
Abstract
Colorectal cancer (CRC) is one of the most common human malignancies. An increasing body of evidence has revealed the important roles long noncoding RNA (lncRNA) plays in the growth dynamics of CRC cells. In this study, we aimed to define the role of LINC00337 in the malignant phenotypes, especially angiogenesis, of CRC and clarify the underlying molecular basis. Bioinformatic analyses identified promoter region methylation of CNN1 in CRC, which was further validated by BSP and MSP assays. Loss- and gain- of function approaches were used to determine the roles of CNN1 and LINC00337 in vitro and in vivo. MTT-based method, Transwell migration/invasion assays, and tube formation assay were adopted to evaluate the cancer cell proliferation, migration/invasion, and proangiogenetic potency respectively in vitro. The tumor growth, microvascular density (MVD) and markers of proliferation (Ki67) and angiogenesis (VEGF) were quantified in nude mice xenografted with CRC cells. It was found that CNN1 downregulation and LINC00337 overexpression occurred in CRC tissues and cells. Besides, the CNN1 promoter region was hypermethylated in CRC. CNN1 overexpression or LINC00337 knockdown restricted CRC cell proliferation, migration/invasion, and proangiogenetic potency in vitro, which was substantiated by the in vivo experiments evidenced by facilitated tumor growth and MVD as well as elevated Ki67 and VEGF. Furthermore, our mechanistic evidence revealed that LINC00337 recruited DNMT1 to the promoter region of CNN1 and restricted the transcription of CNN1. Taken together, this study indicates that LINC00337 facilitates the tumorigenesis and angiogenesis in CRC via recruiting DNMT1 to inhibit the expression of CNN1.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC), one of the leading causes of cancer-related death in the world, accounts for 8% of all cancer deaths [1]. It has been estimated that more than 25% of patients with stage I-III and over 60% of patients at stage IV CRC suffer from recurrence after curative treatment [2]. Moreover, angiogenesis is held responsible for tumor growth and metastasis in CRC, and thus targeted antiangiogenic therapies have been an promising option for CRC treatment [3]. This urgent need has resulted in our focus on the inhibition of angiogenesis, and the knowledge of the pathogenesis of CRC and novel therapeutic targets may contribute to better selection of treatment approaches as well as reduced recurrence [4].
An increasing body of evidence has revealed the critical roles of long noncoding RNAs (lncRNAs) in human cancers [5] due to their modulatory effects on a wide variety of biological and/or pathological processes of human cancers [6]. For instance, lncRNA B4GALT1-AS1 has been demonstrated to promote osteosarcoma cell migration via recruiting RNA-binding protein HuR [7]. Of note, upregulation of lncRNA LINC00337 has been observed in gastric cancers and has been associated with poor clinical outcomes [8]. However, the underlying mechanism of LINC00337 in CRC has not been well developed.
Accumulating evidence has suggested that DNA methyltransferase 1 (DNMT1) functions as an oncogene, which exerts a suppressive effect on the tumor suppressor gene expression, thereby accelerating the tumor progression [9,10,11]. For instance, recruited by lncRNA LINC00337, DNMT1 has been shown to suppress TIMP2 to facilitate the progression of lung cancer [12]. However, whether LINC00337 also accelerated CRC development through recruiting DNMT1 has not been studied.
In this study, we explored the role of LINC00337 in CRC progression in vivo and in vitro and defined the mechanism by which LINC00337 accelerated the progression of CRC. Our experimental data suggested that LINC00337 facilitated the angiogenesis of CRC by suppressing CNN1 expression through recruiting DNMT1. This study furthered our understanding to the modulatory function of LINC00337 in CRC development and identified novel potential therapeutic targets for this disease.
Methods and materials
Bioinformatic analysis
The DNA methylation status of three colorectal tumor samples and corresponding paired normal colorectal tissues were analyzed by reduced representation bisulfite sequencing (RRBS). Differential expressed genes (DEGs) in CRC were analyzed using datasets obtained from Gene Expression Omnibus (GEO) database (GSE24514 and GSE32323). The downregulated DEGs and genes with hypermethylated promoter regions identified through RRBS were intersected to obtain overlapping genes. CRC-related transcriptome data were downloaded from the TCGA database using data transfer tools (provided by GDC Apps) (https://tcga-data.nci.nih.gov/). Among them, CRC-related RNA sequencing data contained 469 cancer samples and 41 normal samples, while CRC-related RNA sequencing data contained 166 cancer samples and 10 normal samples. The expression of CNN1 in CRC was assessed using Wilcox.test.
Patient sample collection
Patients with CRC who received surgeries between January 2016 and December 2018 were recruited in this study (n = 34), where cancer tissues and adjacent normal tissues were harvested. All tumor samples were classified based on the criteria of WHO Classification of Tumors of the Digestive System (4th Edition). Among the patients, there were 19 males and 15 females with the median age of 54.5 years old (33–72 years old).
Cell culture and treatment
Normal human colonic epithelial cell line (HCoEpiC) and CRC cell lines (HCT116, HT29, SW480, SW620, and Lovo), and human umbilical vein cells (HUVECs) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured based on the provided instructions. Briefly, cells were cultured in six-well plates at the concentration of 3 × 105 cells/well. When the cell confluency reached 70~80%, the cells were used for further experiments. HT29 cells at logarithmic growth phase were sub-cultured in six-well plate at the density of 4 × 105 cells/well. When the cell confluency reached 70–80%, HT29 cells were subjected to transfection using Lipofectamine 2000 (11668-019, Invitrogen, Carlsbad, CA) based on the protocols provided by the manufacturer. For each transfection reaction, 250 μl of serum-free Opti-MEM containing 10 μg plasmid (final concentration 50 nM) was mixed with same amount of serum-free Opti-MEM containing 5 μl Lipofectamine 2000. After cell culture for 24 h, the medium was replaced with fresh RPMI1640 medium containing 10% FBS and the cells were further cultured for 24–48 h for further studies.
