
Abstract
Epigenetic mRNA modification is an evolving field. N6-methyladenosine (m6A) is the most frequent internal transcriptional modification in eukaryotic messenger RNAs (mRNAs). This review will discuss the functions of the m6A mRNA machinery, including its “writers” that are components of the methyltransferase complex, its “readers” and its “erasers” (specifically FTO and ALKBH5) in cancer. The writers deposit the m6A and include METTL3, METTL14, WTAP, VIRMA, and RBM15. M6A methylation is removed by the m6A demethylases (FTO and ALKBH5). Lastly, the most diverse members are the readers that can contribute to mRNA splicing, stability, translation, and nuclear export. Many of these functions continue to be elucidated. The dysregulation of this machinery in various malignancies and the associated impact on tumorigenesis and drug response will be discussed herein with a focus on solid tumors. It is clear that, by contributing to either mRNA stability or translation, there are downstream targets that are impacted, contributing to cancer progression and the self-renewal ability of cancer stem cells.
Similar content being viewed by others
Introduction
In the past decade, there have been progressive studies demonstrating that mRNA modification occurs to impact RNA stability and translation, thus impacting the control of gene expression. The most common form of >170 RNA nucleotide modifications is N6-methyladenosine (m6A) [1, 2]. This modification is reversible [3, 4] and has been found to impact >7000 mRNAs in mammalian cell individual transcriptomes [5, 6]. There is additional data demonstrating that m6A modification in mRNAs or non-coding RNAs impact RNA translation and transcript fate/functions. These are critical for many physiologic processes, including the DNA damage response, tissue development (hematopoiesis and neurogenesis), circadian rhythm regulation, sex differentiation, microRNA processes, RNA–protein exchanges, and carcinogenesis [5,6,7,8,9,10,11,12,13,14,15,16,17].
Aberrant cell growth in tumorigenesis has historically been defined by abnormalities in cell division and gene expression as dictated by abnormalities in genetic and epigenetic changes. These abnormalities can be a function of genetic changes (e.g., gene mutations, deletions, amplification, or chromosomal translocations) and/or epigenetic changes, such as DNA or histone modification. In the past 10 years, RNA epitranscriptomics or gene regulation at the RNA level has gained more interest as an additional layer of influence in the development of malignancies. Of the various RNA modifications, m6A has been identified as a reversible RNA modification similar to the well-described histone and DNA modifications that are also reversible. With the development of high-throughput m6A sequencing techniques, there have been identified thousands of mRNA and non-coding RNA transcripts that are associated with m6A modifications with an additional enrichment in the 3’ untranslated regions in close proximity to the stop codons of mRNAs [5, 6].
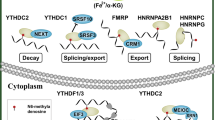
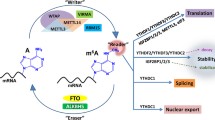
In the framework of mRNA m6A modification, there are methyltransferases and demethylases and in between there are proteins identified as “readers.” The readers can promote decay or enhance RNA stability, promote translation, and impact splicing and nuclear export of various target mRNAs [10,11,12, 18,19,20,21,22]. Therefore, the type of reader protein that recognizes the m6A modification of a given target mRNA can impact the stability of the target mRNA and can affect RNA translation, splicing, or nuclear transport (Fig. 1). This level of regulation—with the concept of mRNA “writers” (methyltransferases), “readers,” and “erasers” (demethylases)—is still a field in its infancy as it pertains to dysregulation in solid tumors.

