
Abstract
Circular RNAs (circRNAs) are involved in the regulation of many pathophysiological processes as non-coding RNAs. This study focuses on the role of circRACGAP1 in the development of non-small cell lung cancer (NSCLC). Expression patterns of circRACGAP1 and miR-144-5p in NSCLC tissues and cell lines were quantified by qRT-PCR analysis. Then, the function of circRACGAP1 on cell proliferation and tumorigenesis were confirmed in vitro and in vivo using CCK-8 assay, colony formation, EdU incorporation, and xenograft technique. The regulation of circRACGAP1 on Gefitinib resistance of NSCLC cells was evaluated by flow cytometry. The regulatory network of circRACGAP1/miR-144-5p/CDKL1 was verified by luciferase reporter assay and RNA pull-down. Western blotting analysis was performed to assess the biomarkers of cell cycle and apoptosis-associated proteins. CircRACGAP1 was highly expressed and miR-144-5p was inhibited both in NSCLC tissues and cell lines, suggesting their negative correlation in NSCLC. Knockdown of circRACGAP1 suppressed cell proliferation via arresting the cell cycle. miR-144-5p was identified as a downstream target to reverse circRACGAP1-mediated cell proliferation. miR-144-5p directly targeted the 3′-UTR of CDKL1 to regulate cell cycle of NSCLC cells. circRACGAP1 knockdown dramatically inhibited the tumor growth and enhanced the sensitivity of NSCLC to Gefitinib in vitro and in vivo. In summary, our study revealed a novel machinery of circRACGAP1/miR-144-5p/CDKL1 for the NSCLC tumorigenesis and development, providing potential diagnostic and therapeutic targets for NSCLC.
Similar content being viewed by others


Introduction
Non-small cell lung cancer (NSCLC), as the most common type of lung cancer, causes the leading death worldwide [1,2,3]. Even the diagnostic and therapeutic strategies for NSCLC have been developed in recent years, the overall 5-year survival rate for NSCLC remains low and most of the NSCLC patients are identified at an advanced stage with metastasis [4,5,6]. Therefore, it is urgent and important to explore the molecular mechanisms related to the pathogenesis of NSCLC, which may provide new biomarkers and/or therapeutic targets to further improve the outcome of NSCLC treatment.
As a class of newly recognized functional non-coding RNAs (ncRNAs), circular RNAs (circRNAs) attracted significant attention in the last decades [7]. CircRNAs are generated from the back-splicing of precursor mRNA (pre-mRNA), from which a covalently closed loop structure was formed without termination of 5′ cap and 3′ poly (A) tails [8, 9]. Due to the unique structure, circRNAs are more stable and abundant in human cells than the linear RNAs. Several studies show that circRNAs are participating in several physiological and pathological processes, such as cancer development [10]. In most cases, circRNAs has a sponge effect to competitively bind with miRNA, recovering the regulation of the miRNA to its targeting mRNA [11]. On the other hand, circRNAs can directly control the target gene transcription by interacting with RNA-binding proteins (RNPs) [12]. In lung cancer, emerging studies have been conducted to investigate the roles of circRNAs on the cancer pathogenesis. For example, circ_0016760 was found as a therapeutic target to promote the cell proliferation, migration in vitro and in vivo [13]. Ma et al. discovered that has_circ_0020123 served as an oncogene to promote the development of NSCLC by suppressing miR-144 [14]. However, only a few circRNAs have been recognized as regulators in NSCLC so far, such as circ_0007382, circ_0014130, and cir_0013958 [15,16,17]. Therefore, the potential roles of novel circRNAs in NSCLC still need further investigation.
Recently, microarray analysis demonstrated that has_circRNA_101053 was significantly upregulated in the NSCLC tissues and cells compared with the adjacent normal tissues and cells. Has_circRNA_101053, which was also designated as circRACGAP1, was generated from the exon of RACGAP1 gene. RACGAP1 was overexpressed in NSCLC tissues, and knockdown of the RACGAP1 could block the cells in G1 phase, inducing cell death ultimately [18]. In a previous work, miR-144-5p was found to regulate the development of colorectal cancer through regulating cyclin-dependent kinase-like protein 1 (CDKL1). CDKL1 was an important member of cell division cycle gene 2-related serine/threonine protein kinase family, whose abnormal expression dysregulated cell cycle in malignant tumors [19]. However, the exact role of circRACGAP1 in the development of NSCLC remains elusive. Therefore, we proposed that circRACGAP1 might regulate the development of NSCLC through regulating miR-144-5p/CDKL1 signaling pathway, which might provide a potential diagnostic and therapeutic target for NSCLC.
Materials and methods
Cell lines and clinical sample
Five NSCLC cell lines, including A549, Calu-3, H1299, H1975, and PC-9, were cultured with RPMI 1640 media supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA, USA). The normal cell of human bronchial epithelial cell line (16HBE) was incubated in DMEM medium with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA, USA). These cell lines were purchased from the Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (Shanghai, China), and had been authenticated before use. Cells were tested without contamination with mycoplasma. Cells were cultured in a humidified 5% CO2 atmosphere at 37 °C.
NSCLC and normal tissues were collected from 30 patients with their signed statement of informed consent. No patients were treated prior to the surgery. The tissues specimens were stored at −80 °C in liquid nitrogen before usage. This study was approved by the Ethics Committee of the Second Xiangya Hospital of Central South University.
Plasmid and transfection
shRNAs for circRACGAP1 and miR-144-5p mimic/inhibitor were obtained from GenePharma (Shanghai, China). Lipofectamine 3000 transfection reagent (Invitrogen) was utilized to mediate these plasmids and RNA oligonucleotides transfect into the NSCLC cells, according to the manufacturer’s instructions.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was obtained from 30 NSCLC samples and adjacent normal tissues using the RNA-Pure kit (Tiangen, Beijing, China). Before reverse transcription of target circRNAs RNA, it was treated with RNAse R (Epicentre, Madison, WI) 30 min at 37 °C. RT-PCR was conducted using GoTaq@qPCR Master Mix (Promega) using SYBR Green (Invitrogen, Carlsbad, CA), which was performed on the StepOne Realtime PCR System (Applied Biosystems, Foster City, CA). The relative gene expression was determined and normalized to GAPDH by the 2−ΔΔct method. The primers used in the study were shown in Table 1.