According to the constructs virus received, the cells were divided into 10 groups: negative control group of overexpression (oe-NC), CNN1 overexpression group (oe-CNN1), negative control group of shRNA transduction (sh-NC), CNN1 knockdown group 1 (sh-CNN1-1), CNN1 knockdown group 2 (sh-CNN1-2), negative control of siRNA transfection (si-NC), LINC00337 knockdown group 1 (si-LINC00337-1), LINC00337 knockdown group 2 (si-LINC00337-2), cells transfected with siRNA targeting LINC00337 and shRNA negative control (si-LINC00337 + sh-NC), cells transfected with siRNA targeting LINC00337 and shRNA targeting CNN1 (si-LINC00337 + sh-CNN1). Vectors and siRNAs were purchased from GenePharma (Shanghai, China). Cells were transfected using Lipofectamine 2000 based on the manufacturer’s instruction.
HT29 cells were adopted to establish the stably lentivirus-transduced cell line for LINC00337 overexpression or CNN1 silencing. Plasmids used in this study were obtained from GenePharma.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissues or cells strictly adhering to protocols provided by TRIZOL reagent (Invitrogen), and RNA concentration was determined. Complementary DNA (cDNA) was synthesized using reverse transcription kit (K1622, Reanta, Beijing China) based on the protocols provided by the manufacturer. The samples were analyzed on a ViiA7 Real-Time PCR system (ViiA7, DAAN GENE, China) GAPDH was used as internal control and the relative expression of genes was calculated using 2-ΔΔCt method [13]. Primers for target genes were synthesized by Takara (Dalian, China) and listed in Table 1.
Western blot analysis
Proteins were extracted from tissues or cells using radio-Immunoprecipitation Assay (RIPA) lysis buffer (Beyotime Biotechnology, Shanghai, China) and the protein concentration of total cell lysate was determined using bicinchoninic acid (BCA) kit (20201ES76, Yeasen, Shanghai, China). The cell lysate was separated in SDS-PAGE gels and transferred onto PVDF membranes (IPVH85R, Millipore, Billerica, MA). The membranes were blocked in 5% bovine serum albumin (BSA) and immunoblots were probed with primary antibodies overnight at 4 °C: rabbit anti-CNN1 (ab46794, Abcam, Cambridge, UK; 1: 5000), rabbit anti-vascular endothelial growth factor (VEGF) (PA5-16754, Thermo Fisher, Austin, Texas, 1: 200). The next day, the membranes were subjected to incubation with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG) (ab205718, Abcam, 1: 20,000) for 1 h at room temperature. The blots were developed, and images were taken. The relative expression of proteins, normalized to β-actin, was represented by gray value ratio of protein bands measured Image J software (National Institutes of Health, Bethesda).
Fluorescent in situ hybridization (FISH)
Subcellular localization of LINC00337 and DNMT1 in HT29 cells were determined by FISH assay. Probes targeting LINC00337 and DNMT1 were prepared using RiboTM lncRNA FISH Probe Mix (Red) (RiboBio, Guangzhou, China) based on the provided instructions. HT29 cells at 80% confluency were fixed with 4% paraformaldehyde, and treated with protease K, glycine, and acetylation solution. The cells were then incubated with pre-hybridization solution for 1 h at 42 °C. After prehybridizaiton, the cells were subjected to incubation with 250 μl of hybridization solution supplemented with probes (300 ng/mL) at 42 °C overnight. The nucleus was stained with DAPI (1: 800) and sealed with antifade mounting medium for photography. Five randomly selected views were photographed under immunofluorescent microscope (Olympus, Tokyo, Japan).
Bisulfite PCR (BSP)
Cellular genomic DNA was isolated using Genomic DNA extraction kit (DP304, TIANGEN) and the isolated genomic DNA was treated with bisulfite to convert unmethylated cytosine to uracil using commercially available kit (ZYMO). The modified DNA was then purified and enriched using AxyPrep PCR clean-up kit (AXY-AP-PCR-250, Axygen). After that, bisulfite-modified promoter regions were amplified using specific primers (Forward primer: TGGGTATGGTGATTTATGTTTGTA; Reverse primer: ATCTCAACCCATCCAAATAATTAAA) for pyrosequencing. Amplified sequences were cloned into pease-t3 vector (TransGen, China) and sequenced using online tools (http://quma.cdb.riken.jp/) to determine the methylation status of CNN1 at the promoter region.
Methylation specific PCR (MSP)
Bisulfite-treated DNA was amplified using specific primers for methylated DNA (M-MSP) and unmethylated DNA (U-MSP), respectively by PCR after unmethylated cytosine was converted to uracil to determine the methylation of CNN1. The amplified DNA fragments were then subjected to 2% agarose gel electrophoresis and images were taken. The primers for Methylated-MSP and Unmethylated-MSP are listed below: Methylated-MSP forward primer: ATTGTTGGTATTTGGAGATAAATTC; Methylated-MSP reverse primer: CACCTAAAAAAACAACCTACGTT; Unmethylated-MSP forward primer: TTGTTGGTATTTGGAGATAAATTTG; Unmethylated-MSP reverse primer: TTCCACCTAAAAAAACAACCTACAT.
Chromatin immunoprecipitation (ChIP)
The enrichment of DNMT1, DNMT3a, and DNMT3b at the promoter region of CNN1 was evaluated by ChIP assay using EZ-Magna ChIPTM A kit (Millipore). DNA and protein in the neuronal cells were crosslinked by 1% formaldehyde. After centrifugation, the pelleted cells were then suspended in lysis buffer at the density of 2 × 106 cells/200 μl. Then, protease inhibitor cocktail was added into the lysis buffer and nucleus were pelleted by centrifugation (5000 × g, 5 min). Next, the DNA-protein complex was sheared into 200–1000 bp fragments by sonication. After centrifugation (14,000 × g, 10 min), the supernatant was then mixed with ChIP dilution buffer. The protein-DNA complex were then precleared by incubating with Protein G magnetic beads. From each sample, 20 μl supernatant was harvested as Input. Mouse anti-DNMT1 (ab13537, Abcam), rabbit anti-DNMT3a (ab2850, Abcam), rabbit anti-DNMT3b (ab2851, Abcam) and isotype IgG (rabbit IgG, ab172730, Abcam; mouse IgG, ab18413, Abcam) were added into supernatants. Meanwhile, 60 μl magnetic protein G beads were subjected to incubation for 2 h at 4 °C followed by centrifugation (700 × g, 1 min). The pelleted complexes were then washed with ChIP washing buffer and de-crosslinked before the DNA was purified and harvested. The interaction was investigated by RT-qPCR using specific primers detecting the promoter region of CNN1.