The MTC is the m6A methyltransferase complex. The MTC is composed of WTAP, VIRMA, METL3, METTL14, and RM15. The MTC serves as methylase or “writer.” FTO and ALKBH5 are demethylases or “erasers.” The “readers” have a variety of functions, including translation (YTHDF1, YTHDF3, YTHDC2), decay (YTHDF2, YTHDF3, YTHDC2), splicing, nuclear export (YTHDC1), and stability (IGF2BP1/2/3).
m6A modification machinery
“Writers” such as methyltransferase-like 3 and 14 (METTL3 and METTL14, respectively) and their respective cofactors RBM15, Wilms tumor1-associated protein (WTAP), RBM15, and VIRMA (KIAA1429) make up the m6A methyltransferase complex (MTC). This grouping of proteins functions as the m6A writer and catalyzes the m6A modification [8, 23,24,25,26,27]. Our recent studies have further characterized how m6A is specifically deposited in the transcriptome. Huang et al. demonstrated that histone H3 trimethylation at Lys36 (H3K36me3), a marker for transcription elongation, guides m6A deposition co-transcriptionally [28]. The mechanism is that H3K36me3 is recognized and bound directly by METTL14, which as noted above is a crucial part of the MTC and thus facilitates binding of the m6A MTC to RNA polymerase II, thus delivering the m6A MTC to actively transcribing RNAs to deposit m6A co-transcriptionally. This work uncovers another layer of gene expression regulation involving the communication between histone modification and RNA methylation [28]. Weng et al. from our group demonstrated that METTL14 is highly expressed in normal hematopoietic stem/progenitor cells and acute myeloid leukemia (AML) cells carrying t(11q23), t(15;17), or t(8, 21) and is downregulated during normal myeloid differentiation [29]. Inhibiting METTL14 induces terminal myeloid differentiation in AML cells and inhibits AML cell survival and growth. The pro-oncogenic role of METTL14 in AML is by regulating its mRNA targets (e.g., MYB and MYC) via m6A modification [29].
Recognized demethylases or “erasers” are FTO (fat mass- and obesity-associated gene) and ALKBH5 [4, 9, 30]. These proteins function to remove the m6A modification from mRNA and create a counter balance to the “writers.”
The “readers” are a functionally more heterogeneous family of proteins with more diverse functionality. The YT521-B homology (YTH) domain family including YTHDC1, YTHDC2, YTHDF1, YTHDF2, and YTHDF3 are considered direct readers [10,11,12, 18,19,20]. YTHDF2, YTHDF3, and YTHDC2 promote degradation of their target mRNAs. YTHDF1, YTHDF3, and YTHDC2 promote translation, while YTHDC1 influences splicing and targets mRNA exportation. Our group has reported that insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs; including IGF2BP 1/2/3) are a distinct family of m6A readers [22]. These target many mRNA transcripts via identifying the consensus GG(m6A)C sequence [22]. In contrast to the mRNA-decay-promoting function of YTHDF2, IGF2BPs promote the stability and storage of their target mRNA (e.g., MYC) and also promote their translation in an m6A-dependent manner and therefore impact gene expression [22]. Additional readers include eukaryotic initiation factor 3 (eIF3) and cytoplasmic METTL3 [13, 31].
Recent work has intimated that antitumor immunity may be in part controlled through mRNA m6A methylation and the “reader” YTHDF1 in dendritic cells [32]. In the context of antitumor immunity, tumor regression often correlates to the neoantigen burden. Han et al. show that durable neoantigen-specific immunity is regulated by m6A methylation via the reader YTHDF1. They demonstrate that Ythdf1-deficient mice showed an amplified antigen-specific CD8+ T cell antitumor response. More specifically, the loss of YTHDF1 in classical dendritic cells amplified the cross-presentation of tumor antigens and the cross-priming of CD8+ T cells in vivo. Transcripts encoding lysosomal proteases are marked by m6A and identified by YTHDF1. The binding of YTHDF1 to these specific transcripts amplifies the translation of lysosomal cathepsins in dendritic cells and inhibition of the cathepsin increases cross-presentation of wild-type dendritic cells. In addition, the therapeutic efficacy of programmed death ligand 1 checkpoint blockade is much more effective in Ythdf1−/− mice, implying that YTHDF1 is a potential target that can amplify anticancer immunotherapy [32].
