
Abstract
Prostate cancer is the second most common cancer in men globally. Prostate cancer patients at advanced stages are usually treated with androgen deprivation therapy (ADT). However, with disease progression, it often becomes the incurable castration-resistant prostate cancer (CRPC). JC polyomavirus (JCPyV) is a human DNA virus. Its virus-like particles (VLPs) exhibit similar tropism to native virions and they are capable of delivering exogenous genes to the target cells for expression. JCPyV has been detected in prostate cells; therefore, prostate cancer cells may be susceptible to JCPyV infection and JCPyV VLPs may be used as a vector for gene therapy against prostate cancer. Here we constructed a plasmid (pPSAtk) that allows expression of the thymidine kinase suicide gene only in androgen receptor (AR) positive prostate cancer cells using the prostate-specific antigen (PSA) promoter, and used JCPyV VLPs as a vector to carry pPSAtk (PSAtk-VLPs) for transcriptional targeting in prostate cancer cells. In this study, we found that PSAtk-VLPs could only kill AR-positive CRPC 22Rv1 cells in vitro and inhibit the growth of tumor nodules in the xenograft mouse model. Our results reveal that PSAtk-VLPs could potentially be used as a new option for treating CRPC patients in the future.
Similar content being viewed by others

Introduction
Prostate cancer is the second most common cancer and the fifth leading cause of cancer-related deaths in men globally [1]. In the United States, prostate cancer is the most prevalent cancer in men and is the second leading cause of death from cancer [2]. Prostate cells depend on the androgen receptor (AR) for signal transduction to support cell growth and survival [3]. Therefore, androgen deprivation therapy (ADT) is used to control androgen levels for tumor suppression [4]. However, the heterogeneity and mutations of cancer cells in patients often lead to progression from castration-sensitive prostate cancer (CSPC) to incurable castration-resistant prostate cancer (CRPC) [5]. Recently, despite the development of next-generation AR-targeted drugs, such as abiraterone and enzalutamide, which are not able to inhibit the activities of androgen receptor variant 7 (AR-V7) in CRPC cells [6], the median progression-free survival (PFS) still has not reached 6 months when compared with the standard therapy [7]. Therefore, there is an urgency to develop new treatment options.
Gene therapy is a powerful tool that delivers genetic material into target cells during cancer treatment to inhibit tumor growth and induce cell apoptosis [8]. Viral vectors are widely used in cancer gene therapy due to their relatively high efficiency of gene transfer [8]. JC polyomavirus (JCPyV) is a human DNA virus. Its virus-like particles (VLPs) can be used as vectors for gene therapy as they not only exhibit tropism that is similar to native virions [9], but are also capable of packaging exogenous DNA that does not contain the viral genome [10]. JCPyV VLP, which carries a suicide gene, can effectively inhibit the growth of susceptible tumor cells [11,12,13,14]. It was previously reported that JCPyV has been detected in prostate cells [15, 16]; therefore, prostate cancer cells may also be susceptible to JCPyV infection, and the JCPyV VLPs may be used as a vector for gene therapy against prostate cancer. Prostate-specific antigen (PSA), an important indicator for clinical diagnosis and monitoring of prostate cancer, is exclusively expressed in prostate epithelium due to its androgen response element (ARE) on the 5′ promoter region that is regulated by AR signaling. The PSA expression level increases proportionally with the degree of tumor malignancy due to the intracellular activity of AR signaling [17]. PSA promoter is a prostate tissue-specific promoter that drives expression of its downstream gene, which is expressed exclusively in AR-positive cells [18], and is a powerful tool for specific gene therapy against prostate cancer. Therefore, in this study, we constructed a plasmid containing the PSA promoter-driven thymidine kinase suicide gene (pPSAtk) and used JCPyV VLP as a gene delivery vector (PSAtk-VLP) to examine the specific cytotoxic effect of PSAtk-VLPs in combination with GCV against AR-positive and AR-negative prostate cancer cells. We also performed tail vein injection of PSAtk-VLPs in combination with GCV in human cancer xenograft mouse models to validate the specificity of PSAtk-VLPs in inhibiting prostate tumor growth, so as to evaluate the potential of PSAtk-VLPs as a new therapeutic option for patients with prostate cancer.