MTT-based method
Cells were seeded into 96-well plates at a density of 5 × 105 cells/well and cultured in 5% CO2 incubator at 37 °C. The cells were incubated with 5 mg/ml MTT (20 μl each well) in 5% CO2 incubator at 37 °C for 4 h. After that, the cells were incubated with dimethyl sulfoxide (DMSO) (200 μl) for 10 min to solubilize the formazan. Spectrophotometric absorbance of the samples at 570 nm was determined by microplate reader. The cell survival rate was calculated based on the formula: survival rate (%) = OD (570 nm) treated group/OD (570 nm) control group.
Transwell migration and invasion assays
Transwell migration and invasion assays were performed as previously described [14]. Transwell chambers (8-μm pore size; Corning, Midland, MI) were adopted in 24-well plates. The Transwell chamber polycarbonate membrane coated with Matrigel (invasion) or without Matrigel (migration). The lower chamber were preadded with 600 mL of 20% FBS RPMI1640 medium. Next, 48 h after transfection, HT29 cells (1 × 106 cells/ml) were seeded to the upper chamber, and incubated in 5% CO2 at 37 °C for 24 h. Transwell chamber were fixed with 5% glutaraldehyde. The cells were then stained with 0.1% crystal violet. Unmigrated cells or uninvaded cells were removed with cottons. Five randomly selected views under an inverted microscope (TE2000, Nikon, Tokyo, Japan) were photographed and analyzed.
Tube formation assay
Forty-eight hours after transfection, the cells were starved for 1 h, and single cell suspension was prepared using DMEM medium. The cells were seeded onto 96-well plates coated with Matrigel (356234, Shanranbio, Shanghai, China) at the density of 1 × 105 cells/ml. The cells were cultured for 18 h and then were photographed under an inverted phase contrast microscope (Leica, Weztlar, Germany). The number of capillary structures was counted by Image-Pro Plus (version 6.0) under at least three views.
Immunofluorescence colocalization
Subcellular localization of LINC00337 and DNMT1 in HT29 cells were determined by immunofluorescence colocalization method. Probes targeting LINC00337 and DNMT1 were prepared using RiboTM lncRNA FISH Probe Mix (Red) (RiboBio, Guangzhou, China). HT29 cells at 80% confluency were fixed with 4% paraformaldehyde, and treated with protease K, glycine, and acetylation solution. After prehybridizaiton, the cells were incubated with 250 μl of hybridization solution supplemented with probes (300 ng/mL) at 42 °C overnight. The nucleus was stained with DAPI, and the cells were sealed with antifade mounting medium for photography. Five randomly selected views were photographed under immunofluorescent microscope (Olympus, Tokyo, Japan).
RNA immunoprecipitation (RIP)
The interaction between lncRNA and DNMT1 was investigated by RIP assay using commercially available kit (Millipore). Briefly, cells were solubilized with RIPA lysis buffer (P0013B, Beyotime). For each binding system, 50 μl of magnetic beads were suspended in 100 μl RIP Wash buffer. Then, 5 μl antibodies were added into the binding system based on the experiment requirement. Beads-antibody complexes were then washed and suspended in 900 μl of RIP Wash Buffer. After that, 100 μl cell lysate was added for binding overnight at 4 °C. The next day, beads-protein complex were immobilized on a magnetic stand. Samples and Input were treated with protease K for RNA extraction, which were used for the detection of lncRNA by RT-qPCR. Antibodies used in this assay are listed as follows: mouse anti-DNMT1 antibody (ab13537, Abcam, 1: 100) or mouse IgG (ab18413, Abcam, 1: 100), which was used as negative control.
Human CRC xenograft in nude mice
Twenty-four BALB/c nude mice (3–5 weeks old, 10–12 g) housed in SPF animal laboratory were used for the establishment of xenograft model. The laboratory humidity was 40–60% and the temperature was kept at 24–26 °C. The laboratory was sterilized with UV periodically, and the cage, bedding, water, and food were sanitized by autoclave. HT29 cells were suspended in PBS and the cell density was adjusted to 1 × 106 cells/ml, and 50 μl of cell suspension was injected into the right flank area of each mice subcutaneously. One week after the cell xenografting, the mice were randomly divided into four groups (n = 6). Meanwhile, four different lentivirus (lv) constructs, lv-sh-NC, lv-sh-CNN1, lv-oe-NC, and lv-oe-LINC00337 were injected into the xenograft of mice. The subcutaneous tumors were measured every 5 days. Mice were euthanized after 30 days using pentobarbital (100 mg/kg, P3761, Sigma, St Louis, MO). Tumor tissues were harvested, and volume was calculated by the formula π(a2b)/6, where a and b are the tumor short and long diameters (in mm), respectively. Tumor volume in the mice was plotted against time and the final tumor weights were determined.
Immunohistochemical staining
Tumor tissue of the xenografts was subjected to paraformaldehyde fixation and paraffin embedding. Antigen retrieval was performed by boiling the sections in citrate buffer (0.01 M sodium citrate, pH 6.0) in a high-pressure container. The endogenous hydrogenase was quenched with 50 μl 3% hydrogen peroxide. After that, the sections were stained with rabbit antibodies against CNN1 (ab46794, Abcam) or Ki67 (ab15580, Abcam) at 4 °C overnight, and normal rabbit serum was added as NC. The next day, the sections were subjected to incubation with HRP-conjugated secondary antibody against rabbit IgG. The sections were then visualized with diaminobenzidine (DAB) solution and nucleus were counterstained with hematoxylin. After that the sections were dehydrated and sealed with mounting media and were subjected to observation under microscope (CKX53, OLYMPUS).
Evaluation of microvascular density (MVD) by immunohistochemistry
For MVD evaluation in the xenografts was evaluated by immunohistochemical staining. The sections were stained with MVD IgG. The determination of microvascular density based on MVD was performed as previously described [15]. In brief, yellow/brown colored cells or areas that can be clearly distinguished from proximal microvessel or grands were defined as neovascular structure. The unconnected branch of the newly form microvessel was also considered as a neovascular structure. Five views were randomly selected on each slide for counting and the mean number was used to represent the MVD of each slide. MVD positive was defined as mean number ≥the threshold of MVD, and MVD negative was defined as mean number <MVD threshold (Note: MVD threshold refers to Weidner standard).
