
Abstract
Mounting evidence suggests that lncRNAs can exert functions in cancer progression in multiple manners. In recent years, competing endogenous RNA (ceRNA) has been widely reported in human cancers as a lncRNA-dominant molecular pathway. The current study aimed at proving the role of lncRNA downregulated RNA in cancer (DRAIC) in breast cancer (BRCA) progression. To be specific, qRT-PCR assay was conducted to measure the expression of DRAIC and other downstream target genes. It was uncovered that DRAIC was expressed at a high level in BRCA cells. Functional analyses, including CCK-8, colony formation, and EdU assays demonstrated that DRAIC depletion suppressed BRCA cell proliferation. In addition, cell apoptosis was promoted due to DRAIC knockdown. The inhibitory effect of DRAIC reduction on BRCA cell migration and invasion was proven by transwell assays. Mechanistically, DRAIC was confirmed to predominantly distribute in the cytoplasm and could interact with miR-432-5p. In addition, stem-loop binding protein (SLBP) was verified to be a downstream target of miR-432-5p and was positively regulated by DRAIC. Taken together, DRAIC sponged miR-432-5p to enhance SLBP expression, by which malignant behaviors of BRCA cells were promoted. Our findings may help to provide a promising therapeutic target for BRCA patients.
Similar content being viewed by others


Introduction
As one of the commonest cancers worldwide, breast cancer (BRCA) is primarily responsible for female cancer deaths [1, 2]. Five-year survival of BRCA patients ranges from 40 to 80% in different countries [3]. Prognosis of BRCA patients without severe metastasis can be good [4]. Once metastasis occurs, spread of BRCA cells can lead to death acceleration [5]. Therefore, detection and treatment of BRCA patients in the early stage is crucial for improving survival rate [6].
Non-coding RNAs are usually classified into two subtypes, which include long non-coding lncRNAs (lncRNAs) and microRNAs (miRNAs) [7]. LncRNAs consist of more than 200 nucleotides [8]. LncRNAs usually function during multiple biological processes [9]. In recent decades, more and more research suggests that lncRNAs have the powerful impact on tumor progression through various gene regulations [10, 11]. LncRNA downregulated RNA in cancer (DRAIC) has been found to play a significant role in promoting nasopharyngeal carcinoma (NPC) [12] and esophageal cancer (EC) [13]. The potential association between DRAIC upregulation and BRCA has been proven [14]. However, detailed functions and underlying mechanism of DRAIC during BRCA development have not yet been explored.
Competing endogenous RNA (ceRNA) network has been reported to be crucial in BRCA progression [15]. In a ceRNA network, lncRNA can serve as a miRNA sponge and enhance mRNA expression. MiR-432-5p has been demonstrated to inhibit progression of various cancers including BRCA [16, 17]. However, the interaction between DRAIC and miR-432-5p remains to be explored. Stem-loop binding protein (SLBP) can facilitate cancer progression [18], but its upstream molecular mechanism still need to be investigated in BRCA. As a whole, this study focused on the role of a DRAIC-dominant ceRNA pathway in BRCA.
Materials and methods
Cell lines and cell culture
The cell lines used in this study, including normal mammary epithelial cell lines (MCF-10A and MCF-12A) and BRCA cell lines (MDA-MB-361, MCF-7, SKBR3, and MDA-MB-231) were purchased from Cell Resources Center of Chinese Academy of Science (Shanghai, China). MCF-10A and MCF-12A cell lines were maintained with the utilization of 100 ml MEGM kit (Lonza, CC-3150; Switzerland) supplemented with 100 ng/ml cholera toxin. MDA-MB-361 cell lines were cultured in Leibovitz medium (Gibco, Grand Island, NY, USA) with 20% fetal bovine serum (FBS). MCF-7 cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco) added with 10% FBS SKBR3 cell lines were cultured in DMEM with 10% FBS. MDA-MB-231 cell lines were cultured in Leibovitz medium (Gibco) containing 10% FBS. All the foregoing cell lines were incubated with atmosphere containing 5% CO2 at 37 °C.
