
Abstract
Background
Angiogenin is a multifunctional secreted ribonuclease that is upregulated in human cancers and downregulated or mutationally inactivated in neurodegenerative diseases. A role for angiogenin in glioblastoma was inferred from the inverse correlation of angiogenin expression with patient survival but had not been experimentally investigated.
Methods
Angiogenin knockout mice were generated and the effect of angiogenin deficiency on glioblastoma progression was examined. Angiogenin and plexin-B2 genes were knocked down in glioblastoma cells and the changes in cell proliferation, invasion and vascular association were examined. Monoclonal antibodies of angiogenin and small molecules were used to assess the therapeutic activity of the angiogenin-plexin-B2 pathway in both genetic and xenograft animal models.
Results
Deletion of Ang1 gene prolonged survival of PDGF-induced glioblastoma in mice in the Ink4a/Arf−/−:Pten−/− background, accompanied by decreased invasion, vascular association and proliferation. Angiogenin upregulated MMP9 and CD24 leading to enhanced invasion and vascular association. Inhibition of angiogenin or plexin-B2, either by shRNA, monoclonal antibody or small molecule inhibitor, decreases sphere formation of patient-derived glioma stem cells, reduces glioblastoma proliferation and invasion and inhibits glioblastoma growth in both genetic and xenograft animal models.
Conclusions
Angiogenin and its receptor, plexin-B2, are a pair of novel regulators that mediate invasion, vascular association and proliferation of glioblastoma cells. Inhibitors of the angiogenin-plexin-B2 axis have therapeutic potential against glioblastoma.
Similar content being viewed by others


Background
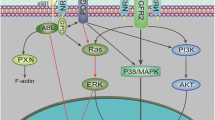
Angiogenin (ANG) is a 14 kDa secreted ribonuclease having regulatory functions in angiogenesis [1, 2], neurogenesis [3], innate immunity [4], hematopoiesis [5] and hematopoietic regeneration [6]. Plexin-B2 (PLXNB2) has recently been identified as the functional ANG receptor that is both necessary and sufficient to mediate the biological functions of ANG in multiple cell types [7]. Several lines of evidence suggest that the ANG-PLXNB2 axis has a role in glioblastoma (GBM) pathogenesis. ANG is expressed in both the vascular and the nervous systems [8], and its expression is upregulated in GBM [9, 10] and downregulated or mutationally inactivated in neurodegenerative diseases including amyotrophic lateral sclerosis (ALS) and Parkinson’s disease (PD) [11]. In the nervous system, ANG is expressed at the highest levels in the granule cell layer of the cerebellum, the dentate granule cells of the hippocampus, and the main olfactory bulb, the cortex and the substantia nigra [12]. PLXNB2 is the second member of the B-type Plexin family which is known to mediate neuronal development and pattern formation. PLXNB2 was originally identified from malignant brain tumours by differential display [13]. It was expressed 8-fold higher in GBM compared to low-grade astrocytomas [13]. PLXNB2 is expressed in the postnatal and adult nervous system, particularly in the subventricular zone (SVZ)-derived neural stem cells [14]. Upregulation of ANG independently predicts worse survival of GBM, in particular the pro-neural subtype [9]. PLXNB2 has been shown to be involved in the invasive growth of GBM [15, 16] and can serve as a biomarker for high-grade glioma [17]. Semaphorin 4 C (Sema4C), the other ligand of PLXNB2, is not upregulated in GBM [15, 17]. These findings prompted us to study the function and mechanism of the ANG-PLXNB2 axis in GBM.
Methods
Generation of Ang1 knockout mice
All animal work was done in accordance with protocols approved by the Institutional Animal Care and Use Committee of Tufts Medical School/Tufts Medical Center and of Harvard Medical School. Mice with a single floxed Ang1 allele were generated by inGenious Targeting Laboratories, Inc. (Stony Brook, NY). Homozygous Ang1LoxP/LoxP mice were obtained by breeding heterozygotes. These mice were crossed with EIIa-Cre mice to generate mosaic mice. The mosaic mice were backcrossed with WT mice to obtain conventional Ang1−/− mice.
Induction and therapy of pro-neural mouse GBM
Nestin-tva;Ink4a/Arf−/−;Pten−/− mice were crossed with Ang1−/− mice to generate experimental animals. For tumour induction, 5-week-old mice (both male and female) were anesthetised and DF-1 chicken fibroblasts infected with Rcas-PDGFB-HA viral vector were delivered intracranially via a Hamilton syringe on a stereotactic fixation device into the SVZ using the following coordinates in reference of bregma: lateral, 1.7 mm; posterior, −0.5 mm; depth, 2.5 mm. Mice were monitored and sacrificed at moribund defined as having significant weight loss (25%) or with severe neurological symptoms. For drug treatment, neomycin or paromomycin was administered i.p. at a daily dose of 10 mg/kg, starting 2 weeks after tumour induction.