Aberrations in the m6A mRNA modification machinery have recently been associated with several malignancies (Table 1), including leukemia, glioblastoma, breast cancer, hepatocellular cancer (HCC), cervical cancer, lung cancer, and gastric cancer (GC) [29, 33,34,35,36]. There is also emerging data that targeting various aspects of this system will lead to novel therapies. The aim of this review is to highlight recent developments on the role of the m6A mRNA modification machinery as it pertains to the development, propagation, and treatment of solid tumors.
The oncogenic “ERASER” FTO: a role in liquid and solid tumors
Approximately 10 years ago, single-nucleotide polymorphisms (SNPs) in FTO were found to be strongly associated with obesity and body mass index (BMI) in humans as determined by genome-wide association studies [37, 38]. More recently, as the m6A mRNA modification proteins have been characterized, there has been more interest in FTO as a demethylase or “eraser.” FTO catalyzes the demethylation of 3-methyl-thymine in single-stranded DNA with Fe(II) and 2-oxoglutarate producing carbon dioxide, formaldehyde, and succinate [3]. Based on its jelly-roll motif protein folding structure, FTO functions with high affinity to m6A in mRNA whereby it functions as an efficient demethylase [4].
The direct causality of FTO and higher BMI or obesity has not been definitively elucidated. However, it is generally believed to be associated with a greater intake of calories perhaps secondary to FTO expression in the hypothalamus [39, 40]. As obesity is associated with several cancers, there are studies that correlate the connection between FTO and some obesity-associated cancers [41, 42]. One mechanism whereby the FTO gene is regulated is by DNA methylation. Hypomethylation of specific CpG sites in the FTO gene leads to increased FTO expression and this correlates with the presence of type 2 diabetes mellitus and some cancers [43].
Initial studies identified some FTO gene SNPs that were associated with the risk of certain cancers, including endometrial cancer and pancreatic cancer [44,45,46]. In breast cancer, SNPs in intron 1 of FTO including rs8047395, rs9939609, and rs7206790 have been identified as having important associations with development of this disease [47]. The precise mechanisms whereby these malignancy risk-associated SNPs in FTO remain to be elucidated.
Our group demonstrated that FTO facilitates oncogenesis in AML [48]. FTO targets genes such as ASB2 and RARA as a demethylase. FTO is overexpressed in certain subtypes of AML and promotes leukemogenesis and prevents all-trans-retinoic acid-induced leukemia cell differentiation. Thus FTO functions as an oncogene in this disease by inhibiting mRNA targets such as ASB2 and RARA by reducing their m6A levels and stability [48]. This work revealed a previously unidentified method of gene regulation in carcinogenesis and highlights the significance of the FTO gene and m6A mRNA demethylation in cancer.
Su et al. from our group has also demonstrated that high levels of FTO sensitize leukemic cells to the oncometabolite R-2-hydroxyglutarate (R-2HG) [34]. R-2HG is produced to relatively high levels by mutant isocitrate dehydrogenase1/2, which is found in 10–20% of AML patients [49]. R-2HG exerts antitumor activity via inhibition of leukemia cell proliferation/viability and induction of cell-cycle arrest and apoptosis. R-2HG inhibits FTO (demethylase) activity and therefore increases m6A mRNA modification. This in turn decreases the stability of MYC/CEBPA transcripts and thus suppresses relevant downstream pro-tumor pathways [34]. These mechanistic findings have been limited in solid tumors; however, there is emerging data of FTO and other members of the m6A mRNA modification machinery and the implications of tumorigenesis. Most recently, Huang et al. developed an effective FTO-specific inhibitor, namely, FB23–2, and showed that targeting FTO by small-molecule inhibitors such as FB23–2 can significantly inhibit AML cell viability/growth, promote apoptosis and inhibit AML progression in vivo [50]. Thus these studies provide proof-of-concept evidence suggesting that FTO is a druggable target and targeting FTO by effective inhibitors holds great therapeutic potential to treat FTO-overexpressing AML [34, 50].