Quantitative real-time PCR (qRT-PCR) assay
QRT-PCR assay was performed to detect expression level of DRAIC, SLBP, miR-432-5p and FOXP3, GAPDH and U6 were respectively used as the internal control of cytoplasm and nucleus. Total RNA was first extracted from the cell lines using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. M-MLV Reverse Transcriptase (Invitrogen) was used to reverse transcribe the extracted RNA to cDNA. Quantification of the RNA expression was made using SYBR Green Master Mix (Thermo Fisher Scientific, Rockford, IL, USA). 2−ΔΔCt method was further utilized for data calculation.
Cell transfection
Lipofectamine™ 3000 (Invitrogen) was used for cell transfection following the manufacturer’s direction. For knockdown of DRAIC, the short hairpin RNAs targeting DRAIC (namely, sh-DRAIC#1 and sh-DRAIC#2) and corresponding internal control (namely, sh-NC) were synthesized by RiboBio (Guangzhou, China). For overexpression or silencing of miR-432-5p, miR-432-5p mimics, miR-432-5p inhibitor and their corresponding controls (NC mimics and NC inhibitor) were synthesized by Genepharma (Shanghai, China). To overexpress SLBP (Gene ID: 7884) or FOXP3 (Gene ID: 50943), the whole sequence of them was cloned into pcDNA3.1 vector (Genepharma) to construct pcDNA3.1/SLBP and pcDNA3.1/FOXP3. Empty pcDNA3.1 vector was used as negative control (NC). Sequences used in this assay were listed in Supplementary Table 1.
Cell counting kit-8 (CCK-8) assay
CCK-8 kit (Beyotime, Shanghai, China) was utilized to evaluate cell viability based on the manufacturer’s protocol. The cell lines were first regularly seeded and incubated in 96-well plates in the atmosphere with 5% CO2 at 37 °C for 48 h. After CCK-8 solution was added into each well, the cells were then incubated for about 2 h at 37 °C. Optical density (OD) value was measured every 24 h using microplate reader (BioTek, Winooski, VT, USA) at 450 nm.
Colony formation assay
Cell lines transfected with indicated plasmids were seeded in six-well plates for about 2-week incubation. Afterwards, the cells were washed using PBS (Thermo Fisher Scientific), fixed in paraformaldehyde and stained with 0.5% crystal violet solution (Gibco). The number of colonies was manually calculated.
Ethynyl deoxyuridine (EdU) assay
Cell lines were seeded into each well of 96-well plates for incubation. And then, the cells were washed with PBS (Thermo Fisher Scientific) and fixed in paraformaldehyde. EdU and DAPI dye (Beyotime) were separately added into wells to stain the proliferative cells and nuclei. After cells were incubated for 30 min, cell proliferation condition was observed using microscope.
Western blot
Cells were lysed by RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA). The protein concentration was measured through the BCA Protein Assay kit (Pierce Biotechnology, Rockford, IL, USA). SDS-PAGE was used to isolate proteins. After the isolation, the proteins were transferred to PVDF membranes (Millipore, Bedford, MA, USA). The membranes were incubated with primary antibodies, including Bax (1:1000, ab32503, Abcam, Cambridge, MA, USA), Bcl-2 (1:2000, ab182858, Abcam), Cleaved caspase 3 (1:500, ab32042, Abcam), Total caspase 3 (1:2000, ab208161, Abcam), SLBP (1:1000, ab181972, Abcam), GAPDH (1:2000, ab9485, Abcam) at 4 °C overnight. Afterwards, the membranes were incubated with secondary antibody Goat Anti-Rat IgG H&L (HRP, 1:2500, ab7097, Abcam) in darkness for about 1 h. GAPDH was regarded as the internal control. The proteins were finally quantified via enhanced chemiluminescence detection kit (Roche, Basel, Switzerland). The original blots of proteins were displayed in Supplementary file 1.
Transwell assays
Transwell migration assay was performed to evaluate cell migratory capabilities. In the assay, the upper chamber was filled with serum-free medium while lower chamber was filled with FBS. The cells placed into the upper chamber were incubated for 24 h at 37 °C. After the incubation, the cells migrated into the lower chamber were fixed with paraformaldehyde and stained by crystal violet. Fluorescence microscope was used to observe stained cells. The steps in transwell invasion assay were similar to those in transwell migration assay, except that the upper transwell chamber was coated with Matrigel membrane (BD Biosciences, Franklin Lakes, NJ, USA).
Subcellular fractionation assay
Subcellular fractionation assay was carried out to validate the location of DRAIC in BRCA cells. In brief, PARIS Kit (Thermo Fisher Scientific) was applied to separate nucleus and cytoplasm according to the manufacturer’s instruction. U6 and GAPDH were separately used as the nuclear and cytoplasmic control.