Brain slice invasion assay
Mice of 20-day-old were sacrificed and the brains were dissected and put in an ice-cold bath of artificial cerebrospinal (ACSF) [18]. Slices of 300 µm were sectioned using a Vibratome 3000 system, incubated in ACSF at 37 °C for 2 h and transferred into Transwell migration inserts (8 µm pores). After rinsing with a culture medium, 50,000 GFP-labeled control (NT) or ANG knockdown (E7) U87 cells were seeded on top of the slices and incubated at 37 °C for 4 h. The slices were washed with PBS, fixed in 4% paraformaldehyde overnight at 4 °C and stained for vasculature with a CD31 antibody. The association of GFP-labeled U87 cells with CD31-positive vessels was examined under a Leica SPE confocal microscopy.
Orthotopic growth of U87 xenograft tumours in athymic mice
Luciferase transfected U87 control (NT) and PLXNB2 knockdown (D2) U87 cells, 1 × 105 cells per mouse in 2 μl, were injected intracranially into the SVZ of 5-week-old female athymic mice (Charles River Laboratories). Tumour growth was examined by a weekly bioluminescent scan on an IVIS Spectrum CT Biophotonic Imager (PerkinElmer). Luciferin was injected 15 min before imaging at 150 mg per kg body weight.
Antibodies, siRNA and chemical reagents
Rabbit polyclonal antibodies against mouse ANG were prepared in house. mAb against human ANG was clone 26-2 F [19]. Antibodies against AKT (Cell Signaling, Cat # 9272 S), BCL-2 (Cell Signaling Cat # 15071), β-tubulin (Santa Cruz, Cat # sc-7342), CD24 (eBioscience, Cat # 14-0247), CD31 (Abcam, Cat # ab28364), E-Cad (Santa Cruz, Cat # E-10), HA (Sigma-Aldrich, Cat # H6908), PCNA (Santa Cruz, Cat # sc-56), MMP9 (Bioss Antibodies, Cat # bs-0397R), N-Cad (Santa Cruz, Cat # 13A9), pAKT-Ser473 (Cell signaling, Cat # 9271) and p44/p42 ERK (Cell Signaling, Cat # 4695) were used at 1:1000 in TBST + 5% BSA for immunoblotting and 1:200 for IF and IHC. Non-immune mouse IgG (Calbiocehem, Cat # NI03) and normal rabbit IgG (Calbiochem, Cat # NI01) were used as negative controls for monoclonal and polyclonal antibodies, respectively. The MEK inhibitor PD98059 (Cayman chemical) and the MMP9 inhibitor CTK8G1150 (Santa Cruz) were used at the concentration recommended by the manufacturers unless otherwise indicated. CellTrackerTM Orange CMTMR dye (Life technologies, C2927) was used according to the manufacturer’s protocol.
Cell Culture and generation of stable knockdown and overexpression cell lines
GS6-22 was derived from an 82-year-old female GBM patient and has been cultured in non-adherent dishes in a sphere medium containing DMEM/F12 (1:1), 1× B27, 10 ng/ml bFGF and 20 ng/ml EGF as described [20]. DF-1, HEK293T, U87 and U251 were obtained from ATCC and cultured in DMEM + 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. HUVEC were obtained from ATCC and were maintained in a human endothelial basal growth medium plus 5 ng/ml bFGF. MEF was prepared from WT and Ang1−/− mice. Cells were screened for mycoplasma contamination every 3 months with e-Myco Mycoplasma PCR Detection Kit (Lilif, Cat # 25235). PCR-based short tandem repeat (STR) profiling of 9 markers (Abmgood, Cat # C287) was performed annually and compared to ATCC and Sigma datasheet to authenticate the identity of HEK293T, U-87 and U-251. For ANG knockdown, pGIPZ vector with a non-targeting (NT) control shRNA construct and 3 ANG-specific shRNA (A4, E4 and E7, Open Biosystems) constructs were used. Lentiviral particles were harvested in HEK293T cell-conditioned medium 72 h after co-transfection with the pCMV-VSV-G and pCMVΔR8.2 plasmids. For PLXNB2 knockdown, pLKO.1-puro vector with an NT control construct and two PLXNB2-specific shRNA constructs (C1 and D2, Open Biosystems) were used. Lentiviral particles were prepared by transient transfection in HEK293T cells using the ViraPower Lentiviral Expression Systems. Lentiviral particles were harvested after 72 h, centrifuged at 781 × g for 15 min and filtered through a 0.45 µm PVDF membrane. The viral particles were then ultra-centrifuged at 83,000 × g for 1.5 h and the pellet was re-suspended in PBS. Stable cell lines were selected with 1.0 μg/ml puromycin.
Cell proliferation, colony formation and AO/EB staining
For proliferation assay, 2000–10,000 cells per well of 24- or 48-well plates were seeded in regular adherent culture dishes in triplicates, incubated at 37 °C for the time indicated, detached by trypsinisation and counted using a Coulter Counter. For anchorage-independent growth assays, 10,000 cells were suspended in a medium containing 0.4% agarose and plated onto a solidified layer of medium containing 0.8% agarose in 6-well plates. Colonies were quantified in five fields in each well 3 weeks post-seeding. AO/EB staining was performed as previously described [21].