In solid tumors such as melanoma, a recent report found that FTO regulates melanoma tumorigenecity and response to anti-programmed death 1 (anti-PD-1) blockade. FTO knockdown increased the m6A methylation of protumorigenic genes, including PD-1, C-X-C chemokine motif receptor 4, and SOX10 that lead to increased RNA decay via the reader YTHDF2. In addition, FTO knockdown sensitized melanoma to interferon gamma and subsequently to anti-PD-1 treatment in mice. This highlights the role of targeting FTO in melanoma to improve sensitivity to checkpoint inhibitors [51].
Glioblastoma
Glioblastoma is the most common and aggressive primary malignant brain tumor, and even with surgical resection, recurrence is common [52]. Cui et al. found that RNA m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells (GSCs) by the regulation of mRNA m6A enrichment and expression [53]. In glioblastoma, similar to in leukemia, R-2HG displays antitumor effects by inhibition of proliferation/survival in FTO-overexpressing cancer cells and targeting of the FTO/m6A/MYC/CEBPA signaling [34]. These pathways are important in cell proliferation and survival. This work demonstrated why it would be reasonable to target FTO in both glioblastoma and AML as noted above.
Zhang et al. queried The Cancer Genome Atlas (TCGA) to assess which components of the m6A machinery were associated with poor patient prognosis [54]. They found that the “eraser” ALKBH5 predicted poor prognosis in all data sets [54]. They showed that targeting ALKBH5 impaired self-renewal and decreased proliferation and tumorigenesis in GSCs. The downstream targets of ALKBH5 in GSCs were evaluated and FOXM1, a key transcription factor important in GSCs, was identified [54]. Mechanistically, ALKBH5 was found to demethylate FOXM1 nascent transcripts leading to increased expression and this may be an avenue to therapy for glioblastoma [54].
RNA methylation in GC
GC is the third most frequent cause of cancer-related mortality and is the fifth most common cancer in the world [55, 56]. The management for early disease is surgery with or without systemic therapy. Advanced disease is managed with chemotherapy and several targeted therapies as a function of tumor characteristics. Xu et al. demonstrated that, by immunohistochemistry and tissue microarray, FTO is markedly increased in GC tissues compared to adjacent non-tumor tissues (56% vs 38%) [57]. FTO expression was significantly associated with poor differentiation and lymph node metastases and positively correlated with worse stage. High FTO expression was also significantly associated with poor prognosis. Downregulation of FTO expression inhibited the proliferation, migration, and invasion of GC cell lines in vitro [57].
Mechanistically, Zhang et al. demonstrated that reduced m6A modification predicts malignant phenotypes and augmented Wnt/phosphoinositide 3-kinase (PI3K)-Akt signaling in GC [33]. Utilizing a proteomics-based GC cohort they had previously generated and the TCGA-GC cohort, they merged the expression of canonical m6A writers (METTL3/METTLE14), readers (YTHDF1/YTHDF2/YTHDF3), and erasers (ALKBH5/FTO), respectively, as W, R, and E signatures to represent the m6A modification. They stratified patients according to those signatures to decipher m6A’s associations with critical mutations, prognosis, and clinical indices. m6A’s biological function in GC was predicted by gene set enrichment analysis and validated via in vitro experiments. W and R were potential tumor-suppressive signatures and E was a potential oncogenic signature in GC. Based on W/R/E stratifications, patients with low m6A were associated with higher mutations of specific genes (CDH1, AR, GLI3, SETBP1, RHOA, MUC6, and TP53) and also demonstrated worse clinical outcomes [33]. Via in vitro experiments, they demonstrated that m6A suppression (as METTL14 knockdown) promoted cell proliferation and invasiveness via activating Wnt and PI3K-Akt signaling, while m6A elevation (i.e., FTO knockdown) reversed these changes [33]. In addition, the findings implied that m6A modification may be involved in interferon signaling and immune responses in GC. These data imply that targeting the “erasers” such as FTO or amplifying the “writers” such as METTL14 may be therapeutic avenues to pursue as it pertains to the m6A machinery in GC [33].