Statistical analysis
All data was analyzed by SPSS 21.0 software (IBM, Armonk, NY, US) and was presented as means ± standard deviation (SD) of at least three independent experiments. Data between two groups were compared using paired (tumor vs. paired adjacent normal tissue) or unpaired t-test. Comparisons among multiple groups were conducted using one-way analysis of variance (ANOVA) with Tukey’s post-hoc test. Comparisons over time were performed with repeated measures ANOVA followed by Bonferroni post-hoc test. The correlation between two factors was analyzed by Pearson analysis. A value of p < 0.05 was considered as statistically significant.
Results
In colorectal tumor, CNN1 is poorly expressed and its promoter region is hypermethylated
The DNA methylation of three colorectal tumor and the paired adjacent normal tissues were retrieved from Sequence Read Archive (SRA) database (SRR5311099, SRR5311100, SRR5311101, SRR5311102, SRR5311103, and SRR5311104). The differentially methylated regions (DMRs) within those tumor tissues and adjacent normal tissues were investigated using R language packages “methylkit” and “edmr” and were annotated the using the annotation information of genes and CpG islands (hg19). We obtained 565 (difference = 25, q value = 0.01) and 8285 (DMC. Q value = 0.05, num. DNMs = 1, num. CpG = 1, DMR.methdiff = 5) DMRs from R packages “methylkit” and “edmr” respectively. We then intersected these two sets and identified 444 common DMR (Fig. 1A). Meanwhile, the annotation of those common DMRs were obtained from R package “methylkit”. After that, the mRNA expression profile related to CRC was analyzed using datasets obtained from GEO database (GSE23514 and GSE32323) (cutoff line: |logFC | >2, adjustP value < 0.05) in order to determine the differential expressed mRNA in CRC. We noticed that, from the dataset GSE24514, 21 genes were upregulated, and 56 genes were downregulated (Fig. 1B, C). In addition, from the other dataset, there were 62 upregulated genes and 122 downregulated genes (Fig. 1D). We then overlapped the downregulated genes in GSE24514 and GSE32323 with genes obtained from DMRs, which identified that CNN1 was in the intersection (Fig. 1E). Meanwhile, the expression of CNN1 was downregulated in CRC and rectal cancer based on the results obtained from GEPIA database (Fig. 1F). With Wilcox.test, the expression of CNN1 in CRC and normal samples from TCGA database was detected. We found that CNN1 was indeed appreciably downregulated in CRC and colorectal adenocarcinoma (Fig. 1G). Moreover, results of RT-qPCR, western blot analysis, and immunohistochemistry identified the significant downregulation of CNN1 in CRC tissues relative to adjacent normal tissues (Fig. 1H–J). Importantly, downregulation of CNN1 has also been observed in CRC [16] and CNN1 has been shown to impair tumor metastasis [17]. Thus, it is possible that downregulation of CNN1 in CRC accelerated the cancer development, and the CNN1 downregulation is caused by hypermethylation at its promoter region. To test the speculation, the CpG island methylation status of CNN1 promoter region (promoter region: upstream 1500 bp, downstream 200 bp) was predicted using online tools (MethPrimer). The result indicated that 1 CpG island at CNN1 promoter region could be hypermethylated (Fig. 1K). Therefore, MSP and BSP analyses were performed to determine the CNN1 promoter region methylation status using three colorectal tumor tissues and adjacent normal tissues. In consistent with the result of bioinformatic analysis, the methylation at CNN1 promoter region was increased in colorectal tumors compared with that in paired adjacent normal tissues (Fig. 1L, M).

A Prediction of differentially methylated genes at promoter region. (left circle: differentially methylated genes predicted by R package “methylkit”; right circle: differentially methylated genes predicted by R package “edmr”; intersection: the intersected genes of differentially methylated genes; B Heatmap of mRNA expression profile of GSE24514; The x-axis represents the sample number, while the y-axis represents the mRNA. The tree diagram on the left represents the clustering of mRNA expression, while each small square in the figure represents the expression level in a sample, and the histogram in the upper right is the color scale. C Volcano plot of mRNA expression profile of GSE24514; green area: downregulated mRNA; red area: upregulated mRNA; black area: mRNA without significant change. D Volcano plot of mRNA expression profile of GSE32323; E Prediction of promoter hypermethylation-mediated downregulated genes; blue circle: downregulated genes identified in GSE24514; red circle: downregulated genes identified in GSE32323; green circle: gene with hypermethylated promoter region commonly predicted by R package “methylkit” and “edmr”, intersection represents the potential downregulated gene(s) caused by promoter hypermethylation; F Expression level of CNN1 in colon adenocarcinoma (COAD) (left) and rectal adenocarcinoma (READ) (right) analyzed by GEPIA, red represents the expression level of CNN1 in tumor, gray indicates the CNN1 expression in normal tissues; G The expression of CNN1 in colorectal adenocarcinoma and rectal adenocarcinoma in TCGA database. The expression in colorectal adenocarcinoma on the left side, rectal adenocarcinoma on the right side, tumor tissue in red and normal tissue in blue. H–J Result of RT-qPCR (n = 34) (H), western blot analysis (I), and immunohistochemistry (J) detecting the expression of CNN1 in CRC and adjacent normal tissues; K Online tool (MethPrimer) predicted potentially hypermethylated CpG islands (blue) within CNN1 promoter region; L, M. Results of MSP (L) and BSP (M) detecting methylation status of CNN1 promoter region using tumor tissues and paired adjacent normal colorectal tissues (n = 34). Data were presented as mean ± standard deviation. *p < 0.05 by paired t-test.
Overexpressing CNN1 suppresses CRC cell growth, mobility, and proangiogenetic potency in vitro
In order to elucidate the function of CNN1 in CRC, in vitro studies were conducted using colorectal epithelial cell lines. RT-qPCR and western blot analysis demonstrated that the expression of CNN1 in CRC cells (HCT116, HT29, SW480, SW620, and lovo) was appreciably lower than that in normal colonic epithelial cells(HCoEpiC) (Fig. 2A, B). Among, the expression of CNN1 was the lowest in HT29 cell line, so HT29 cells were selected for further studies. Transfection efficiency of sh-CNN1 and oe-CNN1 was evaluated by RT-qPCR and Western blot analysis, which validated upregulation of CNN1 in the presence of oe-CNN1 and downregulation in the presence of sh-CNN1. Besides, sh-CNN1-2 achieved optimum knockdown effect. Thus, sh-CNN1-2 was used for further analysis (Fig. 2C).

