073864), Science and Technology Planning Project of Guangzhou (No. 202002030294), Basic and Applied Basic Research Foundation of Guangdong (No. 2021A1515220126, 2018B030311020). In addition, thanks are given to all contributors in this field.
Author information
Authors and Affiliations
Contributions
RA was responsible for writing and revising the paper. RA, Y-QL and Y-LL contributed to data collection. Specially, all TCGA data analysis was provided by Y-QL. ZL was responsible for reviewing and directing the writing of the article. All authors approved the final manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
An, R., Li, YQ., Lin, YL. et al. EZH1/2 as targets for cancer therapy. Cancer Gene Ther 30, 221–235 (2023). https://doi.org/10.1038/s41417-022-00555-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-022-00555-1
- Springer Nature America, Inc.