Western blotting assay
In order to collect proteins from the tissues and cells, a NP-40 buffer (1.0% NP-40, 150 mM sodium chloride, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 50 mM Tris, pH 8.0) supplemented with 1 mM phenylmethylsulfonyl was introduced. Then, the protein lysates were separated on sodium dodecyl sulfate-polyacrylamide gel, followed by transferring to polyvinylidene difluoride membranes, which was blocked with 2% bovine serum albumin (BSA). The primary antibodies, including antibodies for CDKL1 (1:500; Abcam, Cambridge, MA, USA), Cyclin D1 (1:200; Abcam, Cambridge, MA, USA), CDK4 (1:1000; Abcam, Cambridge, MA, USA), Bcl-2 (1:1000; Abcam, Cambridge, MA, USA), Capspase-3 (1:500; Abcam, Cambridge, MA, USA), and Bax (1:1000; Abcam, Cambridge, MA, USA) were incubated followed by appropriate peroxidase-conjugated secondary antibodies (ZSGB-Bio, Beijing, China). GAPDH (1:2000, Santa Cruz) was used as normalized control. Immunoreactive bands were imaged and quantified with an ECL detection kit (Thermo Fisher Scientific, Waltham, MA).
CCK-8 assay
Cell proliferation was tested with the Cell Counting Kit-8 reagent (Beyotime, Beijing, China), according to the manufacturer’s instructions. In brief, cells were seeded in 96-well plates with a density of 3 × 103 cells/well and cultured in RPMI 1640 media supplemented with 10% FBS for a certain period of time. The CCK-8 reagent was added after different time intervals, and then incubated in a CO2 incubator for 1 h. The absorbance of each well was read at 450 nm on a microplate reader to determine the cell viability.
Colony formation assay
The cells after treatment were seed in a fresh 6-well plate with a density of 5 × 103 cells/well in a medium with 10 % FBS. The medium was exchanged every 3 days. At day 15, methanol was used to fix the cells, followed by the staining with 01% crystal violet. The visible colonies were counted, and each group was repeated at least three times.
5-Ethynyl-2′-deoxyuridine (EdU) incorporation assay
Incorporation of EdU into proliferating cells was detected using the Click-iT EdU Alexa Fluor 555 Imaging Kit (Invitrogen), according to the manufacturer’s instructions. In brief, cells were incubated with 10 μM EdU for 30 min and stained with the Click-iT EdU Alexa Fluor 555 Imaging Kit. Images were acquired by Olympus FV1000 confocal laser scanning microscope (Olympus Corporation, Japan).
Cell cycle assay by flow cytometry
The cells in 6-well plate (5 × 104 cells/well) were cultured for 48 h in the complete medium with 10% FBS. Then, the cells were harvested, washed, and fixed with 4% PFA. Before the analysis by FACScan flow cytometer (BD Bioscience, Rockville, MD), the cells were washed and stained with PI (BD Pharmingen) in the dark after incubation with 0.2% Triton X-100 in PBS with RNase A.
Cell apoptosis by flow cytometry
We collected the cells and then stained with Annexin V-FITC (BD Pharmingen, San Diego, CA) and PI (BD Pharmingen) according to the manufacturers’ instructions. Then, a FACScan flow cytometer (BD Bioscience, Rockville, MD) was used to analyze the cells in triplicate.
For cell cycle analysis, the transfected cells were harvested by trypsinization and washed with PBS twice. Before the staining with 20 μg/ml PI in PBS, cells were fixed with 1 ml of ice-cold ethanol (70% (v/v)) at 4 °C overnight. Then, the cells were treated with 0.1% (v/v) Triton X-100 in PBS, followed by the treatment with 200 μg/ml RNase A in PBS at 37 °C for 30 min. Finally, the cells were analyzed with flow cytometry.
Luciferase reporter assay
CDKL1 3′-UTR luciferase reporter (MT-h04084) was purchased from Applied Biological Materials (Richmond, Canada). After seeding H1299 and Calu-3 cells in 96-well plates (1 × 104 cells/well), miR-144-5p mimic or inhibitor (20 nM) and CDKL1 3′-UTR luciferase reporter (100 ng/well) together with the pRLTK Renilla reporter (Promega, Madison, WI) (5 ng/well) were co-transfected using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific). Dual-Glo Luciferase Assay System (Promega) was used to collect the luciferase reporter activity. Each data point was collected from at least triplicate experiment.
RNA pull-down with biotinylated circRACGAP1 probe
A biotinylated probe that can specifically bind with circRACGAP1 was designed and a random probe was used as a control. After the cells were harvested and lysed, the probe-coated beads, which was obtained by incubating the circRACGAP1 probe (Tsingke, Wuhan, China) with streptavidin magnetic beads (Life Technologies, USA), were incubated with cell lysates at 4 °C overnight. The beads were washed, and qRT-PCR assay was used to analyze the amount of the miRNAs that were extracted from the beads using Trizol reagent.
RNA pull-down assay based on MS2-MBP
To investigate the relationship between miR144-5p and CDKL1, we conducted the MS2-MBP based RNA pulldown assay. The MS2-MBP protein was obtained from E. coli, which contains three binding sites with the downstream of CDKL1. A CDKL1-MUT was generated by site-directed mutagenesis, and CDKL1-WT was used as a positive control. With the CDKL1-MUT-MS2 and CDKL1-WT-MS2 plasmids, the H1299 cells were transfected for 48 h before the harvest. The empty vector (MS2) was used as a control. The the cell lysates were followed with the RNA pulldown analysis similar to the previous section. The miR-144-5p associated with CDKL1 were quantified by qRT-PCR analysis.