A, B Results of RT-qPCR (A) and western blot analysis (B) detecting the expression of CNN1 in normal colonic epithelial cells (HCoEpiC) and CRC cells (HCT116, HT29, SW480, SW620, and lovo); C Result of RT-qPCR and Western blot analysis evaluating the transfection efficiency of CNN1 in HT29 cells; D–F Results of MTT-based method (D), Transwell migration assay (E), and Transwell invasion assay (F) demonstrated that CNN1 overexpression restricted CRC cell viability, migration, and invasion while CNN1 knockdown accelerated those cellular activities in vitro. G Result of tube formation assay revealed that CNN1 restricted HUVEC-mediated tumor angiogenesis in vitro. Data were presented as mean ± standard deviation. Comparisons among multiple groups were conducted using one-way ANOVA with Tukey’s post-hoc test. Comparisons over time were performed with repeated measures ANOVA with Bonferroni post-hoc test. *p < 0.05 vs. HCoEpiC cells or oe-NC; #p < 0.05 vs. sh-NC. Cell experiment was repeated in triplicate.
Based on assessment on the malignant phenotypes of CRC cells by MTT-based method, Transwell migration and invasion assay, CNN1 overexpression suppressed the viability, migration, and invasion. However, knockdown of CNN1 resulted in opposite results (Fig. 2D–F). Moreover, we co-cultured HT29 cells with HUVECs and evaluated the angiogenetic potency of HUVECs by tube formation assay. We noticed that HT29 cells with CNN1 overexpression restricted the HUVEC-mediated angiogenesis, while those with CNN1 ablation resulted in enhanced angiogenesis (Fig. 2G). Coherently, CNN1 elevation suppresses CRC cell growth, mobility, and proangiogenetic potency.
Inhibiting CNN1 impairs the tumorigenesis and tumor angiogenesis in vivo
In order to investigate the role of CNN1 in the tumorigenesis and angiogenesis of CRC in vivo, mouse models xenografted with human CRC were established. CNN1 was knocked down in HT29 cells using lentivirus-based transduction system (lv-sh-CNN1 or lv-sh-NC), and the efficiency was validated by western blot in Fig. 3F. Thereafter, HT29 cells transduced with lv-sh-CNN1 or lv-sh-NC were delivered into mice subcutaneously. The mice were sacrificed 30 days after the xenografting. We observed that tumor formed by HT29 cells with CNN1 knockdown displayed higher growth rate (Fig. 3A) and increased tumor volume (Fig. 3B) and weight (Fig. 3C). Furthermore, immunohistochemistry pointed out that CNN1 silencing (Fig. 3D) appreciably elevated the expression of Ki67 in xenograft tumors (Fig. 3E), suggesting that CNN1 knockdown enhanced the tumorigenesis in vivo.

CNN1 was knocked down in HT29 cells using lentivirus-based transduction system (lv-sh-CNN1 or lv-sh-NC). Thereafter, HT29 cells transduced with different lentivirus vectors including lv-sh-CNN1 or lv-sh-NC were injected into nude mice subcutaneously. A Representative xenograft tumors obtained from each group. B Quantitation of tumor volume from indicated groups. C Quantitation of tumor weight from indicated groups. D Representative images and quantitative analysis of immunohistochemistry detecting the expression of CNN1 in colorectal xenografts from indicated groups. E Representative images and analyzed result of immunohistochemistry detecting the expression of Ki67 in colorectal xenografts from indicated groups. F Representative images and quantitative analysis of western blot assay detecting the expression of CNN1 and VEGF in tumors from indicated groups. G Microvascular density evaluation of CRC xenografts from indicated groups. Data were presented as mean ± standard deviation. Comparisons between two groups were conducted using unpaired t-test. Comparisons over time were performed with repeated measures ANOVA followed by Bonferroni post-hoc test. *p < 0.05 vs. Lv-sh-NC.
It is well documented that VEGF functions as a key player in tumor angiogenesis [18], so it is reasonable to speculate that CNN1-silenced CRC cells-regulated angiogenesis is VEGF dependent. Thus, we determined the expression of CNN1 and VEGF in xenograft tumors by western blot analysis and observed that CNN1 knockdown resulted in the elevation of VEGF protein level (Fig. 3F). Meanwhile, MVD analysis suggested that the MVD of tumors from mice injected with CNN1-silenced HT29 cells was appreciably enhanced, as reflected by more yellow/brown stained cells in the tumor tissue (Fig. 3G). Therefore, CNN1 knockdown restricts the tumorigenesis and tumor angiogenesis in vivo.
LINC00337 recruits DNMT1 to promote CNN1 promoter region methylation and inhibit CNN1 transcription
To better understand the mechanism by which CNN1 was downregulated in CRC, we further investigated the molecular activity by which CNN1 was regulated in CRC. As we have shown that CNN1 promoter region has been hypermethylated in CRC, and the DNMT family members, which are DNMT1, DNMT3a, and DNMT3b, have central function on the epigenetic gene regulation [19]. Therefore, we formed a hypothesis that DNMT family members may be involved in the transcriptional inhibition of CNN1 expression. ChIP assay demonstrated that the major DNMT family member recruited to CNN1 promoter region was DNMT1 (Fig. 4A). Further, we noticed that DNMT1 knockdown appreciably elevated the expression of CNN1 (Fig. 4B). Moreover, we predicted 188 CNN1 promoter binding motifs on DNMT1 via Blast analysis, and we presented the first three with the lowest E value (Fig. 4C). These findings indicated that DNMT1 might exert a modulatory effect on CNN1 expression in CRC.