Fluorescence in situ hybridization (FISH) assay
Cells were fixed with 4% paraformaldehyde for about 15 min. After being washed in PBS (Thermo Fisher), cells were cultured with 0.5% TritonX-100 at 4 °C for about 10 min. DRAIC-specific probe (synthesized by Ribobio, Guangzhou, China) and hybridization solution were added to incubate cells overnight. The nuclei of the cells were counterstained with Hoechst dye. The location of DRAIC in BRCA cells was observed under a microscope.
Luciferase reporter assay
The binding sites between DRAIC/SLBP and miR-432-5p were mutated via QuickChang site-directed mutagenesis kit (Agilent Technologies, USA). The DRAIC/SLBP sequences with wild-type (WT) and mutant (MUT) binding sites were cloned into pmirGLO vector (Promega, Madison, WI, USA). Next, DRAIC-WT/SLBP-WT or DRAIC-MUT/SLBP-MUT was co-transfected into BRAC cells with NC mimics or miR-432-5p mimics. Luciferase activity was then measured using the Dual-Luciferase Reporter Gene Assay Kit (Beyotime). Renilla luciferase was used as the internal control.
The DRAIC promoter containing binding sites with or without FOXP3 (DRAIC promoter-WT or DRAIC promoter-MUT) was cloned into pGL3 (Promega) vector and co-transfected into BRCA cells with pcDNA3.1 or pcDNA3.1/FOXP3. Luciferase activity was then measured using the Dual-Luciferase Reporter Gene Assay Kit (Beyotime).
RNA-binding protein immunoprecipitation (RIP) assay
RIP assay was conducted using RNA-binding protein immunoprecipitation kit (Millipore). The lysates were incubated with magnetic beads coated with anti-Ago2 (1:50, ab186733, Abcam) or anti-IgG (1:100, ab190475, Abcam) attached to the magnetic beads. The RNAs were then isolated through immunoprecipitation. Expression levels of the RNAs were finally detected via qRT-PCR.
Chromosome immunoprecipitation (ChIP) assay
ChIP assay was carried out using EZ ChIP Chromatin Immunoprecipitation Kit (Millipore). The cells were fixed in formaldehyde to build DNA cross-links. The DNA fragments were sonicated and incubated with primary antibody anti-FOXP3 (1:50, ab450, Abcam) and NC anti-IgG (1:100, ab190475, Abcam). The DNA fragments isolated from immunoprecipitation was then quantified using qRT-PCR.
Statistical analysis
GraphPad Prism V6.0 software (GraphPad) was used to analyze the statistics obtained from experiments repeated for at least three times. The results were expressed as mean ± standard deviation (SD). Differences between two groups were analyzed using Student’s t test. One-way analysis of variance (ANOVA) or two-way ANOVA was used for comparison among multiple groups. Data was regarded as statistically significant only when p < 0.05.
Results
DRAIC is highly expressed in BRCA cells
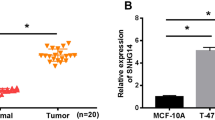
According to the pan-cancer data obtained from starBase (http://starbase.sysu.edu.cn/), expression level of DRAIC was increased in BRCA samples compared to normal breast samples (Fig. 1A). Similarly, data obtained from GEPIA 2 database (http://gepia2.cancer-pku.cn/#analysis) also showed that DRAIC was upregulated in BRCA samples (Fig. 1B). Through qRT-PCR analysis, we validated that DRAIC was upregulated in BRCA cell lines compared to normal mammary epithelial cell lines (Fig. 1C).

A Boxplot generated in starBase Pan-Cancer demonstrated that DRAIC was overexpressed in BRCA samples. Unpaired Student’s t test. B Boxplot generated from GEPIA 2 database showed the high level of DRAIC in BRCA samples (T meant tumor and N represented normal. GEPIA 2 BRCA data were collected from 1085 BRCA tumor tissues and 291 normal tissues.). Unpaired Student’s t test. C Expression levels of DRAIC in normal mammary epithelial cells and BRCA cells were validated by qRT-PCR assay (N = 3). One-way ANOVA. *P < 0.05, **P < 0.01.