Generation of xenografts
0.1 mL H1299 cells (2 × 106) in PBS were subcutaneously injected into six-week-old BALB/c female athymic nude mice (which were randomly divided into n = 6 per group, Vitalriver, Beijing, China) to generate xenografts, within the principle of random allocation. The investigator was blinded to the group allocation during the experiment. The volume of xenografts was recorded every 4 days (tumor volume = (length × width2)/2). After 32 days, and the tumor samples were collected for routine IHC after scarification. All the animal experiments were approved by the Animal Care and Use Committee of the Second Xiangya Hospital of Central South University, and all the mice were housed in a pathogen-free facility in the same University.
Immunohistochemistry (IHC) staining
Tissues were collected and fixed, followed by paraffin embedding and sectioning. The slides were subjected to deparaffinization, rehydration and antigen retrieval. After blocking, slides were then incubated with anti-CDKL1 (1:200, Sigma) at 4 °C overnight. The immunoreactivity was detected using labeled streptavidin biotin (LSAB) method.
Statistical analysis
Quantitative experiments were repeated at least more than three times and presented as the mean ± standard deviation (SD). All data were in a normal distribution, and variance was similar between the groups that are being statistically compared. In selected experiments, a two-tailed Student’s t-test was used for paired comparisons. For the comparison among multiple groups and multiple comparisons were analyzed by one-way analysis of variance (ANOVA). P < 0.05 was recognized as statistically significant differences (*p < 0.05, **p < 0.01, and ***p < 0.001).
Results
circRACGAP1 is upregulated in the NSCLC tissues and cell lines
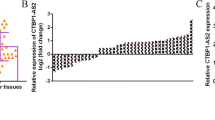
circRACGAP1, playing important role in the development of NSCLC, formed from the exon 10 of RACGAP1 pre-mRNA, as illustrated in the schematic diagram in Fig. 1a. In H1299 cells treated with the transcription inhibitor (actinomycin D), we found that the circular transcript circRACGAP1 was more stable than the linear mRNA transcript through the RNA stability assay (Fig. 1b). The expression of circRACGAP1 in NSCLC and adjacent normal tissues were measured by qRT-PCR analysis. The data showed that circRACGAP1 was significantly upregulated in the NSCLC tissues than that in the adjacent normal tissues (Fig. 1c). Furthermore, we found that the survival rate of NSCLC patients with lower expression of circRACGAP1 was significantly higher than those patients with higher expression of circRACGAP1, which was demonstrated by the prognosis analysis calculated by Kaplan–Meier and log-rank test (Fig. 1d). In addition, qRT-PCR analysis also showed that miR-144-5p was downregulated in the NSCLC tissues (Fig. 1e) compared with the normal tissues. The correlation analysis by Pearson correlation coefficient demonstrated that circRACGAP1 was negatively correlated with miR-144-5p in the NSCLC patient samples (Fig. 1f). Similarly, as shown in Fig. 1g, highly expressed circRACGAP1 and inhibited miR-144-5p were also observed in the five NSCLC cell lines, including A549, Calu-3, H1299, H1975, and PC-9, compared with 16HBE.

a Schematic diagram illustrated the generation of circRACGAP1 from RACGAP1 pre-mRNA (exon 9, 10, 11). b The expression of circRACGAP1 or linear circRACGAP1 mRNA transcripts in H1299 cells with the treatment of transcript inhibitor (actinomycin D) was determined by RNA stability assay. c The expression level of circRACGAP1 in NSCLC and adjacent normal tissues analyzed by qRT-PCR. d Prognosis analysis of the survival rate of NSCLC patients with high or low circRACGAP1 level in the NSCLC tissues, which was performed by Kaplan–Meier and log-rank test. e The expression level of miR-144-5p in NSCLC and adjacent normal tissues analyzed by qRT-PCR. f Pearson correlation showed the correlation between circRACGAP1 and miR-144-5p in the NSCLC patient samples. g The expression level of circRACGAP1 and miR-144-5p in four different NSCLC cell lines, including A549, H1299, PC-9, H1975 and normal 16HBE cell analyzed by qRT-PCR. The mean ± SD is presented for the relative levels from three replications. *p < 0.05, **p < 0.01, ***p < 0.001.
Knockdown of circRACGAP1 inhibits the NSCLC cell proliferation via blocking cell cycle
To explore the biological effects of circRACGAP1 in NSCLC cells, we investigated the cell viability, proliferation, and cell cycle after the circRACGAP1 was knockdown in Calu-3 and H1299 cells. We designed four shRNA for silencing circRACGAP1 and one of them is a control, designated as shRNA-NC. As shown in Fig. 2a, in both H1299 and Calu-3 cells, circRACGAP1-shRNA-1 displayed the most significant suppression on silencing circRACGAP1. Then, we compared the impact of transfection of sh-circRACGAP1-1 on the expression of circRACGAP1 and linear RACGAP1 mRNA. As shown in Fig. 2b, in both H1299 and Calu-3 cells, the expression of circRACGAP1 was dramatically inhibited by sh-circRACGAP1-1, but no impact was observed for the expression of linear RACGAP1 mRNA. The CCK-8 and colony formation assays showed that circRACGAP1 knockdown repressed the cell viability and decreased the numbers of clone formation in H1299 and Calu-3 cells (Fig. 2c, d). Similarly, the EdU incorporation assay also indicated the suppressive effect of circRACGAP1 knockdown on cell proliferation (Fig. 2e). In addition, we further analyzed the impact of circRACGAP1 on the cell cycle by flow cytometry (Fig. 2f). The results demonstrated that knockdown of circRACGAP1 caused the arrest of cell cycle at G1 phase. All these results implied the oncogenic role of circRACGAP1 in NSCLC progression via regulating cell proliferation and cell cycle.