Co-culture of HUVEC and GBM cells
The wells of 48-well plates were coated with 30 μl per well of growth factor-reduced Matrigel (Phenol red free and growth factor reduced, BD Biosciences). CMTMR-stained HUVEC were seeded on the Matrigel at the density of 1.5 × 104 per well, and cultured in an endothelial medium for 4 h to form the tubular network. Stable GFP-expressing U87 cells (2 × 104 per well) were either co-seeded with HUVEC (5 × 103 cells per well) or added 4 h after HUVEC (2 × 104) were seeded. Images were acquired 2–4 h after U87 were seeded.
Ectopic growth of U87 xenograft tumours in athymic mice
U87 cells, 1 × 106 in 50 μl HBSS, were mixed with 50 μl Matrigel and injected s.c. at the right shoulder of the mouse. Treatment with ANG mAb and neomycin (Sigma, Cat # N-6386) started 1-day post tumour inoculation. The dosing regimen for the antibody was daily i.p. injection of 10 mg/kg for 28 days, and that for neomycin was daily i.p. injection of 30 mg/kg for 28 days. The appearance of tumours was examined by palpation until day 40. PBS was used as the control for ANG mAb. Paromomycin (Sigma, Cat # P-5057) was used as a control for neomycin. To examine the effect of PLXNB2 knockdown on ectopic tumour growth, control (NT) and PLXNB2 knockdown (D2) U87 cells (1.5 × 106 cells per mouse, 50% Matrigel) were injected s.c. and animals were sacrificed 3 weeks post-inoculation.
GSC sphere formation
GS6-22 cells were seeded in a sphere medium in an ultra-low adherent 96-well plate at a density ranging from 10 to 400 cells per well and cultured for 7 days. The number of spheres was counted from the entire well. Spheres from the well seeded with 200 cells were also measured for sphere diameters. Test compounds, when present, were added when cells were seeded.
In vivo angiogenesis assay
A cold solution of 0.5 ml Matrigel with or without 0.5 ml of 1 × 106 U87 or U87-E7 cells was injected subcutaneously into C57BL/6 mice. The Matrigel plug was removed 4 days later and processed for IHC analysis of blood vessels with CD31 antibody and for qRT-PCR analysis of the abundance of mouse Cd31 and Cdh5. The primers used in RT-PCR analyses were listed in Supplementary Table 1.
IF and IHC
Animals used for histological analysis were euthanised, and brain tissues were removed and fixed in 10% neutral-buffered formalin for 72 h. Fixed tissues were then embedded in the paraffin, serially sectioned and stained as previously described [22] except that antigen retrieval was achieved by heating the slides in sodium citrate buffer (10 mM Sodium Citrate, 0.05% Tween 20, pH 6.0) in a pressure cooker for 10 min (Cuisinart). Non-immune mouse IgG or normal rabbit IgG was used as controls for background staining. For PCNA staining, positive cells were counted in 3 microscopic fields. For MMP9 and CD24 staining, positive cells were counted in a total of 500 cells. The percentage of positive cells was calculated.
Matrix heterogeneity in H&E stained tissue slides
The extracellular matrix is positively stained by eosin. The standard deviation of binary signals of the eosin staining calculated by ImageJ thus could reflect the level of remodelling of extracellular space or matrix heterogeneity. A total of 15 random lines of equal length from three different fields were drawn and the standard deviation of the eosin signal of each line was calculated via Image J.
qRT-PCR and immunoblotting
Total RNA was extracted using the TRIZOL reagent (Invitrogen) and reverse transcribed using the high capacity cDNA reverse transcription kit (Applied Biosystems). qPCR analysis was performed using the ABI7900HT and SYBR green PCR master mix (Applied Biosystems). The relative mRNA level was determined using the 2−ΔΔCt method with GAPDH as the internal control. The primers used in RT-PCR analyses were listed in Supplementary Table 1. For immunoblot analysis, cells were lysed with RIPA buffer and snap-frozen tissues were homogenised in RIPA buffer with 20 stokes in a tight-fit Dounce homogeniser. Band quantification was done with the ImageJ software (NIH).
Spheroid invasion
Tumour cells under growth conditions were detached with trypsinisation and re-suspended into single-cell suspension (2000 cell/μl) in a complete culture medium. Droplets of 30 μl were dispensed on the lid of 100 mm culture dishes and cultured for 48 h to form spheroids. Spheroids were collected and were mixed with a pre-cold gel solution containing culture medium, collagen (1.5 mg/ml, BD Biosciences), laminin (0.15 mg/ml, Sigma) and test compounds as indicated. While the gel was still in the liquid phase, 50 μl of the mixture containing a single spheroid was transferred into 96-well plate. After the gel was solidified, 100 μl of complete medium with the test compounds was added. For quantification, the number of active invasive cells was counted in an area with a radius that was 1.5 times that of the original spheroid. The total invasion area was measured by ImageJ (NIH) by connecting the farthest invaded cells.