DRAIC promotes proliferation, migration and invasion of BRCA cells
Since DRAIC had relative high expression level in SKBR3 and MDA-MB-231 cells in comparison to other BRCA cells, we designed loss-of-function assays in these two cell lines. At first, transfection efficiency of sh-DRAIC#1 and sh-DRAIC#2 was detected using qRT-PCR (Fig. 2A). After expression of DRAIC was suppressed, proliferation abilities of SKBR3 and MDA-MB-231 cells weakened according to the results of CCK-8 assay, colony formation assay, and EdU assay (Fig. 2B, C and D). In addition, apoptosis-related proteins were measured using western blot. The levels of pro-apoptosis proteins (Bax and Cleaved caspase 3) increased, but the level of anti-apoptosis protein Bcl-2 decreased after DRAIC knockdown (Fig. 2E). Transwell migration and invasion assays were conducted to assess migratory and invasive capabilities of SKBR3 and MDA-MB-231 cells. It turned out that the migratory and invasive capabilities of SKBR3 and MDA-MB-231 cells were weakened by the silencing of DRAIC (Fig. 2F).

A Transfection efficiency of sh-DRAIC#1 and sh-DRAIC#2 was confirmed using qRT-PCR (N = 3). One-way ANOVA. B CCK-8 assay detected the viability of cells with DRAIC silencing (N = 3). One-way ANOVA. C Colony formation assay was used to evaluate the proliferation abilities of DRAIC-silenced BRCA cells (N = 3). One-way ANOVA. D EdU assay measured cell proliferation abilities after DRAIC knockdown (N = 3). One-way ANOVA. Scale bar: 100 μm. E Apoptosis markers were measured using western blot assay after DRAIC silencing (N = 3). One-way ANOVA. F Transwell migration and invasion assays were conducted to estimate cell migratory and invasive capabilities (N = 3). One-way ANOVA. Scale bar: 200 μm. **P < 0.01.
DRAIC serves as miR-432-5p sponge in BRCA cells
Subcellular fractionation assay revealed that majority of DRAIC was distributed in the cytoplasm of SKBR3 and MDA-MB-231 cells (Fig. 3A), which was further proven by FISH assay (Fig. 3B). Cytoplasmic lncRNAs can act as ceRNAs in human cancers. Through searching on DIANA database (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2%2Findex-predicted) (selection condition: threshold value set as 0.9) and starBase database, miR-432-5p was singled out (Fig. 3C). The results of qRT-PCR illustrated that miR-432-5p was expressed at a significantly low level in MCF-7, SKBR3, and MDA-MB-231 cell lines (Fig. 3D). The binding sites between DRAIC and miR-432-5p were obtained and illustrated (Fig. 3E). Afterwards, miR-432-5p was overexpressed for subsequent analyses (Fig. 3F). Through luciferase reporter assay, we identified that the luciferase activity of WT DRAIC significantly reduced in response to the upregulation of miR-432-5p (Fig. 3G).

A Subcellular fractionation assay determined the cytoplasmic location of DRAIC in two BRCA cell lines (N = 3). B FISH assay was carried out to validate cytoplasmic location of DRAIC. Scale bar: 20 μm. C The putative miRNAs binding with DRAIC were searched from DIANA and starBase databases. D Expression levels of miR-432-5p in normal mammary epithelial cells and four BRCA cell lines were measured using qRT-PCR assay (N = 3). One-way ANOVA. E Binding sites between miR-432-5p and DRAIC were predicted via starBase. F Transfection efficiency of miR-432-5p mimics was confirmed by conducting qRT-PCR assay (N = 3). Unpaired Student’s t test. G Luciferase reporter assay was performed to verify that DRAIC sponged miR-432-5p (N = 3). Two-way ANOVA. *P < 0.05, **P < 0.01.