a The expression of circRACGAP1 in H1299 and Calu-3 cells after silencing cricRACGAP1. b qRT-PCR showed the expression of circRACGAP1 and linear RACGAP1 in H1299 and Calu-3 cells after the transfection of sh-NC or sh-circRACGAP1. c The cell viability of H1299 and Calu-3 cells after different treatments measured by CCK-8 assay. d Colony formation of H1299 and Calu-3 cells was analyzed after different treatments. e The EdU incorporation assay was conducted in circRACGAP1 silenced H1299 and Calu-3 cells. f The cell cycle analysis of H1299 and Calu-3 cells after different treatments by flow cytometry. The mean ± SD is presented for the relative levels from three replications. *p < 0.05, **p < 0.01, ***p < 0.001.
circRACGAP1 acts as a sponge for miR-144-5p
In order to explore the regulatory mechanism of circRACGAP1, the potential target site of circRACGAP1 for miR-144-5p was predicted by the TargetScan & miRanda (Fig. 3a). Then, dual-luciferase assay was conducted to investigate the relationship between circRACGAP1 and miR-144-5p. In a dual-luciferase reporter assay, the wild-type circRACGAP1 3′ UTR fragment was inserted into the reporter vector. The results of luciferase reporter assay demonstrated that miR-144-5p mimic could decrease the luciferase activity of the reporter with the circRACGAP1 3′ UTR sequence, but maintained the activity in any group with mutated binding elements for miR-144-5p (Fig. 3b). Subsequently, we applied the biotin-labeled probe pull-down assay to consolidate the interaction between circRACGAP1 and miR-144-5p. The biotinylated probe was used to pull-down circRACGAP1 in H1299 and Calu-3 cell lines (Fig. 3c, d). Further, the level of miR-144-5p was detected and the results demonstrated that miR-144-5p was captured by circRACGAP1 in both H1299 and Calu-3 cell lines. However, silencing of circRACGA1 in these two cell lines significantly suppress the expression of miR-144-5p (Fig. 3e). Moreover, we carried out qRT-PCR assay and demonstrated that knockdown of circRACGAP1 notably increased the expression of miR-144-5p (Fig. 3f). Taken together, all of these results indicated that circRACGAP1 serves as a sponge of miR-144-5p.

a Predicted miR-144-5p binding site on circRACGAP1 sequence and mutation site map. b Luciferase activity in H1299 and Calu-3 cells when miR-144-5p mimics and circRACGAP1 wild-type/mutant luciferase reporter vectors were co-transfected. c, d CircRACGAP1 in the H1299 and Calu-3 cell lysates was pulled down and enriched with circRACGAP1 specific probe, and then analyzed by qRT-PCR. Relative level of circRACGAP1 was normalized to the input. GAPDH was a negative control. e qRT-PCR assay was performed to evaluate the relative levels of miR-144-5p. f The expression of miR-144-5p was assessed using qRT-PCR assay in H1299 and Calu-3 cells transfected with sh-circRACGAP1. The mean ± SD is presented for the relative levels from three replications. *p < 0.05, **p < 0.01, ***p < 0.001.
circRACGAP1 regulates the proliferation of NCSCL cells via targeting miR-144-5p
To investigate whether circRACGAP1 could regulate cell proliferation via targeting miR-144-5p, miR-144-5p inhibitor was applied. Using qRT-PCR assay, we found that the expression of miR-144-5p was significantly inhibited when the cells were co-treated with sh-circRACGAP1 and miR-144-5p inhibitor in both cell lines (Fig. 4a), compared with the cells treated with sh-circRACGAP1 alone. The CCK-8 assay indicated that the inhibition of cell viability induced by circRACGAP1 knockdown was markedly abolished by co-transfection with miR-144-5p inhibitor (Fig. 4b). The colony formation also suggested that knockdown of circRACGAP1 caused the decline of clone formations, while this suppressive effect was reversed by miR-144-5p inhibition (Fig. 4c). Consistently, the downregulation of circRACGAP1 significantly decreased the percentage of the EdU-positive H1299 and Calu-3 cells compared with that of control, and this effect was reversed by co-transfection of miR-144-5p inhibitor (Fig. 4d). Overall, these results demonstrated that miR-144-5p could reverse the effect of circRACGAP1 on the cell proliferation in NSCLC cells.

a The expression of miR-144-5p in H1299 and Calu-3 cells treated with sh-circRACGAP1 and miR-144-5p inhibitor by qRT-PCR. b CCK-8 assay showed the cell proliferation of H1299 and Calu-3 cells with different treatments. c The effects of circRACGAP1 and miR-144-5p on cell proliferation were determined by colony formation assay in H1299 and Calu-3 cells. d The EdU incorporation assay was performed in H1299 and Calu-3 cells with different treatments. The mean ± SD is presented for the relative levels from three replications. *p < 0.05, **p < 0.01, ***p < 0.001.
miR-144-5p directly targets CDKL1
To explore the function of miR-144-5p on cell cycle, we used dual-luciferase assay, flow cytometry and western blot to investigate the underlying molecular mechanism. Through the RAID predication, we found that CDKL1 might be the target of miR-144-5p in NSCLC cells (Fig. 5a), indicating by the binding site between miR-144-5p and 3′-UTR of CDKL1 mRNA at two positions (position 301-8 and position 1695-1701). Therefore, we used the dual-luciferase assay to confirm the interaction between miR-144-5p and CDKL1 at these two positions in H1299 and Calu-3 cells. As shown in Fig. 5b, miR-144-5p could significantly decrease luciferase activity when miR-144-5p mimic and CDKL1-3′-UTR-wt was co-transfected, while the luciferase activity did not change in the CDKL1-3′ UTR-Mut treatment group. This demonstrated that CDKL1 was the target unit of miR-144-5p in NSCLC cells. To further prove the interaction between miR-144-5p and CDKL1, MS2-containing CDKL1 (CDKL1-WT-MS2) constructs were transfected with H1299 cells. Then, RNA pulldown assay based on MS2-maltose-binding protein (MS2-MBP) was utilized to identify the miRNA binds with CDKL1. The analysis through qRT-PCR analysis showed precipitated miR-144-5p was significantly enriched in the CDKL1 WT than that in the group of CDKL1-MUT (Fig. 5c). This demonstrated that miR-144-5p was targeting CDKL1. Furthermore, the qRT-PCR analysis demonstrated that CDKL1 was overexpressed in the NSCLC tissues compared with the adjacent health tissues (Fig. 5d). Correlation analysis through the Spearman’s rank correlation coefficient indicated that miR-144-5p was negatively correlated with the CDKL1 in the NSCLC patient samples (Fig. 5e). Furthermore, the mRNA level and protein level of CDKL1 was significant enhanced in the five NSCLC cell lines (Fig. 5f). After transfecting H1299 and Calu-3 cells with miR-144-5p mimics and miR-144-5p inhibitors, the data of qRT-PCR analysis and western blot analysis verified the negative regulation of miR-144-5p on CDKL1 mRNA and protein levels (Fig. 5g, h). These results suggested that miR-144-5p targeted CDKL1 in NSCLC cells.