Materials and methods
Cell lines and maintenance
All cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA). The human prostate cancer cell lines, 22Rv1 and PC-3, were grown in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS), L-glutamine, nonessential amino acid, HEPES, and penicillin/streptomycin antibiotic solution (Thermo Fisher Scientific, Cambridge, MA). The non-prostate cancer cell lines, lung adenocarcinoma (A549), neuroblastoma (IMR-32) and bladder carcinoma (HT-1197), were maintained in DMEM media with 10% FBS, nonessential amino acid, sodium pyruvate, and penicillin/streptomycin antibiotic solution (Thermo).
Preparation of JCPyV VLPs packaged with exogenous plasmids
The JCPyV VP1 expression plasmid [19] was co-transformed with one of the following plasmids into JM109 E. coli (Promega, Madison, WI): pEGFP-N3 (Clontech, Mountain View, CA), pUMVC1-tk (Aldevron, Fargo, ND), and pPSAtk. The transformed E. coli strains were grown in LB medium with chloramphenicol to select for JCPyV VP1 expression plasmid and kanamycin to select for pEGFP-N3, pUMVC1-tk, or pPSAtk. VP1 protein expression was induced by the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 16 h at 30 °C. IPTG-induced bacteria were lysed, and a supernatant containing soluble proteins was collected by centrifugation. Subsequently, JCPyV VLPs containing packaged plasmid DNA were purified by 10–30% sucrose gradients and concentrated by filtration in Centricon units (Millipore). VLPs packaged with pEGFP-N3, pUMVC1-tk, and pPSAtk are designated as gfp-VLPs, CMVtk-VLPs, and PSAtk-VLPs.
Transduction of human prostate cancer cells with gfp-VLPs
22Rv1 cells in 35 mm dish were washed with PBS and incubated with 10 μg of control VLPs or gfp-VLPs at 4°C for 1 h. Afterward, the cells were washed with cool PBS to remove free VLPs, placed in the complete medium, and then incubated at 37 °C in 5% CO2 for 72 h.
Green fluorescent protein (GFP) expression in the transduced cells was detected by fluorescence microscopy (Carl Zeiss, Germany).
Analysis of human prostate tumor targeting by gfp-VLP in a xenograft mouse model
Five-week-old male nude mice (National Laboratory Animal Center, Taipei, Taiwan) were housed and maintained in controlled, specific pathogen-free airflow cabinets. Animal care and treatment were done in accordance with the guidelines for laboratory animals of the National Chung Cheng University Institutional Animal Care and Use Committee. To establish a xenograft model, the nude mice were subcutaneously inoculated with 5 × 105 22Rv1 cells. 1 week later, the mice were intravenously injected with gfp-VLPs at 70 μg per injection every other day, six times in total. At 20 days after implantation of the tumor cells, the mice were anesthetized, and their tumor nodules were removed and embedded in an optimum cutting temperature compound (Sakura Finetek, Torrance, CA). GFP expression was detected with a fluorescence microscope (Carl Zeiss).