SLBP is a direct target of miR-432-5p in BRCA cells
To screen out the potential downstream targets of miR-432-5p, we applied three online bioinformatics tools (miRDB: http://www.mirdb.org/index.html), starBase: Pan-Cancer was set to ten cancer types and CLIP-Data was set to strict stringency ≥=5, TargetScan: http://www.targetscan.org/vert_72/). Four mRNAs were selected out for qRT-PCR analysis (Fig. 4A). Boxplots generated via GEPIA 2 database showed the expression of MRPL34 (Fig. S1A), SLBP (Fig. S1B), SNRPD2 (Fig. S1C) and MLLT6 (Fig. S1D) in BRCA tissues and normal tissues. It turned out that only SLBP and SNRPD2 were highly expressed in BRCA tissues. Importantly, silencing of DRAIC led to the significant downregulation of SLBP (Fig. 4B). Thus, we chose SLBP to be the research target. Two binding fragments between SLBP and miR-432-5p were predicted (Fig. 4C). After co-transfection, the luciferase activity of three vectors (SLBP-WT, SLBP-MUT1, SLBP-MUT2) were all reduced by the upregulation of miR-432-5p while that of SLBP-MUT1 + 2 had no obvious change (Fig. 4D), indicating that both two binding sequences were responsible for the interaction between SLBP and miR-432-5p. Next, the inhibitory efficiency of miR-432-5p inhibitor was identified by qRT-PCR (Fig. 4E). The mRNA and protein levels of SLBP decreased by DRAIC silencing were restored by the inhibition of miR-432-5p (Fig. 4F and G). The statistics of Ago2-RIP assay indicated that DRAIC, miR-432-5p, and SLBP all existed in the RISC complexes (Fig. 4H).

A MRNAs that could bind with miR-432-5p were predicted from miRDB, Targetscan, and starBase. B QRT-PCR detected expression of four candidate mRNAs in SKBR3 and MDA-MB-231 cells with DRAIC knockdown (N = 3). One-way ANOVA. C The predicted binding sites between SLBP and miR-432-5p were searched from starBase. D Luciferase reporter assay proved the interaction between SLBP and miR-432-5p (N = 3). Two-way ANOVA. E Transfection efficiency of miR-432-5p inhibitor was detected via qRT-PCR (N = 3). Unpaired Student’s t test. F QRT-PCR analyzed SLBP mRNA level in DRAIC-silenced cells after inhibition of miR-432-5p (N = 3). One-way ANOVA. G Western blot analyzed SLBP protein level in DRAIC-silenced cells after inhibition of miR-432-5p. H Ago2-RIP assay was conducted to verify the existence of DRAIC, miR-432-5p and SLBP in RISC (N = 3). Unpaired Student’s t test. *P < 0.05, **P < 0.01.
SLBP is involved in miR-432-5p-mediated BRCA cellular processes
According to the results of CCK-8 assay, colony formation assay, and EdU assay, the proliferation ability of SKBR3 cells suppressed by miR-432-5p overexpression was recovered after co-overexpression of SLBP (Fig. 5A, B and C). Meanwhile, miR-432-5p mimics-induced apoptosis was reversed by the upregulation of SLBP (Fig. 5D). Finally, the migratory and invasive capabilities of SKBR3 cells were discovered to decline after expression of miR-432-5p increased but this tendency was restored by the overexpression of SLBP (Fig. 5E).

SKBR3 cells were transfected with NC mimics, miR-432-5p mimics and miR-432-5p mimics+pcDNA3.1/SLBP for rescue assays. A CCK-8 assay detected cell viability (N = 3). One-way ANOVA. B Colony formation assay was used to evaluate colony formation ability (N = 3). One-way ANOVA. C EdU assay measured cell proliferation ability (N = 3). One-way ANOVA. Scale bar: 100 μm. D Apoptosis-related proteins were measured by means of western blot assay (N = 3). One-way ANOVA. E Transwell migration and invasion assays were conducted to estimate cell migratory and invasive capabilities (N = 3). One-way ANOVA. Scale bar: 20 μm. **P < 0.01.
DRAIC promotes BRCA cell growth by sponging miR-432-5p to upregulate SLBP
Transfection efficiency of pcDNA3.1/SLBP in SKBR3 cells was detected using qRT-PCR and western blot assays (Fig. 6A). Through CCK-8 assay, colony formation assay, and EdU assay, we observed that DRAIC knockdown-induced inhibition on cell proliferation was rescued by the inhibition of miR-432-5p or the upregulation of SLBP (Fig. 6B, C and D). Moreover, cell apoptosis induced by the silencing of DRAIC was reduced again by the knockdown of miR-432-5p or the upregulation of SLBP (Fig. 6E). Transwell assays revealed that silencing of miR-432-5p or overexpression of SLBP could abolish the effect of DRAIC silencing on cell migration and invasion (Fig. 6F).