Writers and erasers in breast cancer
Breast cancer is the most prevalent malignancy in women. Although there has been great progress in this disease, the primary cause of mortality is secondary to distant metastases [58]. The population of tumor-initiating cells or breast cancer stem cells (BCSCs) have the capability of self-renewal [59]. The phenotype of these cells is characterized by the expression of several core pluripotency proteins, including Kruppel-like factor 4 (KLF4), Octamer-binding transcription factor 4 (OCT4), sex-determining region Y (SRY)-box 2 (SOX2), and NANOG [60,61,62,63,64]. In the context of metastatic disease, intratumor hypoxia leads to the expression of the transcription factor hypoxia-inducible factor (HIF)-1α [65]. There is recent work indicating that HIFs are necessary for the maintenance of BCSCs via transcriptional regulation of genes encoding the pluripotency-associated genes NANOG, SOX2, and KLF4 [66]. Pluripotency factors have been associated with changes in mRNA stability as dictated by m6A mRNA methylation. Zhang et al. have reported that exposure of breast cancer cells to hypoxia induces m6A demethylation and stabilization of NANOG mRNA, thus supporting the BCSC phenotype [67]. In addition, they showed that downregulating the expression of the ALKBH5 (coding a demethylase or eraser) or HIF-1s (which activate ALKBH5 gene transcription in hypoxic breast cancer cells) led to decreased NANOG expression and growth inhibition of BCSCs in vivo [67]. Further work is needed to assess whether competitive agonists of ALKBH5 may be useful as therapy that targets BCSCs.
In other work, Cai et al. demonstrated that METTL3 overexpression in breast cancer drives the progression of breast cancer via inhibiting tumor-suppressor let-7g [68]. Initial observations were that the overexpression of both METTL3 and the oncoprotein mammalian hepatitis B X-interacting protein (HBXIP) were associated with breast cancer [68,69,70]. Mechanistically, they were able to demonstrate that HBXIP modulates METTL3 by inhibiting miRNA let-7g, which downregulates the expression of METTL3. Interestingly, they found that METTL3 promoted the expression of HBXIP via m6A modification essentially creating a feedback loop [70]. These findings provided new insights into the mechanism of m6A mRNA modification in the progression of breast cancer and more work remains to be done in this field.
m6A machinery in the liver: non-alcoholic steatohepatitis (NASH) and HCC
NASH is a rising etiology of liver failure worldwide. With the rise of obesity, the prevalence of NASH continues to increase and also correlates with the incidence of HCC in this population [71]. At present, there are no Food and Drug Administration-approved medications for the treatment of NASH. NASH is histologically characterized by hepatocyte ballooning, inflammation, focal fibrosis, and steatosis [72]. It is further described as lipotoxicity in hepatocytes. There are limited treatment options. Lim et al. demonstrate that the expression of FTO is significantly increased in the livers of NASH patients as well as in a rodent model [72]. They demonstrated that genetic silencing of FTO protects against palmitate-induced oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis in vitro [72]. These results indicate that FTO overexpression may have a deleterious role in hepatocytes, and perhaps this contributes to the increased liver damage in NASH. Studies need to be done further to explore FTO targeting as a route to mitigate NASH and perhaps decrease the risk of HCC development in the future in these patients. In addition, it is not clear whether the lipotoxicity is inducing the FTO expression or FTO expression is facilitating lipotoxicity.