a Online bioinformatic prediction (http://starbase.sysu.edu.cn/) suggested the target of miR-144-5p was 3′-UTR of CDKL1 mRNA. b Luciferase activity in H1299 and Calu-3 cells when miR-144-5p mimic and CDKL1 wild-type/mutant luciferase reporter vector were co-transfected. c RNA pull-down assay based on MS2-MBP followed by miRNA qRT-PCR to analyze miR-144-5p endogenously associated with CDKL1. The schematic diagram on the left reveals the strategies of the experiment. d qRT-PCR showed the CDKL1 level in the NSCLC tissue. e Spearman’s rank correlation coefficient demonstrated the correlation between the miR-144-5p and CDKL1 in the NSCLC patient samples. f The expression level of CDKL1 in A549, H1299, H1975, PC-9 and normal cell line BEAS-2B analyzed by qRT-PCR and western blotting analysis. g, h The mRNA and protein expression level of CDKL1 in H1299 and Calu-3 cells with the transfection of miR-144-5p mimics or miR-144-5p inhibitor. The mean ± SD is presented for the relative levels from three replications. *p < 0.05, **p < 0.01, ***p < 0.001.
circRACGAP1 facilitates NSCLC cell proliferation through miR-144-5p/CDKL1
Next, we studied how the circRACGAP1 regulate the proliferation of NSCLC cells by targeting miR-144-5p/CDKL1 pathway. With the CCK-8 assay, we found that the cell proliferation rate was significantly reduced within circRACGAP1 knockdown, whereas, this effect could be reversed by either knockout of miR-144-5p or overexpression of CDKL1 (Fig. 6a). We also investigated the impact of circRACGAP1 on the cell cycle. Flow cytometry showed that knockdown of circRACGAP1 increased the proportion of cells in G1 phase and decreased the proportion of cells in S pahse (Fig. 6b). Similarly, either silencing of miR-144-5p or overexpression of CDKL1 in these circRACGAP1 silenced cells could restore the proportion of cells in G1 phase and increasement of the S phase cells (Fig. 6b and Fig. S1). Moreover, we discovered that the upregulated miR-144-5p markedly suppressed expression of CDKL1, Cyclin D1 and CDK4 (Fig. 6c) in both cell lines. However, overexpression CDKL1 in the highly expressed miR-144-5p cells reversed the effects of miR-144-5p (Fig. 6c). These data suggested that circRACGAP1 promoted the cell proliferation of NSCLC cells through miR-144-5p/CDKL1 pathway.

a CCK-8 assay showed the cell proliferation of H1299 and Calu-3 cells after the treatment of sh-circRACGAP1, sh-circRACGAP1 + anti-miR-144-5p, sh-circRACGAP1 + pcDNA-CDKL1. b The quantitative histogram of cell cycle in H1299 and Calu-3 cells with different treatment was presented, and the corresponding original data have been shown in Supplementary Fig. 1. c The impact of miR-345-5p on the expressions of CDKL1 by western blotting analysis in H1299 and Calu-3 cells. The mean ± SD is presented for the relative levels from three replications. *p < 0.05, **p < 0.01, ***p < 0.001.
circRACGAP1 promotes the NSCLC cell proliferation and drug resistance in vitro
Furthermore, the impact of circRACGAP1 on the drug resistance of NSCLC cells was also investigated. We treated H1299 and Calu-3 cells with Gefitinib, and then followed with silencing circRACGAP1 with sh-circRACGAP. Cell cycle analysis by flow cytometry showed that the proportion of G1 phase cells in the group with only Gefitinib treatment had a little increase compared with the control group (Fig. 7a). However, silencing circRACGAP1 could significantly increase the proportion of cells in G1 phase, blocking the cell cycle (Fig. 7a). We also investigated the cell apoptosis using flow cytometry in these different treatment groups. The group with treatment of Gefitinib and silencing circRACGAP1 together showed significant cell apoptosis, indicating that silencing circRACGAP1 could enhance the cell sensitivity to drug treatment (Fig. 7b). Meanwhile, the protein analysis through western blot suggested that the cell cycle regulating proteins, such as (CDKL1, Cyclin D1, CDK4) and Bcl-2 were significantly decreased by treating the cells with Gefitinib and silencing circRACGAP1, while the expression of pro-apoptotic proteins (Bax and Caspase3) exerted the opposite data (Fig. 7c, d). The above results demonstrated that circRACGAP1 could promote the drug resistance of NSCLC cells to Gefitinib.