A QRT-PCR and western blot assays detected transfection efficiency of pcDNA3.1/SLBP (N = 3). Unpaired Student’s t test. For rescue assays, SKBR3 cells were transfected with sh-NC, sh-DRAIC#1, sh-DRAIC#1+miR-432-5p inhibitor, sh-DRAIC#1 + pcDNA3.1/SLBP. B CCK-8 assay detected cell viability (N = 3). One-way ANOVA. C Colony formation assay was used to evaluate colony formation ability (N = 3). One-way ANOVA. D EdU assay measured cell proliferation ability (N = 3). One-way ANOVA. Scale bar: 100 μm. E Apoptosis-related proteins were detected in western blot (N = 3). One-way ANOVA. F Transwell migration and invasion assays were conducted to estimate cell migratory and invasive capabilities (N = 3). One-way ANOVA. Scale bar: 200 μm. **P < 0.01.
FOXP3 activates the transcription of DRAIC in BRCA cells
Searching from online databases UCSC (http://genome.ucsc.edu/) and PROMO (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3), we obtained several potential upstream regulators for DRAIC. Among which, ER-alpha (Fig. S2A), YY1 (Fig. S2B), and FOXP3 (Fig. S2C) were overexpressed in BRCA samples according to GEPIA 2 database. To determine the actual transcriptional regulator for DRAIC, we overexpressed ER-alpha, YY1, and FOXP3 separately in BRCA cells (Fig. 7A) and detected the expression of DRAIC. The results revealed that the level of DRAIC was obviously enhanced responding to FOXP3 overexpression (Fig. 7B). Enrichment of DRAIC promoter could be tested in anti-FOXP3 protein complexes through ChIP assay (Fig. 7C), which indicated the binding of FOXP3 to DRAIC promoter. The binding site predicted from JASPAR (http://jaspar.genereg.net/) was illustrated (Fig. 7D). After the SKBR3 and MDA-MB-231 cells were transfected with pcDNA3.1/FOXP3, the luciferase activity of WT DRAIC promoter was significantly raised while that of MUT DRAIC promoter was barely changed (Fig. 7E), suggesting FOXP3 activated DRAIC transcription through binding to its promoter on this site.

A Transfection efficiency of pcDNA3.1/ER-alpha, pcDNA3.1/YY1, and pcDNA3.1/FOXP3 was detected using qRT-PCR assay (N = 3). Unpaired Student’s t test. B DRAIC expression was tested after overexpression of ER-alpha, YY or FOXP3 (N = 3). One-way ANOVA. C ChIP assay was conducted to determine whether FOXP3 could combine with DRAIC promoter. Unpaired Student’s t test. D JASPAR predicted binding sites of FOXP3. E Luciferase reporter assay illustrated that FOXP3 could combine with DRAIC promoter (N = 3). Two-way ANOVA. **P < 0.01.
Discussion
BRCA is divided into several subtypes, which includes Luminal A, Luminal B, Her2-enriched, Basal-like, and Normal-like BRCA [19]. Differentially expressed genes can be useful for the diagnosis of different subtypes of BRCA [20]. Due to distant metastasis and high recurrence rate, surgery and chemotherapy are not very effective [21, 22]. Therefore, exploring more potential effective therapeutic targets is urgent [23].
LncRNAs have been widely reported in human cancers due to their oncogenic role in BRCA [24,25,26,27]. Upregulation of DRAIC has been proven to be correlated with adverse features of BRCA [14]. It can act as miR-122 sponge to facilitate NPC progression [12]. In this study, we determined that DRAIC was highly expressed in BRCA cells and promoted BRCA cell proliferation, migration and invasion but inhibited apoptosis.
In cytoplasm, lncRNAs usually exert functions in a ceRNA manner through sponging miRNAs to upregulate mRNAs [28, 29]. Therefore, expression of ceRNAs can be regulated to promote or suppress tumor development [30]. Similarly, this study revealed the cytoplasmic location of DRAIC and explored its underlying molecular mechanism. Based on bioinformatics analysis and mechanism experiments, we demonstrated that DRAIC could interact with miR-432-5p in BRCA cells. According to previous reports, miR-432-5p can act as a tumor suppressor in human cancers by interacting with lncRNAs [17, 31,32,33]. Here, we firstly uncovered the interaction between miR-432-5p and DRAIC in BRCA. Moreover, miR-432-5p could be involved in DRAIC-mediated BRCA cellular processes. SLBP can be targeted by miR-384 and promotes osteosarcoma metastasis [18]. To our knowledge, our study was the first to reveal the functions and mechanism of SLBP in BRCA. In this study, we verified that SLBP was a downstream target of miR-432-5p and was positively regulated by DRAIC in BRCA cells. More importantly, SLBP was involved in BRCA cellular processes mediated by DRAIC and miR-432-5p. At last, we investigated the upstream molecular mechanism of DRAIC in BRCA cells and identified that FOXP3 was a transcriptional activator for DRAIC.