In liver cancer, it has been reported that the writer components function as a tumor suppressors. Ma et al. found that, in HCC, METTL14 and m6A levels were decreased relative to normal or paratumor controls with unchanged levels of the other writers METTL3 and WTAP [36]. In addition, they found that METTL14 expression correlated with poor prognosis. METTL14 knockdown facilitated HCC metastasis and overexpression-amplified tumor invasion and metastasis via m6A-dependent modulation of microRNA (i.e., mir-126) processing via interaction with DGCR8 [36]. In another study, Chen et al. found that METTL3 was actually overexpressed in HCC compared to normal control, with WTAP levels unchanged [73]. METTL3 overexpression in this cohort correlated with worse prognosis. Furthermore, they demonstrated that overexpression of METTL3 augmented growth of HCC both in vitro and in vivo while downregulation of METTL3 inhibited tumorigenesis and lung metastasis in vivo [73]. These findings were associated with negative regulation of SOCS2 expression by an m6A- and YTHDF2-dependent mechanism [73]. These data therefore imply that potential therapeutic targets in HCC development include FTO in the context of NASH and the writers METTL3 and METTL14 as they pertain to HCC growth and metastasis.
Although the focus of this review is primarily on m6A methylation of mRNA, there is developing data signifying the importance of m6A methylation of ribosomal RNA (rRNA). Recently, a new m6A methyltransferase, ZCCHC4, which functions to target human 28S rRNA for methylation, was identified [74]. When ZCCHC4 is knocked out, there is less global translation and cell proliferation. In HCC in particular, ZCCHC4 protein is overexpressed and knockdown of this gene allows for HCC tumor regression in xenograft models.
m6A mRNA methylation in endometrial cancer: an METTL14 hotspot
Sequencing studies have identified that 70% of endometrial tumors have reduced total m6A mRNA methylation compared to adjacent normal cells [75]. However, the functional significance was largely unknown. The specific METTL14 (R298P) mutation occurs at the RNA-binding groove and leads to inhibiting m6A mRNA methylation in tumors [75]. In addition, endometrial tumors have significantly reduced METTL3 m6A methyltransferase. It appears that these abnormalities are mutually exclusive [75]. Liu et al. have found that either METTL14 mutation or reduced expression of METTL3 led to increased proliferation and tumorigenicity of endometrial cancer cells via activation of the AKT pathway [75]. Similar to several other solid and liquid tumors, they conclude that change in m6A methylation is an oncogenic mechanism in endometrial cancer. A great deal needs to be studied about these mechanisms in order to develop relevant therapeutics for the treatment of endometrial cancer.
RNA modification in pancreatic cancer
Pancreatic ductal adenocarcinoma is a fatal malignancy with a 5-year survival of 9% [76]. He et al. found that ALKBH5 was downregulated in pancreatic cancer cells, in which a long non-coding RNA, KCNK15-AS1 is a direct target of ALKBH5 and thus is also downregulated; forced expression of ALKBH5 or KCNK15-AS1 could inhibit pancreatic cancer cell migration and invasion [77]. More recently, Zhang et al. reported that cigarette smoke condensate could cause hypomethylation in the METTL3 promoter region and thereby upregulate the expression of METTL3, which in turn promotes the maturation process of primary microRNA-25 (miR-25) in pancreatic duct epithelial cells. The excessive miR-25-3p maturation results in the activation of the oncogenic AKT-p70S6K signaling, which promotes malignant phenotypes of pancreatic cancer cells [76]. This study revealed a previously unappreciated link between cigarette smoke, m6A modification, microRNA maturation, and the pathogenesis of pancreatic cancer.
Colon cancer: METTLE3 is associated with tumor progression
The role of m6A methylation in colorectal cancer (CRC) remains largely unexplored. Li et al. have found via the TCGA that METTL3 expression correlated with poor prognosis in CRC [78]. METTL3 knockdown led to decreased CRC cell self-renewal, stem cell frequency, and migration in vitro and inhibited growth and metastases in vivo [78]. METTLE3 was also found to target SOX2. Mechanistically, they found that, when SOX2 transcripts were methylated, they were then recognized by a specific m6A reader, IGF2BP2, to prevent SOX2 mRNA degradations [78]. In addition, they found that the combination of “writer” METTL3, “reader” IGF2BP2 and “target” SOX2 correlated with better prognostic accuracy for CRC patients than each individual component [78, 79]. More work is needed to assess the efficacy of targeting components of this combination as a therapeutic strategy.