A Result of ChIP assay evaluating the recruitment of DNMT1, DNTM3a, DNMT3b at CNN1 promoter region; B Expression of CNN1 in HT29 cells in response to DNMT1, DNMT3a, or DNMT3b knockdown; C Result of blast predicting the binding motif, which interacted with CNN1 promoter region, on DNMT1; the figure displays first three with lowest E values; D LINC00337 expression in CRC analyzed by GEPIA; E Subcellular localization of LINC00337 in CRC cell determined by FISH assay. F Result of RT-qPCR measuring the expression of LINC00337 in CRC tissues and paired adjacent normal tissues (n = 34); G Result of Pearson correlation analysis evaluating the relationship between the expression of LINC00337 and CNN1; H Result of immunofluorescence assay investigating the localization of LINC00337 and DNMT1; I Result of RIP assay detecting the interaction between LINC00337 and DNMT1; J Result of RT-qPCR measuring the expression of LINC00337 under indicated conditions; K Results of RT-qPCR and western blot analysis investigating the expression of CNN1 under indicated conditions; L Result of ChIP assay evaluating the recruitment of DNMT1 at CNN1 promoter region under indicated conditions; M Result of BSP assay detecting the methylation status of CNN1 promoter region. Data were presented as mean ± standard deviation. Comparisons between two groups were conducted using paired or unpaired t-test. Comparisons among multiple groups were conducted using one-way ANOVA with Tukey’s post-hoc test. The correlation between two factors was analyzed by Pearson analysis. *p < 0.05 vs. sh-NC, si-NC, adjacent normal tissues, IgG or si-LINC00337-2. Cell experiment was repeated in triplicate.
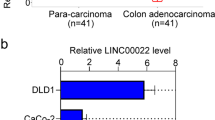
Accumulating evidence has suggested that LINC00337 suppresses gene transcription through recruiting DNMT1 to the promoter region of target genes [8, 12]. Thus, it is reasonable to postulate that DNMT1 mediates CNN1 suppression in a LINC00337-dependent manner in CRC. To test this speculation, we evaluated the expression level of LINC00337 in CRC in GEPIA database. The result suggested that the expression of LINC00337 was elevated in CRC (Fig. 4D). Moreover, we performed FISH assay to detect the subcellular localization of LINC00337 in CRC cells. The results showed that LINC00337 was mainly localized in the nucleus (Fig. 4E). Meanwhile, the result of RT-qPCR also validated the upregulation of LINC00337 in CRC tissues versus adjacent normal colorectal tissue (Fig. 4F). Furthermore, the Pearson correlation analysis revealed that the expression of LINC00337 was negatively related with that of CNN1 in CRC (Fig. 4G). These observations further highlighted the possibility that the hypermethylation of CNN1 promoter region was mediated by LINC00337-recruited DNMT1. We then confirmed the localization of LINC00337 and DNMT1 by immunofluorescence assay, which validated the prediction of LncATLAS website, suggesting the nuclear localization of LINC00337 (Fig. 4H). RIP assay result demonstrated that LINC00337 could be precipitated by DNMT1 (Fig. 4I).
In order to further identify the function of LINC00337 in the development of CRC, the expression of LINC00337 was altered in HT29 cells (Fig. 4J). The result indicated that compared with si-LINC00337-2, si-LINC00337-1 achieved higher knockdown efficiency. Thus, si-LINC00337-1 was used for further studies. We then measured the expression of CNN1 in LINC00337-silenced cells and control cells where LINC00337 expression was not changed. We noticed that LINC00337 silencing led to elevated CNN1 expression at both protein and mRNA levels (Fig. 4K). Meanwhile, the recruitment of DNMT1 at CNN1 promoter region was also evaluated and the result indicated that the recruitment of DNMT1 at CNN1 promoter region was impaired by LINC00337 knockdown (Fig. 4L). As expected, results of BSP analysis revealed that LINC00337 silencing mitigated the methylation status of CNN1 promoter region (Fig. 4M). These findings suggested that LINC00337 inhibits CNN1 transcription through recruiting DNMT1.
Silencing LINC00337 restricts CRC cell growth, mobility, and tube formation ability via upregulating CNN1 expression
Of note, our data have demonstrated that LINC00337 suppresses CNN1 expression in CRC cells. To evaluate whether LINC00337 regulates the activities of CRC cells via inhibiting CNN1, we silenced LINC00337 and/or CNN1 in HT29 cells and validated the knockdown efficiency via RT-qPCR and western blot analysis (Fig. 5A, B). The result of MTT-based method indicated that LINC00337 knockdown impaired the viability of HT29 cells while the trend was reversed by LINC00337- and CNN1-silenced HT29 cells (Fig. 5C). Moreover, LINC00337 knockdown restricted the CRC cell mobility as reflected by the reduced migrated and invaded HT29 cells, which was rescued by further CNN1 silencing (Fig. 5D, E, Supplementary Fig. 1a, b). Meanwhile, knockdown of LINC00337 restricted tube formation and then angiogenetic ability of co-cultured HUVECs, but further CNN1 knockdown rescued the tube formation and angiogenetic ability (Fig. 5F, Supplementary Fig. 1c). Taken together, LINC00337 knockdown inhibits CRC cell proliferation, migration, invasion, and tube formation ability in vitro.

A Result of RT-qPCR detecting the expression of CNN1 and LINC00337 in HT29 cells under indicated conditions; B Representative images of western blot analysis detecting the expression of CNN1 in HT29 cells under indicated conditions; C Result of MTT-based method evaluating the viability of HT29 cells under indicated conditions; D, E Quantitative data of Transwell migration assay (D) and Transwell invasion assay (E) investigating the mobility potency of HT29 cells under indicated conditions; F Quantitative data of tube formation assay evaluating the proangiogenetic potency of HUVECs under indicated conditions. Data were presented as mean ± standard deviation. Comparisons among multiple groups were conducted using one-way ANOVA with Tukey’s post-hoc test. Comparisons over time were performed with repeated measures ANOVA with Bonferroni post-hoc test. *p < 0.05 vs. si-NC; #p < 0.05 vs. si-LINC00337 + sh-NC. Cell experiment was repeated in triplicate.