Construction of expression plasmids containing the PSA promoter
To construct a PSA promoter-driven GFP plasmid (pPSAgfp), the human PSA enhancer/promoter fragment was amplified from the pDRIVE-PSA-hPSA vector (InvivoGen, San Diego, CA) by PCR using the primers 5′-GGTTCATATGCCTGCAGGCCTCTAGAAATC-3′ and 5′-CCGCTCGAGTGACACAGCTCTCCGGGT-3′. The PCR product was digested with NdeI and XhoI (NEB, Ipswich, MA) and ligated using T4 DNA ligase (NEB) with the pEGFP-N3 plasmid DNA, from which the CMV promoter was removed by digestion with AseI and XhoI. The resulting plasmid, pPSAgfp, was confirmed by DNA sequencing. The PSA enhancer/promoter fragment described above was also amplified by PCR using the primers 5′- GTTAATTAATGTACACCTGCAGGCCTCTAG-3′ and 5′-GGTGACGTCGACACAGCTCTCC-3′. The PCR product was digested with BsrGI and SalI (NEB) and ligated using T4 DNA ligase with the pUMVC1-tk plasmid DNA (Aldevron), from which the CMV promoter was removed by digestion with BsrGI and SalI. The resulting plasmid, pPSAtk, was confirmed by DNA sequencing.
Transfection of pPSAgfp into prostate and non-prostate cancer cells
When confluency reached approximately 80%, cells including two prostate cancer cells, PC-3 (AR-negative cell) and 22Rv1 (AR-positive cell), lung adenocarcinoma (A549), neuroblastoma (IMR-32) and bladder carcinoma (HT-1197) were transfected with 3 μg pPSAgfp using Lipofectamine 3000 (Thermo) following the manufacturer’s instructions. GFP expression was detected after 72 h post-transfection using a confocal microscope (LSM 510, Carl Zeiss).
Cytotoxicity assay
The growth-inhibiting effect of the thymidine kinase suicide gene on tumor cells was assessed with the Cell Counting Kit-8 (CCK-8) (Sigma-Aldrich, St. Louis, MO) as follows. Cells were seeded in 96-well flat-bottom microtiter plates and either transfected or infected with 0.2 μg DNA or 0.5 μg VLPs per well, respectively. The cells were maintained at 37 °C and 5% CO2 with or without treatment with 10 μg/mL ganciclovir (GCV) (InvivoGen). At 72 h post-transfection or post-infection, the culture medium was changed to 100 μL per well of fresh medium containing 10 μL of CCK-8 solution, and the cells were incubated at 37 °C and 5% CO2 for 1 h. Finally, absorbance at 450 nm was measured using a microplate reader (Thermo), and cell viability was calculated.
Analysis of specific inhibition of human prostate tumor growth by PSAtk-VLPs in a xenograft mouse model
Four-week-old male CAnN.Cg-Foxn1nu/CrlNarl mice were purchased from the National Laboratory Animal Center (NLAC), Taiwan. All animal experiments were approved by the Institutional Animal Care and Use Committee of National Chung Cheng University (IACUC Approval No. 1050401). Twenty nude mice were injected subcutaneously in the right flank either with 5 × 105 22Rv1 cells or 106 HT1197 cells. A week after injection, the 22Rv1 or HT1197 tumor-bearing mice were randomized into the treatment group and control group each with five mice. The treatment group was injected with 70 μg of PSAtk-VLPs through the tail vein every other day, ten times in total. GCV was administered by intraperitoneal injection (50 mg kg−1) every other day after the first PSAtk-VLP injection. On day 28, subcutaneous tumor nodules were surgically removed and photographed, and their weights were measured.
Results
Gene delivery by using JCPyV VLPs into prostate cancer cells in vitro and in vivo
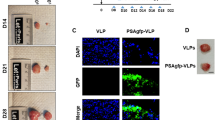
In order to confirm that JCPyV VLPs could be used as vectors for delivering and expressing exogenous genes in prostate cancer cells, VLPs packaged with the green fluorescence plasmid (gfp-VLPs) were purified for gene transduction. 22Rv1 cells, a CRPC cell line, were used as the target cells. Fluorescence microscopy of 22Rv1 at 72-hour post-infection with gfp-VLPs showed that VLPs are capable of delivering plasmid DNA into prostate cancer cells and expression of the green fluorescent protein (GFP) achieving nearly 98% transduction rate. This fluorescent signal was absent in cells infected with control VLPs (Fig. 1). The main goal of cancer gene therapy is to achieve accurate transduction of transgenes to tumor sites using an effective delivery system. The blood circulatory system offers great possibilities for achieving this goal. In order to investigate whether JCPyV VLPs could accurately deliver the transgene to the prostate tumor sites, we subcutaneously inoculated 22Rv1 into nude mice to establish a prostate tumor xenograft mouse model. Once the solid tumor nodule was formed, the mice were subjected to tail vein injections with gfp-VLPs in order to deliver JCPyV VLPs to the prostate tumor site for transduction through the blood circulation. As shown in Fig. 2, the tumor section of the gfp-VLPs-treated group showed green fluorescence, indicating that after entering the blood circulatory system, VLPs could reach the prostate tumor sites and transduce the exogenous gene into the cells for expression. These results demonstrate that JCPyV VLP is able to deliver the exogenous gene into prostate cancer cells in vivo.