Transwell migration and invasion assays
Twenty four-well plates with 8.0 μm Transwell inserts were used. A total of 5 × 104 cells, re-suspended in serum-free medium, was added to the top chamber and the lower chamber was filled with a medium containing 10% FBS. After 24 h incubation, cells that had migrated to the bottom surface of the inserts were fixed and stained with 0.1% crystal violet, followed by dissolving in acetic acid for quantification.
TUNEL assay
The apoptosis rate was determined by In Situ Cell Death Detection Kit (Roche) per the manufacturer’s instruction. Formalin-fixed, paraffin-embedded tumour sections were dewaxed, rehydrated and permeabilised. TUNEL reaction mixture (50 μl) was added and incubated for 60 min at 37 °C in a humidified chamber in the dark. Negative control was set up simultaneously and went through the same procedures. The slides were rinsed three times with PBS and mounted with cover glasses using the anti-fade mounting solution, and analysed under a Nikon eclipse Ti fluorescence microscope.
Statistics and reproducibility tests
Data were presented as the means ± SD. The significance was analysed using Student’s t-test, one-way ANOVA with Kruskal–Wallis test, or two-way ANOVA for multiple comparisons. Kaplan–Meier survival curves were analysed using log-rank test. Animal numbers were determined based on empirical estimation. No methods were used to determine whether the data met the assumptions of the statistical approach. No randomisation method was used. No specific inclusion and exclusion criteria were used for any data or subjects. Investigator was blinded during the experiment and when assessing the outcome. For all analyses, *p < 0.05; **p < 0.01; ***p < 0.001; ns not significant.
Results
ANG and PLXNB2 are inversely correlated to the survival of GBM patients
Gene expression of profiling interactive analysis (GEPIA) showed that ANG and PLXNB2 transcript levels in GBM samples (TCGA, Cell 2013) are 18.7- and 3.1-fold higher, respectively, than that in the matched normal brain tissues (TCGA/GTEx) (Supplementary Fig. 1a). Survival analysis showed that the 50% survival time was 13.1 and 19.1 months for patients with high and low ANG expression, and was 13.0 and 18.0 months for patients with high and low PLXNB2 expression, respectively. The 1-year survival was 58.7 and 84.1% for patients with high and low ANG expression, and was 58.5 and 83.8% for patients with high and low PLXNB2 expression, respectively (Supplementary Fig. 1b). Further, low transcript levels of both ANG and PLXNB2 were found to be associated with better overall survival of the same GBM patient cohort (Supplementary Fig. 1c). ANG and PLXNB2 expressions were also positively correlated in GBM patients (Supplementary Fig. 1d). Among the three subtypes of GBM, high ANG expression is more significantly correlated with worse survival in the pro-neural/neural subtype than in the mesenchymal and classic subtypes (Supplementary Fig. 1e). According to the new classification of GBM subtypes [23], pro-neural and neural subtypes were combined in these analyses. These bioinformatics analyses show that expression of both ANG and PLXNB2 is inversely correlated to the overall survival of GBM patients, and in particular of the pro-neural/neural subtype.
Ang1 deficiency attenuates the oncogenic transformation of mouse embryonic fibroblasts (MEF)
To explore the role of ANG in GBM, we generated Ang1 knockout (Ang1−/−) mice (Supplementary Fig. 2) and characterised the effect of Ang1 deficiency on transformation susceptibility. Ang1−/− mice under the pure C57BL/6 genetic background were viable and developed normally in laboratory conditions. MEF isolated from wild-type (WT) and Ang1−/− mice had similar morphologies, however, upon transformation with K-Ras-V12 and SV40LT, the transformed MEF (tMEF) of the Ang1−/− genotype displayed a reduction in motility (Supplementary Fig. 3a), invasiveness (Supplementary Fig. 3b) and expression of invasion-related matrix metalloproteinases (MMPs) including Mmp2, Mmp3 and Mmp9 but not Col1a2, a protein that was reported to suppress glioma growth and invasion [24] (Supplementary Fig. 3c). The ability of Ang1−/− tMEF to form colonies in soft agar was decreased as compared to that of WT tMEF, as indicated by a 7-fold decrease in colony number and 1-fold reduction in colony size (Supplementary Fig. 3d), indicating that Ang1 deficiency impaired anchorage-independent growth. Moreover, there was a 9-fold increase of apoptotic cells in colonies of Ang1−/− tMEF than in that of WT tMEF as shown by acridine orange (AO) and ethidium bromide (EB) staining (Supplementary Fig. 3e). Furthermore, Ang1−/− tMEF failed to establish xenograft tumours in athymic mice. None of the 12 mice inoculated with 5 × 106 Ang1−/− tMEF developed palpable tumours in 50 days, whereas all mice inoculated with the same number of WT tMEF developed sizable tumours in the same time period (Supplementary Fig. 3f). Together, these data demonstrate that deletion of Ang1 gene in mice decreased the transformation susceptibility of MEF to oncogenic stimuli, suggesting that ANG plays a role in malignant transformation.