Collectively, our current study firstly revealed the functions of DRAIC in BRCA and uncovered a novel ceRNA pathway, which may contribute to a promising therapeutic target for BRCA patients. Lack of animal study is a shortcoming of this study, we will establish animal model to prove the effect of DRAIC on in vivo tumor growth and metastasis. More importantly, we will conduct clinical study in our future study.
Conclusion
DRAIC is transcriptionally activated by FOXP3 and promotes BRCA progression via miR-432-5p/SLBP axis. Our findings may help to find a novel potential therapeutic target for BRCA patients.
References
Kolak A, Kamińska M, Sygit K, Budny A, Surdyka D, Kukiełka-Budny B, et al. Primary and secondary prevention of breast cancer. Ann Agric Environ Med. 2017;24:549–53.
Ganz PA, Goodwin PJ. Breast cancer survivorship: where are we today? Adv Exp Med Biol. 2015;862:1–8.
Coleman MP, Quaresma M, Berrino F, Lutz JM, De Angelis R, Capocaccia R, et al. Cancer survival in five continents: a worldwide population-based study (CONCORD). Lancet Oncol. 2008;9:730–56.
Merrill AY, White A, Howard-McNatt M. Paget’s Disease of the Breast: an Institutional Review and Surgical Management. Am Surg. 2017;83:e96–8.
Hunter KW, Crawford NP, Alsarraj J. Mechanisms of metastasis. Breast Cancer Res. 2008;10:S2. Suppl 1Suppl 1
Akram M, Iqbal M, Daniyal M, Khan AU. Awareness and current knowledge of breast cancer. Biol Res. 2017;50:33.
Wei JW, Huang K, Yang C, Kang CS. Non-coding RNAs as regulators in epigenetics (Review). Oncol Rep. 2017;37:3–9.
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21.
Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9.
Evans JR, Feng FY, Chinnaiyan AM. The bright side of dark matter: lncRNAs in cancer. J Clin Investig. 2016;126:2775–82.
Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46.
Liao B, Wang Z, Zhu Y, Wang M, Liu Y. Long noncoding RNA DRAIC acts as a microRNA-122 sponge to facilitate nasopharyngeal carcinoma cell proliferation, migration and invasion via regulating SATB1. Artif Cells Nanomed Biotechnol. 2019;47:3585–97.
Li F, Zhou X, Chen M, Fan W. Regulatory effect of LncRNA DRAIC/miR-149-5p/NFIB molecular network on autophagy of esophageal cancer cells and its biological behavior. Exp Mol Pathol. 2020;116:104491.
Zhao D, Dong JT. Upregulation of Long Non-Coding RNA DRAIC Correlates with Adverse Features of Breast Cancer. Non-coding RNA. 2018;4:39.
Sun M, Wu D, Zhou K, Li H, Gong X, Wei Q, et al. An eight-lncRNA signature predicts survival of breast cancer patients: a comprehensive study based on weighted gene co-expression network analysis and competing endogenous RNA network. Breast Cancer Res Treat. 2019;175:59–75.
Liu Y, Lu C, Zhou Y, Zhang Z, Sun L. Circular RNA hsa_circ_0008039 promotes breast cancer cell proliferation and migration by regulating miR-432-5p/E2F3 axis. Biochem Biophys Res Commun. 2018;502:358–63.
Yang G, Han B, Feng T. ZFAS1 knockdown inhibits viability and enhances cisplatin cytotoxicity by up-regulating miR-432-5p in glioma cells. Basic Clin Pharmacol Toxicol. 2019;125:518–26.
Wang Y, Huang H, Li Y. Knocking down miR-384 promotes growth and metastasis of osteosarcoma MG63 cells by targeting SLBP. Artificial cells. Nanomed Biotechnol. 2019;47:1458–65.
Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52.