Gene delivery using JCPyV VLPs into prostate cancer cells for expression. 22Rv1 cells were infected with control VLPs or gfp-VLPs. GFP expression in the infected cells was visualized at 72 h post-infection using fluorescence microscopy

Gene delivery using JCPyV VLPs into prostate cancer cells in the xenograft mouse model. Human prostate carcinoma–xenografted mice were administered with control VLPs or gfp-VLPs intravenously. The resulting tumor nodule from each mouse was cryosectioned and examined for GFP expression by fluorescence microscopy
Specific expression of PSA promoter-driven GFP gene in prostate cancer cells
The PSA promoter with an upstream enhancer sequence can enhance the expression level of its downstream genes [20]. Therefore, we replaced the CMV promoter in pEGFP-N3 (Clontech) with the PSA enhancer/promoter amplified via polymerase chain reaction (PCR) from the pDRIVE-PSA-hPSA (InvivoGen) vector to construct the PSA enhancer/promoter-driven green fluorescent protein plasmid (pPSAgfp). To demonstrate the function of the PSA promoter to transcriptionally target prostate cells, the plasmid was then transfected into different cell lines, including lung adenocarcinoma (A549), neuroblastoma (IMR-32), bladder carcinoma (HT-1197), and two prostate cancer cell lines [PC-3 (AR-negative cell) and 22Rv1 (AR-positive cell)]. Fluorescence microscopy showed that the green fluorescence could only be detected in 22Rv1 but could not be detected in other non-prostate cells (Fig. 3). PC-3 is a prostate cancer cells, but it is an AR-negative cell that could not activate the PSA promoter and GFP expression (Fig. 3). The result indicates that the PSA enhancer/promoter used in this study exhibited transcriptional targeting in prostate cells due to the reliance of the PSA promoter on AR signaling.

Expression of pPSAgfp in prostate and non-prostate cancer cells. Lung adenocarcinoma (A549), neuroblastoma (IMR-32), bladder carcinoma (HT-1197), AR-negative prostate carcinoma (PC-3), and AR-positive prostate carcinoma (22Rv1) cells were transfected with pPSAgfp DNA. Cells were fixed 48 h after transfection and visualized by fluorescence confocal microscopy
Specific cytotoxicity of PSA promoter-driven TK gene in prostate cancer cells by transfection
The HSV-tk/GCV system is one of the common strategies to induce DNA single-strand break by intervening in the DNA replication process during cell division, leading to tumor cell death [21]. In order to achieve the therapeutic strategy of transcriptional targeting against prostate cancer cells using the HSV-tk/GCV system, we constructed the pPSAtk plasmid by replacing the CMV promoter in the pUMVC1-tk (Aldevron) vector, which can express HSV-TK, with the PSA enhancer/promoter fragment. Subsequently, we transfected different cell lines with pUMVC1-tk or pPSAtk. The transfected cells were treated with GCV and the cell survival rate was assessed. Results showed that the CMVtk/GCV treatment had cytotoxic effects against all different cancer cells (Fig. 4), indicating the effectiveness of the CMV-tk/GCV system without tissue specificity. In contrast, the pPSAtk/GCV treatment only had cytotoxic effects against 22Rv1 cells, suggesting that the pPSAtk construct in this study showed specific cytotoxicity against AR-positive prostate cancer cells (Fig. 4).