Ang1 deficiency prolongs survival of PDGF-induced pro-neural GBM mice
To explore the role of ANG in GBM, the Ang1−/− mice were crossed with Nestin-tva;Ink4a/Arf−/−;Pten−/− mice to generate compound Ang1−/−;N-tva;Ink4a/Arf−/−;Pten−/− mice and the effect of Ang1 deficiency on the progression of PDGF-induced GBM was examined. The Nestin-tva;Ink4a/Arf−/−;Pten−/− mice have been shown to develop pro-neural GBM [25] upon PDGFB expression in neuronal precursor cells. Specific expression of PDGFB in neuronal precursor cells was achieved by the Racs/tva system in which tva expression was controlled by the Nestin promoter and PDGFB was delivered through intracranial injection of DF-1 chicken fibroblasts that had been infected with and producing the Rcas-HA-PDGFB vector [25]. The median survival time of GBM mice under the Ang1−/− the background was 48 days, 10 days longer than that under the WT background (Fig. 1a), representing a 26% increase in survival. The median survival of GBM mice under the Ang1+/− background was 43 days, in-between that of the WT and Ang1−/− mice. These results indicate that Ang1 deficiency prolonged the survival of PDGF-induced pro-neural GBM mice.

a Survival plots of wild-type (WT) (n = 15), Ang knockout heterozygous (Ang1+/−) (n = 22) and homozygous (Ang1−/−) (n = 19) GBM mice. The numbers shown are median survival. P-values are in comparison to the WT group. b Representative gross histology of fixed brain tissues (sagittal) of sham-operated control mice (normal) and GBM mice of three different genotypes (WT, Ang1+/− and Ang1−/−). Tumour boundaries were marked by dotted white lines, and injection sites were indicated by crosses. c Representative H&E staining of coronal and sagittal brain sections of WT and Ang1−/− GBM mice. Tumour boundaries were marked by dotted white lines. In the coronal sections, high-magnification images of the tumour core and the brain midline were shown. Bar = 50 μm. In the sagittal sections, high-magnification images of the brain cortex and the olfactory bulb were shown. Bar = 50 μm. d IHC of PCNA for proliferating cells. The percentage of PCNA-positive cells was calculated based on cells counted in 3 microscopic fields (×400) at the tumour edge (invasive front), tumour core and perivascular region. Bar = 100 μm. e Necrotic area revealed by H&E staining, indicated by arrows. Bar = 200 μm. f IHC of CD31 for blood vessels (indicated by arrows). Vessels were counted in 3 microscopic fields (×400). Bar = 100 μm. g Apoptotic cells revealed by TUNEL staining (indicated by arrows). Percentage of apoptosis was calculated from apoptotic cells counted in 3 microscopic fields (×400). Bar = 100 μm. Data shown in the bar graphs are means ± SD of three replicates. **p < 0.01; ***p < 0.001.
Postmortem examination revealed that the WT GBM were more invasive, as shown by a distorted midline, than Ang1+/− and Ang1−/− GBM (Fig. 1b, c), indicating that Ang1 deficiency slowed disease progression. These data provide the first experimental evidence that ANG expression is inversely correlated with GBM survival.
Ang1 deficiency decreases the malignant phenotype of mouse GBM
Histological examination (Fig. 1c) revealed that cellularity at the tumour core and brain cortex in Ang1−/− lesions were decreased than in WT lesions, indicating a lower proliferation rate of the Ang1−/− GBM cells. PCNA staining confirmed decreased cell proliferation throughout the entire area of Ang1−/− a tumour with the most significant reduction observed in the perivascular region where the PCNA-positive cells were decreased by 8.2-fold as compared to that in WT tumour (Fig. 1d). Ang1−/− GBM cells displayed a less invasive phenotype as shown by fewer cells invading across the midline into the other hemisphere and into the olfactory blub (Fig. 1c). Ang1−/− tumours also had a 2.2-fold larger necrotic area (Fig. 1e), 2-fold reduced angiogenesis (Fig. 1f), and 20-fold increased apoptosis (Fig. 1g). These results indicate that the slower progression of Ang1 GBM is characterised by a phenotype of lower proliferation, less invasiveness, decreased angiogenesis and increased apoptosis.