Conclusion
In summary, it is clear that the m6A mRNA machinery is an important mechanism in gene regulation and expression. In nearly all malignancies studied, there appears to be a role in contributing to cancer stem cell self-renewal. Targeting the various functions of “writers,” “readers,” and “erasers” is a field of great interest and the oncogenic roles of the m6A RNA methylation machinery needs to be further elucidated. There is a great deal to be learned by this novel epigenetic regulation at the RNA level. Development of effective and selective small-molecule compounds or other agents/tools targeting the dysregulated m6A machinery components is urgently needed as these may provide more effective novel therapies for cancer treatment.
References
Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46:D303–7.
Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361:1346–9.
Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013;29:108–15.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–46.
Deng X, Su R, Feng X, Wei M, Chen J. Role of N(6)-methyladenosine modification in cancer. Curr Opin Genet Dev. 2018;48:1–7.
Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16:191–8.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–99.
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115–27.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5’ UTR m(6)A promotes cap-independent translation. Cell. 2015;163:999–1010.
Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–5.
Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–4.
Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543:573–6.
Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, et al. m(6)A modulates haematopoietic stem and progenitor cell specification. Nature. 2017;549:273–6.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–19.
Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27:444–7.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27:315–28.
Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS. Regulation of m(6)A transcripts by the 3’->5’ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68:374.e12–87.e12.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3:1233–47.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–5.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89.
Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 2014;8:284–96.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–73.
Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, et al. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature. 2019;567:414–9.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22:191.e9–205.e9.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18:31–42.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62:335–45.
Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566:270–4.
Zhang C, Zhang M, Ge S, Huang W, Lin X, Gao J, et al. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 2019;8:4766–81.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172:90.e23–105.e23.
Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28:507–17.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary microRNA processing. Hepatology. 2017;65:529–43.
Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–6.
Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115.
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–94.
McTaggart JS, Lee S, Iberl M, Church C, Cox RD, Ashcroft FM. FTO is expressed in neurones throughout the brain and its expression is unaltered by fasting. PLoS ONE. 2011;6:e27968.
Kang Y, Liu F, Liu Y. Is FTO gene variant related to cancer risk independently of adiposity? An updated meta-analysis of 129,467 cases and 290,633 controls. Oncotarget. 2017;8:50987–96.
Deng X, Su R, Stanford S, Chen J. Critical enzymatic functions of FTO in obesity and cancer. Front Endocrinol. 2018;9:396.
Melnik BC. Milk: an epigenetic amplifier of FTO-mediated transcription? Implications for Western diseases. J Transl Med. 2015;13:385.
Delahanty RJ, Beeghly-Fadiel A, Xiang YB, Long J, Cai Q, Wen W, et al. Association of obesity-related genetic variants with endometrial cancer risk: a report from the Shanghai Endometrial Cancer Genetics Study. Am J. Epidemiol. 2011;174:1115–26.
Huang X, Zhao J, Yang M, Li M, Zheng J. Association between FTO gene polymorphism (rs9939609 T/A) and cancer risk: a meta-analysis. Eur J Cancer Care. 2017. https://doi.org/10.1111/ecc.12464.
Lurie G, Gaudet MM, Spurdle AB, Carney ME, Wilkens LR, Yang HP, et al. The obesity-associated polymorphisms FTO rs9939609 and MC4R rs17782313 and endometrial cancer risk in non-Hispanic white women. PLoS ONE. 2011;6:e16756.
Kaklamani V, Yi N, Sadim M, Siziopikou K, Zhang K, Xu Y, et al. The role of the fat mass and obesity associated gene (FTO) in breast cancer risk. BMC Med Genet. 2011;12:52.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. 2017;31:127–41.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 2019;35:677.e10–91.e10.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782.