H1299 and Calu-3 cells were treated with control, Gefitinib, sh-NC with Gefitinib or sh-circRACGAP1 with Gefitinib. a The cell cycle analysis by flow cytometry for H1299 and Calu-3 with different treatments. b The cell apoptosis analysis using flow cytometry in H1299 and Calu-3 cells with different treatments. c The expression levels of cell cycle-related proteins in H1299 and Calu-3 cells with different treatments by western blotting assay. d The expression levels of apoptosis-associated proteins in H1299 and Calu-3 cells with different treatments by western blotting assay. The mean ± SD is presented for the relative levels from three replications. *p < 0.05, **p < 0.01.
circRACGAP1 promoted NSCLC tumor growth and enhanced drug resistance in vivo
In order to support our in vitro findings, we conducted in vivo experiment to test the role of circRACGAP1 in NSCLC. After injecting H1299 cells into the right armpit of nude Balb/C mice subcutaneously, an in vivo xenograft model was established, which was then treated by silencing circRACGAP1, treatment of Gefitinib, or combination. As demonstrated in Fig. 8a, b, Gefitinib treatment or silencing circRACGAP1 inhibited the tumor growth to some extent, whereas, the combined treatment of silencing circRACGAP1 and Gefitinib demonstrated a significant tumor growth inhibition compared with other groups. The results indicated that knockdown of circRACGAP1 could enhance the sensitivity of NSCLC tumor to Gefitinib. The measurement of mRNA of circRACGAP1 and miR-144-5p in these treated groups demonstrated that the treatment of Gefitinib alone could inhibit the expression of circRACGAP1 and promote the expression of miR-144-5p, which was very similar with the results in the group treated with sh-circRACGAP1. Moreover, silencing circRACGAP1 could enhance the effect of the treatment of Gefitinib (Fig. 8c).

The tumor was treated with control, sh-NC, sh-circRACGAP1, sh-NC with Gefitinib, or sh-circRACGAP1 with Gefitinib. a The volumes of tumor in the mice with different treatments. The tumor volume was recorded every 5 days. b The tumor weight of the mice with different treatment after 30 days. c The expressions of circRACGAP1 and miR-144-5p in the tumor with different treatments by qRT-PCR. d Immunohistochemical staining was performed to evaluate the expression of CDKL1. e The protein levels of CDKL1, Cyclin D1, and CDK4 in the xenograft tumors with different treatments by western blotting. The mean ± SD is presented for the relative levels from three replications. *p < 0.05, **p < 0.01.
Furthermore, the expression level of CDKL1 was also analyzed by IHC (Fig. 8d) and WB (Fig. 8e). All the results suggested that knockdown of circRACGAP1 and treatment of Gefitinib separately could inhibit the expression of CDKL1 in tumor tissues, but the co-treatment of silencing circRACGAP1 and Gefitinib strengthened this impact more apparent. With western blot analysis, we observed that the expressions of CDKL1, Cyclin D1, and CDK4 in the combination treatment of Gefitinib and silencing circRACGAP1 were significantly inhibited compared with other groups (Fig. 8e). Taken together, these results suggested that circRACGAP1 could promote NSCLC tumor growth and drug resistance by regulating miR-144-5p/CDKL1 pathway.
Discussion
As a notorious malignant tumor, lung cancer is the major cause of the cancer-related death in many countries with increased incidence and mortality [20,21,22]. Lung cancer composes of two main sub-types, including small cell lung cancer (SCLC) and NSCLC. About 85% of the patients are diagnosed as NSCLC, with overall five-year survival rate less than 7% [23]. Studies by our group and other researchers indicated that ncRNA was one of key elements during the progress of lung cancer [24,25,26,27]. circRNAs were found to be involved in cancer development, progression, and metastasis in several different types of cancer as a novel type of ncRNAs [28,29,30]. Due to the extremely abundant binding sites of circRNAs for miRNAs, circRNAs served as a sponge by absorbing miRNAs, which regulated the targeting gene expression [31]. Compared with the linear RNAs, high conservation and stability were the two most important properties of circRNAs, which made them to be ideal biomarkers for diagnosis of diseases. Recently, circRNAs were demonstrated abnormal expression in a variety of human cancers, such as colorectal cancer, hepatocellular carcinoma, breast cancer, glioma, lung cancer, etc. [13, 30, 32,33,34,35]. circRNAs might be upregulated or deregulated in lung cancer, compared to the normal tissues. For instance, circRNA-ITCH was found to be downregulated in lung cancer [36], and Has_circ_0000064 was upregulated in lung cancer, which promoted the proliferation and migration of tumor cells [37]. In this study, we found that the circRACGAP1 was both upregulated in the NSCLC tissues and cell lines compared with the normal tissues and cell line. This result implied that circRACGAP1 might also promote the tumor development in NSCLC. Moreover, we tested the expression level of miR-144-5p at the same time. The results showed that circRACGAP1 and miR-144-5p had a reverse correlation in NSCLC tissues and cell lines. The bioinformatics analysis demonstrated that miR-144-5p was the target of circRACGAP1, which was also proved by luciferase reporter assay and RNA pull-down assay. Therefore, we aimed to elucidate the mechanism of circRACGAP1 in NSCLC in this work.
circRNAs are considered as miRNA sponges to regulate the targeting genes in cancer [10, 16, 30]. Through binding to miRNA, circRNA can control the mRNA expression at post-transcription level. miRNA, as a type of small single-stranded ncRNAs, usually negatively regulates the targeting gene expression through binding to the 3′-UTR. In some cancers, miR-144-5p works as an oncogene, such as in breast cancer, gastric cancer, prostate cancer, and NSCLC [14, 38,39,40]. It is found to promote tumor growth and metastasis through targeting CDKL1 [19]. However, no reports are available to investigate the association between circRACGAP1 and miR-144-5p in NSCLC so far. In this study, we demonstrated that the knockdown of circRACGAP1 in H1299 and Calu-3 cells could significantly inhibit the cell proliferation through blocking the cell cycle in the G1 phase. However, Yang et al. reported that knockdown of RACGAP1 in hepatocellular carcinoma increased the percentage of cells in G2/M phase but with reduced G0/G1 phase [41]. The difference might be caused by the difference between the cells we were investigated. As reported previously, miR-144-5p was found to suppress tumor development and progression via targeting CDKL1 in colorectal cancer, which is a cell cycle blocking related gene [42]. By bioinformatics analysis, luciferase reporter assay, western blot and flow cytometry analysis, we found that miR-144-5p blocked the NSCLC cell cycle in G1 phage through regulating CDKL1, which is an inhibitor of cell-cycle and recognized as a major p53 target gene. Overall, circRACGAP1 could promote the NSCLC tumor growth and progression by interacting with miR-144-5p via regulating the CDKL1 axis.