Cytotoxicity assays for pPSAtk in prostate and non-prostate cancer cells. The viability of human cells—A549, IMR-32, HT-1197, PC-3, and 22Rv1—was assessed by the CCK-8 method 72 h after various treatments. The treatment combinations included PBS, pCMVtk, or pPSAtk followed by PBS (PBS/PBS, pCMVtk/PBS, or pPSAtk/PBS) and PBS, pCMVtk, or pPSAtk followed by GCV (PBS/GCV, pCMVtk/GCV or pPSAtk/GCV). Experiments were performed in triplicate and repeated three times. Student’s t-test, *P-value < 0.005
Specific cytotoxicity of PSA promoter-driven TK gene in prostate cancer cells by JCPyV VLP infection
To further demonstrate specific cytotoxicity of PSAtk delivered by JCPyV VLPs, three JCPyV VLP susceptible cell lines, lung adenocarcinoma (A549), neuroblastoma (IMR-32) and bladder carcinoma (HT-1197), and two prostate cancer cell lines (PC-3 and 22Rv1), were used to examine the specific cytotoxicity of PSAtk-VLPs. The CMVtk-VLPs- or PSAtk-VLPs-infected cells were treated with GCV. The results showed significant cytotoxic effects of CMVtk-VLP/GCV treatment against the all cancer cell lines, while the treatment with PSAtk-VLP/GCV only showed significant cytotoxic effects against 22Rv1 (Fig. 5), indicating that the suicide gene carried by JCPyV VLPs shows selective cytotoxicity against cancer cells when its expression is driven by tissue-specific promoters. To further confirm the specificity of PSAtk-VLPs in animal, we established xenograft mouse models for human bladder cancer and human prostate cancer. These models were subject to tail vein injections with PSAtk-VLPs, followed by intraperitoneal injections with GCV. The results found that the PSAtk-VLPs/GCV treatment resulted in 39% inhibition of prostate tumor growth compared with the control (Fig. 6). However, the same strategy did not show any effects against bladder cancer cells, which are also JCPyV-susceptible cells (Fig. 6). The findings indicated that JCPyV VLPs with the prostate-specific promoter are capable of transcriptional targeting against prostate cancer cells, and they could effectively inhibit prostate tumor growth. Furthermore, PSAtk-VLP was effectively delivered to the tumor site through the blood circulatory system, indicating that PSAtk-VLP may potentially exhibit cytotoxicity against metastatic prostate cancer cells.

Cytotoxicity assays for pPSAtk delivered by JCPyV VLPs in prostate and non-prostate cancer cells. The viability of human cells—A549, IMR-32, HT-1197, PC-3, and 22Rv1—was assessed by the CCK-8 method 72 h after various treatments. The treatment combinations included PBS, VLP, CMVtk-VLP, or PSAtk-VLP followed by PBS (PBS/PBS, VLP/PBS, CMVtk-VLP/PBS, or PSAtk-VLP/PBS) and PBS, VLP, CMVtk-VLP, or PSAtk-VLP followed by GCV (PBS/GCV, VLP/GCV, CMVtk-VLP/GCV, or PSAtk-VLP/GCV). Experiments were performed in triplicate and repeated three times. Student’s t-test, *P-value < 0.005

Specific inhibition of human prostate carcinoma tumor nodule growth by PSAtk-VLPs in a xenograft mouse model. Human bladder carcinoma (a) and prostate carcinoma (c)—xenografted mice were intravenously administered with PSAtk-VLPs followed by GCV or no treatment. b, d Quantification of tumor nodule weights. Student’s t-test, *P < 0.05. a Scale bar = 50 mm. b Scale bar = 100 mm
Discussion
In the current study, we demonstrated that the human JCPyV VLPs could serve as vectors for gene transfer to prostate cancer cells. The tissue-specific suicide gene expressing plasmid (pPSAtk) that showed specific cytotoxic effects in AR-positive prostate cancer cells has been constructed. JCPyV VLPs packaged with pPSAtk (PSAtk-VLPs) are capable of transcriptional targeting, and they exhibit selective cytotoxicity against prostate cancer cells in the presence of GCV. We further examined the specific cytotoxicity of PSAtk-VLPs in human prostate carcinoma–xenografted mice. The results demonstrated that PSAtk-VLPs could reach the tumor sites through blood circulation, thus effectively and selectively inhibiting prostate tumor growth. Mutation or amplification of AR is one of the most common genomic alteration events in prostate cancer [22]. AR is a nuclear receptor composed of an amino-terminal transactivation domain (NTD), a DNA-binding domain (DBD), and a