Garcia CR, Slone SA, Dolecek TA, Huang B, Neltner JH, Villano JL. Primary central nervous system tumor treatment and survival in the United States, 2004–15. J Neurooncol. 2019;144:179–91.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–34.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606 e596.
McGuire S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv Nutr. 2016;7:418–9.
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
Xu D, Shao W, Jiang Y, Wang X, Liu Y, Liu X. FTO expression is associated with the occurrence of gastric cancer and prognosis. Oncol Rep. 2017;38:2285–92.
O’Shaughnessy J. Extending survival with chemotherapy in metastatic breast cancer. Oncologist. 2005;10(Suppl 3):20–9.
Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69:1302–13.
Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507.
Hu T, Liu S, Breiter DR, Wang F, Tang Y, Sun S. Octamer 4 small interfering RNA results in cancer stem cell-like cell apoptosis. Cancer Res. 2008;68:6533–40.
Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–72.
Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31:1354–65.
Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014;135:2741–8.
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408.
Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71:4640–52.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. 2016;113:E2047–56.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–9.
Wang Y, Cui M, Cai X, Sun B, Liu F, Zhang X, et al. The oncoprotein HBXIP up-regulates SCG3 through modulating E2F1 and miR-509-3p in hepatoma cells. Cancer Lett. 2014;352:169–78.
Yue L, Li L, Liu F, Hu N, Zhang W, Bai X, et al. The oncoprotein HBXIP activates transcriptional coregulatory protein LMO4 via Sp1 to promote proliferation of breast cancer cells. Carcinogenesis. 2013;34:927–35.
Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142:711.e6–25.e6.
Lim A, Zhou J, Sinha RA, Singh BK, Ghosh S, Lim KH, et al. Hepatic FTO expression is increased in NASH and its silencing attenuates palmitic acid-induced lipotoxicity. Biochem Biophys Res Commun. 2016;479:476–81.
Xu K, Yang Y, Feng GH, Sun BF, Chen JQ, Li YF, et al. Mettl3-mediated m(6)A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 2017;27:1100–14.
Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, et al. N(6-)methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15:88–94.
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20:1074–83.
Zhang J, Bai R, Li M, Ye H, Wu C, Wang C, et al. Excessive miR-25-3p maturation via N(6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun. 2019;10:1858.
He Y, Hu H, Wang Y, Yuan H, Lu Z, Wu P, et al. ALKBH5 inhibits pancreatic cancer motility by decreasing long non-coding RNA KCNK15-AS1 methylation. Cell Physiol Biochem. 2018;48:838–46.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18:112.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–70.
Acknowledgements
We apologize to colleagues whose work could not be included owing to space limitations.
Funding
This work was supported in part by the National Institute of Health (NIH) Grants R01 CA214965 (to JC), R01 CA236399 (to JC), R01 CA211614 (to JC), R56 DK120282 (to JC), as well as the Norman and Sadie Lee Grant City of Hope (to LM) and PanCan Translational Grant 2019 (to LM). JC is a Leukemia & Lymphoma Society (LLS) Scholar.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
JC has a patent filed based on his R-2HG/FTO work. JC is a scientific founder and the chief scientific officer of Genovel Biotech Corp. and also holds equity with this company.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Melstrom, L., Chen, J. RNA N6-methyladenosine modification in solid tumors: new therapeutic frontiers. Cancer Gene Ther 27, 625–633 (2020). https://doi.org/10.1038/s41417-020-0160-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-020-0160-4
- Springer Nature America, Inc.
This article is cited by
-
The ALKBH5/SOX4 axis promotes liver cancer stem cell properties via activating the SHH signaling pathway
Journal of Cancer Research and Clinical Oncology (2023)
-
The role of m6A modification in the biological functions and diseases
Signal Transduction and Targeted Therapy (2021)