circRNA was also found to involve in drug resistance of cancer. For instance, Wu et al. reported that circPAN3 mediated drug resistance by targeting miR-153-5p/miR-183-5p-XIAP axis in acute myeloid leukemia [43]. Therefore, it is interesting to uncover the possible impact of circRACGAP1 on the drug resistance in NSCLC. After treating NSCLC cells with Gefitinib or silencing circRACGAP1, we found that silencing circRACGAP1 could significantly promote the expressions of Bax and Caspase-3 as pro-apoptotic proteins and inhibit the expression of Bcl-2 in vitro and in vivo. Moreover, silencing of circRACGAP1 significantly blocked the cell cycle coupled with the treatment of Gefitinib in vitro and in vivo. Our results suggested that gefitinib could suppress the expression of circRACGAP1 and promote the expression of miR-144-5p in NSCLC cells. Even though the detail mechanism of Gefitinib on the expression of circRACGAP1 is not clear, which need further investigation, many circRNAs have been identified differently expressed in the Gefitinib effective and non-effective patients, which might be used as biomarker for predicting the efficacy of Gefitinib therapy in NSCLC patients [44]. For example, Pan et al. found that the tyrosine kinase inhibitor (TKI) resistant patients showed an upregulated expression of circBA9.3 in leukemic cells [45]. Therefore, we propose that Gefitinib might affect the expressions of circRACGAP1 in NSCLC cells through its impact on the TKI pathway, which will need further investigations.
In conclusion, we firstly presented the role of circRACGAP1 and its interaction with miR-144-5p in NSCLC. Our findings demonstrated that circRACGAP1 contributed to NSCLC progression by regulating the miR-144-5p/CDKL1 axis. These results suggested that circRACGAP1 might be used as a potential target for NSCLC diagnosis and therapy.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86.
Wang L, Tong X, Zhou Z, Wang S, Lei Z, Zhang T, et al. Circular RNA hsa_circ_0008305 (circPTK2) inhibits TGF-beta-induced epithelial-mesenchymal transition and metastasis by controlling TIF1gamma in non-small cell lung cancer. Mol Cancer. 2018;17:140.
Zang H, Wang W, Fan S. The role of microRNAs in resistance to targeted treatments of non-small cell lung cancer. Cancer Chemother Pharm. 2017;79:227–31.
Feng J, Zhang X, Zhu H, Wang X, Ni S, Huang J. High expression of FoxP1 is associated with improved survival in patients with non-small cell lung cancer. Am J Clin Pathol. 2012;138:230–5.
DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252–71.
Tian J, Luo Y, Xiang J, Tang J. Combined treatment for non-small cell lung cancer and breast cancer patients with brain metastases with whole brain radiotherapy and temozolomide: a systematic review and meta-analysis. J Neurooncol. 2017;135:217–27.
Zhang M, Xin Y. Circular RNAs: a new frontier for cancer diagnosis and therapy. J Hematol Oncol. 2018;11:21.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–8.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–57.
Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58:870–85.
Piwecka M, Glazar P, Hernandez-Miranda LR, Memczak S, Wolf SA, Rybak-Wolf A, et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science. 2017;357. https://doi.org/10.1126/science.aam8526.
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22:256–64.
Li Y, Hu J, Li L, Cai S, Zhang H, Zhu X, et al. Upregulated circular RNA circ_0016760 indicates unfavorable prognosis in NSCLC and promotes cell progression through miR-1287/GAGE1 axis. Biochem Biophys Res Commun. 2018;503:2089–94.
Danhua Qu BY, Rui Xin, Tianggang MA. A novel circular RNA hsa_circ_0020123 exerts oncogenic properties through suppression of miR-144 in non-small cell lung cancer. Am J Cancer Res. 2018;8:1387–402.
Zou Q, Wang T, Li B, Li G, Zhang L, Wang B, et al. Overexpression of circ-0067934 is associated with increased cellular proliferation and the prognosis of non-small cell lung cancer. Oncol Lett. 2018;16:5551–6.
Yao JT, Zhao SH, Liu QP, Lv MQ, Zhou DX, Liao ZJ, et al. Over-expression of CircRNA_100876 in non-small cell lung cancer and its prognostic value. Pathol Res Pr. 2017;213:453–6.
Zhang S, Zeng X, Ding T, Guo L, Li Y, Ou S, et al. Microarray profile of circular RNAs identifies hsa_circ_0014130 as a new circular RNA biomarker in non-small cell lung cancer. Sci Rep. 2018;8:2878.
Liang Y, Liu M, Wang P, Ding X, Cao Y. Analysis of 20 genes at chromosome band 12q13: RACGAP1 and MCRS1 overexpression in nonsmall-cell lung cancer. Genes Chromosomes Cancer. 2013;52:305–15.
Tang L, Gao Y, Yan F, Tang J. Evaluation of cyclin-dependent kinase-like 1 expression in breast cancer tissues and its regulation in cancer cell growth. Cancer Biother Radiopharm. 2012;27:392–8.
Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30.
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32.
Rimkunas VM, Crosby KE, Li D, Hu Y, Kelly ME, Gu TL, et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res. 2012;18:4449–57.
Byers LA, Rudin CM. Small cell lung cancer: where do we go from here? Cancer. 2015;121:664–72.
Hu W, Bi ZY, Chen ZL, Liu C, Li LL, Zhang F, et al. Emerging landscape of circular RNAs in lung cancer. Cancer Lett. 2018;427:18–27.
Jiang MM, Mai ZT, Wan SZ, Chi YM, Zhang X, Sun BH, et al. Microarray profiles reveal that circular RNA hsa_circ_0007385 functions as an oncogene in non-small cell lung cancer tumorigenesis. J Cancer Res Clin Oncol. 2018;144:667–74.
Liu W, Xiao P, Wu H, Wang L, Kong D, Yu F. MicroRNA-98 plays a suppressive role in non-small cell lung cancer through inhibition of SALL4 protein expression. Oncol Res. 2017;25:975–88.