carboxy-terminal ligand-binding domain (LBD). Upon androgen binding, AR undergoes conformational changes and enters the nucleus to regulate the expression of downstream genes. In prostate cancer cells, over-activation of the AR signaling pathway promotes uncontrolled growth and proliferation of cancer cells [23]. Therefore, ADT, controlling AR signaling pathway in prostate cancer cells, is an effective systematic therapy for patients with prostate cancer [4]. However, the prostate cancer cells become resistant to ADT as the disease progresses to CRPC. Next-generation AR-targeted agents (i.e., enzalutamide and abiraterone), which could extend patient survival, have been used in recent years, but they still could not completely cure prostate cancer patients [7].
Among the many different splice variants of AR, androgen receptor variant 7 (AR-V7) is commonly and highly expressed in CRPC cells. AR-V7 remains active and can promote the expression of downstream genes without ligand binding due to the lack of a C-terminal LBD. This might be the reason for the suboptimal efficacy of abiraterone (which blocks androgen production) and enzalutamide (which inhibits AR activity by blocking its C-terminal LBD) [6]. Prostate tissue-specific promoter (PSA promoter) used in this study contains ARE [24], which can be recognized and regulated by the DBD of AR, including AR-V7. Therefore, we added a suicide gene sequence following the PSA promoter, so that it can still effectively inhibit the growth of prostate cancer cells in CRPC 22Rv1 cells that highly express AR-V7 [25]. Through systematic transportation, the transcriptional targeting effect of PSAtk-VLPs can also be observed in the xenograft mouse model (Fig. 6). PSAtk-VLPs are expected to achieve additional therapeutic outcomes in patients with different AR variant forms of prostate cancer if they have been made possible via future studies to be used in combined with other therapeutic strategies.
The success of gene therapy depends on the efficacy of the therapeutic gene and the appropriate choice of delivery vector. According to previous reports, the host of JCPyV is limited to humans [26]. JCPyV VLPs exhibit tropism, which is similar to native JCPyV [9, 10], and can selectively deliver exogenous genetic material into susceptible cells [11,12,13,14]. In addition, it has also been shown that JCPyV VLP is not able to deliver genes into other organs such as brain, heart, lung, liver, spleen, and kidney, for expression in a mouse model [27]. JCPyV VLPs can be used for different applications by carrying DNA fragments with different characteristics. In our previous studies, JCPyV VLPs have been used to deliver shRNA expression plasmids into human kidney cells to inhibit BKPyV replication [28]. However, development of a gene delivery vector by using JCPyV VLPs has been discussed in our previous publication [29]. In this study, we used JCPyV VLPs as vectors for gene therapy against prostate cancer for the first time, and we used the PSA promoter-driven-suicide gene as a cargo to allow specific inhibition of prostate tumor growth by PSAtk-VLPs in a mouse model. Previous laboratory-based studies have also proposed the use of different viral vectors that carry PSA promoter-driven genes to achieve specific gene therapeutic strategies against prostate cancer [30, 31]. Viral vectors have the advantage of highly efficient transduction over non-viral vectors, but their safety require further discussion as they still carry their own viral DNA sequences [32]. JCPyV VLPs, which exhibit highly efficient transduction similar to native viruses but lack viral DNA sequences, may have a broader scope of applications [33]. JCPyV VLPs can be used for different therapeutic strategies depending on the properties of DNA fragments they carry. In addition to transcriptional targeting, JCPyV VLPs can also be used to track specific cancer cells when viral attachment proteins (VAP) are modified, such as the linkage of tissue-specific peptides or antibodies. Taken together, our results show the potential application of PSAtk-VLPs as a new therapeutic option against both CSPC and CRPC in patients with AR-positive prostate cancer.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30.
Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308.
Nguyen PL, Alibhai SM, Basaria S, D’Amico AV, Kantoff PW, Keating NL, et al. Adverse effects of androgen deprivation therapy and strategies to mitigate them. Eur Urol. 2015;67:825–36.
Debes JD, Tindall DJ. Mechanisms of androgen-refractory prostate cancer. N Engl J Med. 2004;351:1488–90.
Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–38.
Buttigliero C, Tucci M, Bertaglia V, Vignani F, Bironzo P, Di Maio M, et al. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat Rev. 2015;41:884–92.
Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351–60.
Goldmann C, Petry H, Frye S, Ast O, Ebitsch S, Jentsch KD, et al. Molecular cloning and expression of major structural protein VP1 of the human polyomavirus JC virus: formation of virus-like particles useful for immunological and therapeutic studies. J Virol. 1999;73:4465–9.
Ou WC, Wang M, Fung CY, Tsai RT, Chao PC, Hseu TH, et al. The major capsid protein, VP1, of human JC virus expressed in Escherichia coli is able to self-assemble into a capsid-like particle and deliver exogenous DNA into human kidney cells. J Gen Virol. 1999;80(Pt 1):39–46.
Chao CN, Huang YL, Lin MC, Fang CY, Shen CH, Chen PL, et al. Inhibition of human diffuse large B-cell lymphoma growth by JC polyomavirus-like particles delivering a suicide gene. J Transl Med. 2015;13:29.
Fang CY, Tsai YD, Lin MC, Wang M, Chen PL, Chao CN, et al. Inhibition of human bladder cancer growth by a suicide gene delivered by JC polyomavirus virus-like particles in a mouse model. J Urol. 2015;193:2100–6.
Chao CN, Lin MC, Fang CY, Chen PL, Chang D, Shen CH, et al. Gene therapy for human lung adenocarcinoma using a suicide gene driven by a lung-specific promoter delivered by JC virus-like particles. PLoS One. 2016;11:e0157865.
Chao CN, Yang YH, Wu MS, Chou MC, Fang CY, Lin MC, et al. Gene therapy for human glioblastoma using neurotropic JC virus-like particles as a gene delivery vector. Sci Rep. 2018;8:2213.
Delbue S, Matei DV, Carloni C, Pecchenini V, Carluccio S, Villani S, et al. Evidence supporting the association of polyomavirus BK genome with prostate cancer. Med Microbiol Immunol. 2013;202:425–30.
Anzivino E, Rodio DM, Mischitelli M, Bellizzi A, Sciarra A, Salciccia S, et al. High frequency of JCV DNA detection in prostate cancer tissues. Cancer Genom Proteom. 2015;12:189–200.
Lilja H, Ulmert D, Vickers AJ. Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. Nat Rev Cancer. 2008;8:268–78.
Pang S, Taneja S, Dardashti K, Cohan P, Kaboo R, Sokoloff M, et al. Prostate tissue specificity of the prostate-specific antigen promoter isolated from a patient with prostate cancer. Hum Gene Ther. 1995;6:1417–26.
Fang CY, Lin PY, Ou WC, Chen PL, Shen CH, Chang D, et al. Analysis of the size of DNA packaged by the human JC virus-like particle. J Virol Methods. 2012;182:87–92.