a GSC spheres of GS6-22 cells infected with non-targeting (NT) control and E7 ANG-specific shRNA. Cells were seeded at various density in 96-well plates and cultured in sphere medium for 7 days. The numbers of spheres with a diameter >30 µm were counted from the entire well. Sphere diameters shown were from the wells seeded with 200 cells. Bar = 500 μm. b GS6-22 spheres in the presence of a non-immune control IgG (30 μg/ml), ANG protein (1 μg/ml) and anti-ANG IgG 26-2 F (30 µg/ml) for 7 days. Spheres of >30 µm in diameter were counted from the entire well. Bar = 500 µm. c Proliferation of control or ANG knockdown U251 cells. ANG and RNaseA proteins were added at 1 μg/ml when cells were seeded. Cell numbers were determined by a Coulter counter. d 47 S rRNA in U251 cells determined by qRT-PCR using ACTB (β-actin) as the internal control. The product of a representative qRT-PCR reaction was analysed by agarose gel electrophoresis. e ERK and AKT phosphorylation in U251 cells induced by ANG (1 µg/ml). Band intensities of p-ERK and p-AKT were determined by Image J and normalised to the total ERK and AKT, respectively, of the corresponding time points. f Colony of control and ANG knockdown U87 cells formed in the presence of an isotype control IgG or 26-2 F (50 μg/ml), or in the presence of exogenous ANG (1 μg/ml). Colony numbers and diameters were counted and measured in 5 microscopic areas (×40). Bar = 500 μm. g Apoptotic cells in colonies of control (NT) or ANG knockdown (E7) U87 in the presence of isotype control IgG or 26-2 F (50 μg/ml), or in the presence of exogenous ANG (1 μg/ml). Live and apoptotic cells were stained green and red by AO and EB, respectively. The percentage of apoptotic cells was determined from 500 cells examined in each well. Bar = 200 μm. h Effect of ANG on stress-induced apoptosis of GBM cells. Control or ANG knockdown U87 cells were cultures in oxidative (SA, 0.25 mM), ER (tunicamycin, 12.5 µg/ml) and nutrient deprivation (serum-free) stresses in the absence or presence of ANG (1 µg/ml) for 5 days. Apoptotic cells were determined by AO/ER staining. The percentage of apoptotic cells was determined from 500 cells examined in each well. Data shown in the bar graphs are means ± SD of three replicates. *p < 0.05; **p < 0.01; ***p < 0.001, ns not significant.
ANG regulates sphere formation of patients-derived glioma stem cells (GSCs) and proliferation of GBM cells
We next examined the effect of ANG inhibition on patient-derived GSCs as well as on established GBM cell lines. Three ANG-specific shRNA sequences (A4, E4 and E7) were used to knockdown ANG expression using a non-targeting (NT) shRNA as a control. Among the three ANG-specific shRNA constructs, E7 was the most efficient in knocking-down ANG expression (Supplementary Fig. 4a) and was used in subsequent experiments. First, we performed Matrigel plug angiogenesis assay to examine the effect of ANG knockdown on the angiogenic potential of U87 GBM cells. The density of CD31-positive vessels was decreased by 48%, and the mRNA levels of mouse Cd31 and mouse VE-Cadherin (Cdh5), the two mouse endothelial cell markers, were decreased by 58 and 75%, respectively, in Matrigel inoculated with cells transfected with E7 as compared to that transfected with NT (Supplementary Fig. 4b), demonstrating the reduced angiogenic potential of ANG knockdown cells.
The effect of ANG knockdown on cancer stem cell (CSC) sphere formation was examined with GS6-22, a GSC line of primary GBM propagated in undifferentiated sphere format exhibiting high tumour-initiating potential [20]. Knockdown of ANG significantly decreased the ability of GS6-22 cells to form spheres as indicated by decreased number and size of spheres (Fig. 2a). The number of spheres formed from 200 NT control GS6-22 was 142 with an average diameter of 72 μm, and that from the same number of ANG knockdown GS6-22 was 79 with an average diameter of 46 μm, indicating that knockdown of ANG in GS6-22 cells resulted in a reduction of sphere number and size by 44% and 36%, respectively, suggesting a regulatory function ANG in GSCs. Exogenous ANG protein was able to enhance the sphere formation of GS6-22 cells (Fig. 2b), confirming the role of ANG in regulating GSC sphere formation. Consistently, an ANG-specific monoclonal antibody (mAb), 26-2 F, inhibited the sphere formation of GS6-22 cells (Fig. 2b). In these experiments, an isotype control mouse IgG1κ was used as a control that had no effect on sphere formation. Together, these results demonstrate that inhibition of ANG attenuates the ability of GSCs to form spheres, suggesting a role of ANG in maintaining the self-renewal capacity of GSCs.
The effect of ANG knockdown on GBM cell proliferation and colony formation was examined using U251 and U87 cells. Knockdown of ANG expression inhibited proliferation of both U251 (Fig. 2c) and U87 cells (Supplementary Fig. 4c), which could be partially rescued by exogenous ANG but not by RNase A, a biologically inactive homologous protein that has 78% amino acid similarity to ANG. Knockdown of ANG decreased the level of 47 S rRNA precursor in U251 cells, which could also be partially rescued by exogenous ANG (Fig. 2d), supporting previous reports that the proliferation effect of ANG is related to the promotion of rRNA transcription [22, 26,27,28,29,30,31,32,33]. ANG also enhanced phosphorylation of AKT and ERK in U251 cells in a time-dependent manner (Fig. 2e). ANG has been shown previously to activate ERK and AKT in endothelial cells [34,35,36] and neuronal cells [37,38,39] thereby inducing a variety of cellular activities including proliferation and survival. Furthermore, ANG inhibition either by mAb 26-2 F or shRNA E7 resulted in a decrease in both the number and size of U87 colonies formed in soft agar (Fig. 2f). Again, the inhibitory effect of knocking down ANG could be partially rescued by exogenous ANG (Fig. 2f). These results demonstrate the role of ANG in GBM proliferation.