Wang R, Yan B, Li Z, Jiang Y, Mao C, Wang X, et al. Long non-coding RNA HOX transcript antisense RNA promotes expression of 14-3-3sigma in non-small cell lung cancer. Exp Ther Med. 2017;14:4503–8.
Dai X, Zhang N, Cheng Y, Yang T, Chen Y, Liu Z, et al. RNA-binding protein trinucleotide repeat-containing 6A regulates the formation of circular RNA 0006916, with important functions in lung cancer cells. Carcinogenesis. 2018;39:981–92.
Zhu X, Wang X, Wei S, Chen Y, Chen Y, Fan X, et al. hsa_circ_0013958: a circular RNA and potential novel biomarker for lung adenocarcinoma. FEBS J. 2017;284:2170–82.
Zhong Y, Du Y, Yang X, Mo Y, Fan C, Xiong F, et al. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Mol Cancer. 2018;17:79.
Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009;23:1743–8.
Begum S, Yiu A, Stebbing J, Castellano L. Novel tumour suppressive protein encoded by circular RNA, circ-SHPRH, in glioblastomas. Oncogene. 2018;37:4055–7.
Meng J, Chen S, Han JX, Qian B, Wang XR, Zhong WL, et al. Twist1 regulates vimentin through Cul2 circular RNA to promote EMT in hepatocellular carcinoma. Cancer Res. 2018;78:4150–62.
Wu J, Jiang Z, Chen C, Hu Q, Fu Z, Chen J, et al. CircIRAK3 sponges miR-3607 to facilitate breast cancer metastasis. Cancer Lett. 2018;430:179–92.
Ma Y, Zhang X, Wang YZ, Tian H, Xu S. Research progress of circular RNAs in lung cancer. Cancer Biol Ther. 2019;20:123–9.
Wan L, Zhang L, Fan K, Cheng ZX, Sun QC, Wang JJ. Circular RNA-ITCH suppresses lung cancer proliferation via inhibiting the wnt/beta-catenin pathway. Biomed Res Int. 2016;2016:1579490.
Luo YH, Zhu XZ, Huang KW, Zhang Q, Fan YX, Yan PW, et al. Emerging roles of circular RNA hsa_circ_0000064 in the proliferation and metastasis of lung cancer. Biomed Pharmacother. 2017;96:892–8.
Matsushita R, Seki N, Chiyomaru T, Inoguchi S, Ishihara T, Goto Y, et al. Tumour-suppressive microRNA-144-5p directly targets CCNE1/2 as potential prognostic markers in bladder cancer. Br J Cancer. 2015;113:282–9.
Yamada Y, Arai T, Kojima S, Sugawara S, Kato M, Okato A, et al. Regulation of antitumor miR-144-5p targets oncogenes: Direct regulation of syndecan-3 and its clinical significance. Cancer Sci. 2018;109:2919–36.
Song L, Peng L, Hua S, Li X, Ma L, Jie J, et al. miR-144-5p enhances the radiosensitivity of non-small-cell lung cancer cells via targeting ATF2. Biomed Res Int. 2018;2018:5109497.
Yang XM, Cao XY, He P, Li J, Feng MX, Zhang YL, et al. Overexpression of Rac GTPase activating protein 1 contributes to proliferation of cancer cells by reducing hippo signaling to promote cytokinesis. Gastroenterology. 2018;155:1233–49. e1222
Pei J, Xiao W, Zhu D, Ji X, Shi L, Deng X. LncRNA DSCAM-AS1 promotes proliferation, migration and invasion of colorectal cancer cells via modulating miR-144-5p/CDKL1. Life Sci. 2019:117050.
Shang J, Chen WM, Wang ZH, Wei TN, Chen ZZ, Wu WB. CircPAN3 mediates drug resistance in acute myeloid leukemia through the miR-153-5p/miR-183-5p-XIAP axis. Exp Hematol. 2019;70:42–54. e3.
Liu YT, Han XH, Xing PY, Hu XS, Hao XZ, Wang Y, et al. Circular RNA profiling identified as a biomarker for predicting the efficacy of Gefitinib therapy for non-small cell lung cancer. J Thorac Dis. 2019;11:1779–87.
Chandran RK, Geetha N, Sakthivel KM, Aswathy CG, Gopinath P, Raj TVA, et al. Genomic amplification of BCR-ABL1 fusion gene and its impact on the disease progression mechanism in patients with chronic myelogenous leukemia. Gene. 2019;686:85–91.
Author information
Authors and Affiliations
Contributions
Guarantor of integrity of the entire study: PL; Study concepts: C-HH, PL; Study design: PL, C-HH; Definition of intellectual content: ML, HX; Literature research: ML, HX, FW, H-XZ; Clinical studies: ML, HX, Z-KX; Experimental studies: ML, HX, BL, Z-KX; Data acquisition: ML, HX, BL, Z-KX; Data analysis: ML, HX, BL, Z-KX; Statistical analysis: FW, H-XZ; Manuscript preparation: HX, FW, Z-KX; Manuscript editing: ML, PL, H-XZ; Manuscript review: PL.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Ethics Committee of the Second Xiangya Hospital of Central South University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lu, M., Xiong, H., Xia, ZK. et al. circRACGAP1 promotes non-small cell lung cancer proliferation by regulating miR-144-5p/CDKL1 signaling pathway. Cancer Gene Ther 28, 197–211 (2021). https://doi.org/10.1038/s41417-020-00209-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-020-00209-0
- Springer Nature America, Inc.
This article is cited by
-
CircUBA2 promotes the cancer stem cell-like properties of gastric cancer through upregulating STC1 via sponging miR-144-5p
Cancer Cell International (2024)
-
A novel circ_MACF1/miR-942-5p/TGFBR2 axis regulates the functional behaviors and drug sensitivity in gefitinib-resistant non-small cell lung cancer cells
BMC Pulmonary Medicine (2022)
-
The emerging roles of circular RNA-mediated autophagy in tumorigenesis and cancer progression
Cell Death Discovery (2022)