Latham JP, Searle PF, Mautner V, James ND. Prostate-specific antigen promoter/enhancer driven gene therapy for prostate cancer: construction and testing of a tissue-specific adenovirus vector. Cancer Res. 2000;60:334–41.
Karjoo Z, Chen X, Hatefi A. Progress and problems with the use of suicide genes for targeted cancer therapy. Adv Drug Deliv Rev. 2016;99(Pt A):113–28.
Schoenborn JR, Nelson P, Fang M. Genomic profiling defines subtypes of prostate cancer with the potential for therapeutic stratification. Clin Cancer Res. 2013;19:4058–66.
Mills IG. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat Rev Cancer. 2014;14:187–98.
Riegman PH, Vlietstra RJ, van der Korput JA, Brinkmann AO, Trapman J. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Mol Endocrinol. 1991;5:1921–30.
Ma Y, Luk A, Young FP, Lynch D, Chua W, Balakrishnar B, et al. Droplet Digital PCR Based Androgen Receptor Variant 7 (AR-V7) Detection from Prostate Cancer Patient Blood Biopsies. Int J Mol Sci. 2016;17.
DeCaprio JA, Garcea RL. A cornucopia of human polyomaviruses. Nat Rev Microbiol. 2013;11:264–76.
Chen LS, Wang M, Ou WC, Fung CY, Chen PL, Chang CF, et al. Efficient gene transfer using the human JC virus-like particle that inhibits human colon adenocarcinoma growth in a nude mouse model. Gene Ther. 2010;17:1033–41.
Lin MC, Wang M, Fang CY, Chen PL, Shen CH, Chang D. Inhibition of BK virus replication in human kidney cells by BK virus large tumor antigen-specific shRNA delivered by JC virus-like particles. Antivir Res. 2014;103:25–31.
Chang CF, Wang M, Ou WC, Chen PL, Shen CH, Lin PY, et al. Human JC virus-like particles as a gene delivery vector. Expert Opin Biol Ther. 2011;11:1169–75.
Yoshimura I, Ikegami S, Suzuki S, Tadakuma T, Hayakawa M. Adenovirus mediated prostate specific enzyme prodrug gene therapy using prostate specific antigen promoter enhanced by the Cre-loxP system. J Urol. 2002;168:2659–64.
Yu D, Chen D, Chiu C, Razmazma B, Chow YH, Pang S. Prostate-specific targeting using PSA promoter-based lentiviral vectors. Cancer Gene Ther. 2001;8:628–35.
Ibraheem D, Elaissari A, Fessi H. Gene therapy and DNA delivery systems. Int J Pharm. 2014;459:70–83.
Teunissen EA, de Raad M, Mastrobattista E. Production and biomedical applications of virus-like particles derived from polyomaviruses. J Control Release. 2013;172:305–21.
Acknowledgements
This research was supported by the Ministry of Science and Technology [grant number MOST 106-2320-B-194-002-MY3], Taiwan; and through Ditmanson Medical Foundation Chiayi Christian Hospital [grant number R104-023].
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lin, MC., Wang, M., Chou, MC. et al. Gene therapy for castration-resistant prostate cancer cells using JC polyomavirus-like particles packaged with a PSA promoter driven-suicide gene. Cancer Gene Ther 26, 208–215 (2019). https://doi.org/10.1038/s41417-019-0083-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-019-0083-0
- Springer Nature America, Inc.
This article is cited by
-
Inhibition of orthotopic castration-resistant prostate cancer growth and metastasis in mice by JC VLPs carrying a suicide gene driven by the PSA promoter
Cancer Gene Therapy (2023)
-
Suppression of bone metastatic castration-resistant prostate cancer cell growth by a suicide gene delivered by JC polyomavirus-like particles
Gene Therapy (2023)
-
Peptide-guided JC polyomavirus-like particles specifically target bladder cancer cells for gene therapy
Scientific Reports (2021)
-
CRPC-specific gene therapy
Nature Reviews Urology (2019)