Inhibition of ANG exacerbates stressed-induced apoptosis of GBM cells
ANG inhibition resulted in not only a decrease in colony formation but also an increase in apoptosis within the U87 colonies. AO and EB staining showed that the percentage of apoptotic cells in the colonies formed from NT control cells in the presence of mAb 26-2 F was 12%, which was 3-fold higher than that (4%) in the presence of a non-immune isotype IgG control (Fig. 2g). More robust apoptosis (62%) was found in the colonies of ANG knockdown cells (E7), representing a 15.5-fold increase as compared to that of NT control cells (Fig. 2g). The addition of exogenous ANG protein to ANG knockdown U87 cells decreased apoptosis within the colonies from 62 to 27%, suggesting that deficiency of endogenous ANG could be partially rescued by exogenous ANG. Next, we examined the ability of ANG to prevent stress-induced apoptosis and found that the apoptosis rate in ANG knockdown cells (E7) was increased by 10.1-, 32.6- and 2.3-fold, as compared to the NT control cells, under oxidative, endoplasmic reticulum (ER) and starvation stresses inflicted by sodium arsenite (SA), tunicamycin and serum-free medium, respectively (Fig. 2h, Supplementary Fig. 4d). Exogenous ANG protein was able to partially rescue stress-induced apoptosis in both WT and ANG knockdown cells. These results, together with the observation that there was a dramatic increase in apoptosis in Ang1−/− mouse GBM tissues (Fig. 1g), indicate that ANG prevents GBM cell apoptosis and mediates GBM survival. Pathway enrichment analysis revealed that ANG expression has the highest association (P < 10−8) with the anti-apoptosis gene signatures in GBM patients (Supplementary Fig. 4e).
ANG upregulates MMP9 to promote GBM invasion
The finding that Ang1−/− mouse GBM tumour was less invasive (Fig. 1c) suggested a role of ANG in GBM invasion. This contention was supported by the observation that the extracellular matrix (ECM) of GBM tumours in WT mice was more heterogeneous as compared to that in Ang1−/− mice (Fig. 3a), indicating a lower remodelling process in the ECM of Ang1−/− GBM. We therefore examined the effect of ANG on MMP9, a matrix metalloproteinase that has been shown to be a pro-invasive factor of GBM [40] and to regulate perivascular invasion of GBM [41]. Immunohistochemistry (IHC) (Fig. 3b) analysis revealed that Ang1−/− mouse GBM tissue had a lower level of MMP9 as compared to the WT. Consistently, ANG was able to enhance the mRNA level of MMP9 by 2.9- and 1.8-fold, respectively, in U87 and U251 cells (Fig. 3c), and the proteinase activity in the conditioned medium of U87 cells by 2-fold (Fig. 3d). PD98059, a MEK inhibitor, abolished ANG-induced MMP9 upregulation (Fig. 3e), suggesting a role of the MEK/ERK pathway in ANG-stimulated MMP9 expression.

a H&E staining of WT and Ang1−/− mouse GBM tissues. Bar = 50 μm. Pixels along 15 lines in 3 fields were determined and the standard deviation was used as indication of matrix heterogeneity. b IHC of MMP9 in WT and Ang1−/− mouse GBM tissues. MMP9 positive cells were counted among 500 total cells in each section. Bar = 50 μm. c qRT-PCR analysis of MMP9 mRNA normalised to GAPDH in U87 and U251 cells treated with or without exogenous ANG (1 μg/ml). d MMP9 zymography. The conditioned medium of U87 cells treated with or without ANG (1 μg/ml) was subjected to zymography. Image shown was from a representative experiment. e Effect of MEK inhibitor on ANG-induced MMP9 expression. U251 cells were cultured in the presence or absence of PD98059 (50 nM) and treated with or without ANG (1 μg/ml). Band intensity of MMP9 were determined by Image J and normalised to that of tubulin. f U251 spheroid invasion in response to ANG (1 µg/ml) or anti-ANG IgG 26-2 F (50 μg/ml). Tumour cells invaded into the matrix were recorded for 2 days. Invasive cells were defined as those that had migrated out of the initial spheroid area. The invasive area was defined by the area where invaded tumour cells were found. Data shown are fold changes of invasive area in comparison to the spheroid area at 0 h. Bar = 50 μm. g Effect of MMP9 inhibition on ANG-mediated invasion. U87 spheroids were treated with ANG (1 μg/ml), MMP9 inhibitor CTK8G1150 (50 nM) or both for 48 h. Bar = 200 μm. h Effect of MMP9 inhibition on ANG mAb-mediated inhibition of U87 spheroids invasion. Spheroids were treated with a non-immune isotype control IgG or ANG-specific IgG 26-2 F (50 μg/ml) in the presence or absence of MMP9 inhibitor CTK8G1150 (50 nM). Bar = 200 μm. Data shown in the bar graphs are means ± SD of three replicates. *p < 0.05; **p < 0.01; ***p < 0.001; ns not significant.