LINC00337 promotes tumorigenesis in vivo by inhibiting CNN1 transcription
To evaluate the role of LINC00337 in the tumorigenesis of CRC in vivo, human CRC xenograft model was established. Mice were injected with HT29 cells transduced with lv-oe-NC or lv-oe-LINC00337 respectively, with LINC00337 overexpression validated by qRT-PCR in Fig. 6A. It was established that LINC00337 overexpression appreciably increased the tumorigenesis of CRC, as reflected by increased larger tumor volume (Fig. 6B), and higher tumor weight (Fig. 6C). Furthermore, LINC00337 overexpression appreciably diminished the expression of CNN1, indicating that LINC00337 suppressed the CNN1 expression at transcription level (Fig. 6D). Besides, we evaluated the methylation status of CNN1 promoter region and found that LINC00337 overexpression resulted in enhanced methylation at CNN1 promoter region (Fig. 6E). Western blot analysis confirmed that LINC00337 overexpression diminished protein expression of CNN1 but elevated that of VEGF in xenograft tumors (Fig. 6F). Moreover, immunohistochemistry results revealed that overexpressing LINC00337 facilitated the expression of Ki67 (Fig. 6G). Meanwhile, we evaluated the MVD in xenografts and observed that LINC00337 upregulation led to an increase in the neovascular structure in xenografts as reflected by increased MVD-positive structures (Fig. 6H). This observation suggested that LINC00337 accelerated the proangiogenetic potency of xenograft tumors of CRC cells, at least partially, by inhibiting the expression of CNN1 and enhancing the expression of VEGF.

A Result of RT-qPCR detecting the efficiency of lentiviral oe-LINC00337 transduction; B Quantitation of tumor volume from indicated groups; C Quantitation of tumor weight from indicated groups; D Result of RT-qPCR detecting the expression of CNN1 in response to lentiviral oe-LINC00337. E Result of BSP assay detecting the methylation of CNN1 promoter region. F Representative images and quantitative analysis of western blot assay detecting the expression of CNN1 and VEGF in tumors from indicated groups; G. Result of immunohistochemistry assay evaluating the expression of Ki67 in tumors from indicated groups; H. Result of MVD assay evaluating the MVD in tumors from indicated groups. Data were presented as mean ± standard deviation. Comparisons between two groups were conducted using unpaired t-test. Comparisons over time were performed with repeated measures ANOVA followed by Bonferroni post-hoc test. *p < 0.05. n = 6.
Discussion
CRC, with a high capacity for tumor metastasis, is one of the leading causes of cancer-related death in both men and women [20]. Despite the advanced achieved on treatment, majority of patients are often diagnosed at advanced stages resulting in bad clinical outcome of this malignancy [21]. Therefore, novel and effective therapeutic strategy is urgently needed, which requires better understanding to the pathogenesis of CRC. It is interesting that lncRNAs have been suggested to modulate gene expression associated with tumor etiology and development of variable malignancies, such as angiogenesis in CRC [22], which may offer clues for improved understanding of underlying molecular mechanisms. Our findings demonstrated that oncogenic LINC00337 recruited DNMT1 to promote CNN1 promoter region methylation and inhibit CNN1 transcription in CRC.
It has been well documented that CNN1 functions as a tumor suppressor [17] and it has been shown to be downregulated in tumors [23, 24]. The experimental observations obtained indicated corroborating findings that CNN1 was poorly expressed in colorectal tumor and its promoter region was hypermethylated. Our in vivo experiments also revealed that CNN1 knockdown induced not only higher growth rate, increased tumor volume and weight of tumor, but also accelerated proliferation and angiogenesis, as evidenced by elevated Ki67 and VEGF. Since VEGF is a pivotal modulator of angiogenesis in various malignancies, where oncogenes elevate its expression [25], inhibition of VEGF induced by CNN1 therefore halts angiogenesis and tumor growth in CRC.
Further mechanistic studies suggested that LINC00337 recruits DNMT1 to augment CNN1 promoter region methylation and inhibit CNN1 transcription. DNMT1 has been shown to be recruited by LINC00337 to the promoter region of metalloproteinase inhibitor (TIMP2) to suppress its expression [12]. In alignment with our study, most recent study has demonstrated that LINC00337 exerts an inhibitory effect on p21 expression by recruiting EZH2 at its promoter region [8]. Moreover, our results that LINC00337 knockdown suppressed the CRC proliferation, invasion, migration and proangiogenetic potency. Consistently, most recent studies have demonstrated that knockdown of LINC00337 impairs the growth and mobility of lung cancer cells and gastric cancer cells [12, 26]. Besides, we found that LINC00337 overexpression, by reducing CNN1, not only increased tumor volume and weight of tumor, but also accelerated proliferation and angiogenesis evidenced by elevated Ki67 and VEGF. Therefore, our future direction is to explore how LINC00337 is involved in mediating the cell–cell communication between CRC cells and endothelial cells.
In conclusion, our study demonstrates that LINC00337 recruits DNMT1 to the promoter region of CNN1. The LINC00337-recruited DNMT1 then methylates the CNN1 promoter region and inhibits its transcription, which consequently promotes the tumor growth and angiogenesis in CRC (Fig. 7). The current study has revealed the oncogenic role of LINC00337 in the malignant phenotypes of CRC and defined the underlying molecular mechanism by which LINC00337 facilitated the tumor growth and angiogenesis. More importantly, we have identified the critical molecules that play important roles in CRC cell growth, mobility, and proangiogenetic potency. These findings will contribute to the development of novel therapeutic approach. Besides, the regulation of CNN1 on VEGF is indeed worthy of further investigation. Further experiments are required to explore the modulatory mechanism of CNN1 on VEGF and large-scale cohorts should be performed to validate findings obtained in this study.

In CRC cells, LINC00337 recruits DNMT1 to the promoter region of CNN1. The LINC00337-recruited DNMT1 then methylates the CNN1 promoter region and inhibits its transcription, which consequently promotes tumor angiogenesis.
Data availability
The datasets generated/analysed during the current study are available.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29.
van der Stok EP, Spaander MCW, Grunhagen DJ, Verhoef C, Kuipers EJ. Surveillance after curative treatment for colorectal cancer. Nat Rev Clin Oncol. 2017;14:297–315.
Sun D, Zhang F, Qian J, Shen W, Fan H, Tan J, et al. 4’-hydroxywogonin inhibits colorectal cancer angiogenesis by disrupting PI3K/AKT signaling. Chem Biol Interact. 2018;296:26–33.
Wu C, Zhu XT, Xia L, Wang L, Yu W, Guo Q, et al. High expression of long noncoding RNA PCNA-AS1 promotes non-small-cell lung cancer cell proliferation and oncogenic activity via upregulating CCND1. J Cancer. 2020;11:1959–67.