Yeo SK, Guan JL. Breast Cancer: multiple subtypes within a tumor? Trends Cancer. 2017;3:753–60.
Zhang N, Cao C, Zhu Y, Liu P, Liu L, Lu K, et al. Primary breast lymphoma: a single center study. Oncol Lett. 2017;13:1014–8.
Zubor P, Kubatka P, Dankova Z, Gondova A, Kajo K, Hatok J, et al. miRNA in a multiomic context for diagnosis, treatment monitoring and personalized management of metastatic breast cancer. Future Oncol (Lond, Engl). 2018;14:1847–67.
Tang Y, Wang Y, Kiani MF, Wang B. Classification, Treatment Strategy, and Associated Drug Resistance in Breast Cancer. Clin Breast Cancer. 2016;16:335–43.
Zhao W, Geng D, Li S, Chen Z, Sun M. LncRNA HOTAIR influences cell growth, migration, invasion, and apoptosis via the miR-20a-5p/HMGA2 axis in breast cancer. Cancer Med. 2018;7:842–55.
Dong H, Hu J, Zou K, Ye M, Chen Y, Wu C, et al. Activation of LncRNA TINCR by H3K27 acetylation promotes Trastuzumab resistance and epithelial-mesenchymal transition by targeting MicroRNA-125b in breast Cancer. Mol Cancer. 2019;18:3.
Müller V, Oliveira-Ferrer L, Steinbach B, Pantel K, Schwarzenbach H. Interplay of lncRNA H19/miR-675 and lncRNA NEAT1/miR-204 in breast cancer. Mol Oncol. 2019;13:1137–49.
Xiu B, Chi Y, Liu L, Chi W, Zhang Q, Chen J, et al. LINC02273 drives breast cancer metastasis by epigenetically increasing AGR2. Transcription 2019;18:187.
Zhang K, Li Q, Kang X, Wang Y, Wang S. Identification and functional characterization of lncRNAs acting as ceRNA involved in the malignant progression of glioblastoma multiforme. Oncol Rep. 2016;36:2911–25.
Qi X, Zhang DH, Wu N, Xiao JH, Wang X, Ma W. ceRNA in cancer: possible functions and clinical implications. J Med Genet. 2015;52:710–8.
Ning S, Zhang J, Wang P, Zhi H, Wang J, Liu Y, et al. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2016;44:D980–5. D1
Xu T, Lei T, Li SQ, Mai EH, Ding FH, Niu B. DNAH17-AS1 promotes pancreatic carcinoma by increasing PPME1 expression via inhibition of miR-432-5p. World J Gastroenterol. 2020;26:1745–57.
Zhang J, Xu C, Gao Y, Wang Y, Ding Z, Zhang Y, et al. A Novel Long Non-coding RNA, MSTRG.51053.2 Regulates Cisplatin Resistance by Sponging the miR-432-5p in Non-small Cell Lung Cancer Cells. Front Oncol. 2020;10:215.
Hu C, Jiang R, Cheng Z, Lu Y, Gu L, Li H, et al. Ophiopogonin-B Suppresses Epithelial-mesenchymal Transition in Human Lung Adenocarcinoma Cells via the Linc00668/miR-432-5p/EMT Axis. J Cancer. 2019;10:2849–56.
Acknowledgements
We want to thank our laboratory staff for their contributions.
Funding
This study was supported by Construction of biological sample bank and clinical diagnosis and treatment information database of breast cancer, 2016YFC0901304.
Author information
Authors and Affiliations
Contributions
SL, HJ and ZZ designed the study. SL, HJ and DW contributed to implementation of experiments. ZZ worked out also all technical details. DW was responsible for the numerical calculations. ZZ and DW analyzed the data. All authors participated in the discussion of the results and contributed to the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, S., Jia, H., Zhang, Z. et al. DRAIC promotes growth of breast cancer by sponging miR-432-5p to upregulate SLBP. Cancer Gene Ther 29, 951–960 (2022). https://doi.org/10.1038/s41417-021-00388-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-021-00388-4
- Springer Nature America, Inc.
This article is cited by
-
The novel circFKBP8/miR-432-5p/E2F7 cascade functions as a regulatory network in breast cancer
Hereditas (2024)
-
Circ-TRIO promotes TNBC progression by regulating the miR-432-5p/CCDC58 axis
Cell Death & Disease (2022)