Xiong T, Huang C, Li J, Yu S, Chen F, Zhang Z, et al. LncRNA NRON promotes the proliferation, metastasis and EMT process in bladder cancer. J Cancer. 2020;11:1751–60.
Wang H, Zhang Y, Yang H, Qin M, Ding X, Liu R, et al. In vivo SELEX of an inhibitory NSCLC-specific RNA aptamer from PEGylated RNA library. Mol Ther Nucleic Acids. 2018;10:187–98.
Li Z, Wang Y, Hu R, Xu R, Xu W. LncRNA B4GALT1-AS1 recruits HuR to promote osteosarcoma cells stemness and migration via enhancing YAP transcriptional activity. Cell Prolif. 2018;51:e12504.
Hu B, Wang X, Li L. Long noncoding RNA LINC00337 promote gastric cancer proliferation through repressing p21 mediated by EZH2. Am J Transl Res. 2019;11:3238–45.
Guo Y, Wang M, Jia X, Zhu H, Zhi Y, Yuan L. Wnt signaling pathway upregulates DNMT1 to trigger NHERF1 promoter hypermethylation in colon cancer. Oncol Rep. 2018;40:1165–73.
Seo JS, Choi YH, Moon JW, Kim HS, Park SH. Hinokitiol induces DNA demethylation via DNMT1 and UHRF1 inhibition in colon cancer cells. BMC Cell Biol. 2017;18:14.
Ning X, Shi Z, Liu X, Zhang A, Han L, Jiang K, et al. DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Lett. 2015;359:198–205.
Zhang X, Gong J, Lu J, Chen J, Zhou Y, Li T, et al. Long noncoding RNA LINC00337 accelerates the non-small-cell lung cancer progression through inhibiting TIMP2 by recruiting DNMT1. Am J Transl Res. 2019;11:6075–83.
Ayuk SM, Abrahamse H, Houreld NN. The role of photobiomodulation on gene expression of cell adhesion molecules in diabetic wounded fibroblasts in vitro. J Photochem Photobio B. 2016;161:368–74.
Wang S, Wu Y, Hou Y, Guan X, Castelvetere MP, Oblak JJ, et al. CXCR2 macromolecular complex in pancreatic cancer: a potential therapeutic target in tumor growth. Transl Oncol. 2013;6:216–25.
Takagi K, Takada T, Amano H, Yoshida M, Miura H, Toyota N, et al. Analysis of microvessels in pancreatic cancer: by light microscopy, confocal laser scan microscopy, and electron microscopy. J Hepatobiliary Pancreat Surg. 2008;15:384–90.
Drew JE, Farquharson AJ, Mayer CD, Vase HF, Coates PJ, Steele RJ, et al. Predictive gene signatures: molecular markers distinguishing colon adenomatous polyp and carcinoma. PLoS ONE. 2014;9:e113071.
Yamane T, Asanoma K, Kobayashi H, Liu G, Yagi H, Ohgami T, et al. Identification of the critical site of calponin 1 for suppression of ovarian cancer properties. Anticancer Res. 2015;35:5993–9.
Kim TK, Park CS, Jang J, Kim MR, Na HJ, Lee K, et al. Inhibition of VEGF-dependent angiogenesis and tumor angiogenesis by an optimized antibody targeting CLEC14a. Mol Oncol. 2018;12:356–72.
Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19:81–92.
Han G, Wei Z, Cui H, Zhang W, Wei X, Lu Z, et al. NUSAP1 gene silencing inhibits cell proliferation, migration and invasion through inhibiting DNMT1 gene expression in human colorectal cancer. Exp Cell Res. 2018;367:216–21.
Ji Q, Liu X, Han Z, Zhou L, Sui H, Yan L, et al. Resveratrol suppresses epithelial-to-mesenchymal transition in colorectal cancer through TGF-beta1/Smads signaling pathway mediated Snail/E-cadherin expression. BMC Cancer. 2015;15:97.
Jin KT, Yao JY, Fang XL, Di H, Ma YY. Roles of lncRNAs in cancer: focusing on angiogenesis. Life Sci. 2020;252:117647.
Wang Z, Li TE, Chen M, Pan JJ, Shen KW. miR-106b-5p contributes to the lung metastasis of breast cancer via targeting CNN1 and regulating Rho/ROCK1 pathway. Aging (Albany NY). 2020;12:1867–87.
Liu Y, Wu X, Wang G, Hu S, Zhang Y, Zhao SCALD1. CNN1, and TAGLN identified as potential prognostic molecular markers of bladder cancer by bioinformatics analysis. Med (Baltim). 2019;98:e13847.
Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005;69:4–10.
Navaneethan U, Zhu X, Parsi MA, Varadarajulu S. Pre-operative biliary drainage is associated with shortened survival time in patients with cholangiocarcinoma. Gastroenterol Rep. (Oxf). 2019;7:185–92.
Acknowledgements
We acknowledge and appreciate our colleagues for their valuable efforts and comments on this paper.
Author information
Authors and Affiliations
Contributions
XX and JN designed the study. LL and CD collated the data, FM and DS carried out data analyses and produced the initial draft of the manuscript. XX and JN contributed to drafting the manuscript. All authors have read and approved the final submitted manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was performed under the approval of the Ethics Committee of Linyi People’s Hospital and all participated patients have submitted written informed consents. All animal studies were accomplished under the protocol approved by the Institutional Animal Care and Use Committee of Linyi People’s Hospital.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Xu, X., Nie, J., Lu, L. et al. LINC00337 promotes tumor angiogenesis in colorectal cancer by recruiting DNMT1, which suppresses the expression of CNN1. Cancer Gene Ther 28, 1285–1297 (2021). https://doi.org/10.1038/s41417-020-00277-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-020-00277-2
- Springer Nature America, Inc.
This article is cited by
-
Genome-wide screening for differentially methylated long noncoding RNAs identifies LIFR-AS1 as an epigenetically regulated lncRNA that inhibits the progression of colorectal cancer
Clinical Epigenetics (2022)
-
DNMT1-induced miR-378a-3p silencing promotes angiogenesis via the NF-κB signaling pathway by targeting TRAF1 in hepatocellular carcinoma
Journal of Experimental & Clinical Cancer Research (2021)