We next examined if MMP9 mediates ANG-induced 3-dimentional invasion of GBM cells into a gel composed of collagen and laminin [42], which mimics the in vivo extracellular environment of the brain. While exogenous ANG was able to stimulate spheroid invasion of U251 and U87 cells, ANG-specific mAb 26-2 F inhibited invasion of GBM spheroid derived from U 251 cells (Fig. 3f) and U87 cells (Supplementary Fig. 5a), confirming a role of ANG in promoting GBM invasion. MMP9 inhibitor CTK8G1150 completely abolished the stimulatory activity of ANG (Fig. 3g) and did not have an additional inhibitory effect in combination with 26-2 F (Fig. 3h), demonstrating that MMP9 mediates ANG-induced GBM invasion.
ANG upregulates CD24 to promote the vascular association of GBM
CD24, a cell surface protein that has been shown to be associated with invasive growth [43] and poor prognosis of GBM [44], was found to be downregulated in Ang−/− mouse GBM (Fig. 4a). CD24 has been reported to promote GBM invasion in vivo but not in vitro [43], suggesting that the in vivo microenvironment plays a decisive role in CD24-mediated GBM invasion. To determine if CD24 regulates ANG-induced association of GBM with the vasculature, we first confirmed that the mRNA level of CD24 could be upregulated by ANG in both U87 and U251 cells (Fig. 4b). Next, we examined the effect of CD24 overexpression on GBM vessel co-option, a movement of cancer cells along the pre-existing vasculature and an alternative to angiogenesis to gain access to nutrients, which is a distinct feature of GBM invasion [45]. In a co-culture experiment of U87 and human umbilical endothelial cells (HUVEC), we found that overexpression of CD24 in U87 cells increased the percentage of HUVEC-associated GBM cells from 56 to 84% (Fig. 4c) and decreased the circularity of HUVEC-associated GBM cells from 0.91 to 0.42 (Fig. 4d), indicating that CD24 overexpression enhanced association of GBM cells with endothelial cells. Enhanced association of GBM cells to a preformed HUVEC network was also notable upon CD24 overexpression (Fig. 4e). Moreover, exogenous ANG protein also increased the association of GBM cells to a preformed HUVEC network, but had no further effect on that of CD24-overexpressing cells (Fig. 4f), suggesting that CD24 mediates ANG-enhanced association between GBM cells and endothelial cells. This mechanism was further supported by the observation that ANG knockdown decreased the association of GBM cells to HUVEC network, which could be fully rescued by overexpressing CD24 (Fig. 4g). We also examined the effect of ANG knockdown (E7) on GBM cell invasions in mouse brain slices by an overlay culture (Fig. 4h). While the majority of control cells infiltrated into the brain slice, most ANG knockdown cells stayed on the seeding surface (Fig. 4h). Further, ANG knockdown cells that had invaded the brain slices were less interactive with the vasculature as compared to the control GBM cells (Fig. 4h). Taken together, these results suggest that ANG promotes vascular association of GBM by upregulating CD24.

a IHC of CD24 in WT and Ang1−/− mouse GBM tissues. CD24-positive cells were counted among 500 total cells. Bar = 50 μm. b qRT-PCR analysis of CD24 mRNA in U87 and U251 cells treated with or without exogenous ANG (1 μg/ml). c, d Effect of CD24 overexpression in GBM cells on endothelial cell association (c) and cell circularity (d). U87 cells stably expressing GFP were transfected with an empty vector control or a vector encoding CD24 (CD24), co-cultured with CMTMR-stained HUVEC (red) for 3 h. Arrows indicate tumour cells that are associated with endothelial cells. Percentage and circularity of tumour cells associated with endothelial cells was quantified. Circularity = 4pi (area/perimeter2). A circularity value of 1.0 indicates a perfect circle. A decrease in circularity indicates an increasingly elongated polygon. Bar = 20 μm. e Association of vector and CD24-overexpressing U87-GFP cells to established HUVEC network. Images were taken 8 h after tumour cells were added to the pre-established HUVEC network. Bar = 50 μm. f Effect of ANG on association of GBM cells to HUVEC network. Vector or CD24-overexpressing U87-GFP cells were seeded on an established HUVEC network and cultured in the presence or absence of ANG (1 µg/ml) for 2 h. Percentage of HUVEC-associated GBM cells were counted in 3 microscopic fields (×400). Bar = 50 µm. g Effect of ANG knockdown on endothelial association of vector and CD24-overexpressing GBM cells. Vector and CD24-overexpressing U87-GFP cells were infected with control (NT) or ANG-specific shRNA (E7), seeded on HUVEC network and cultured for 2 h. Percentage of endothelial cell-associated GBM cells was calculated from 3 microscopic fields (×400). Bar = 50 µm. h ANG knockdown inhibits U87 invasion in mouse brain tissues. Control (NT) and ANG knockdown (E7) U87-GFP cells (50,000) were cultured on mouse brain slices (300 µm) for 4 h. Images were taken with a Leica SPE confocal microscopy. Top panel, Z section showing tumour cells (green) invaded into the tissue. Bottom panel, high-magnification images showing interactions between invaded tumour cells (green) with mouse brain vessels (red). Data shown in the bar graphs are means ± SD of three replicates. *p < 0.05; **p < 0.01; ***p < 0.001.
PLXNB2 mediates GBM proliferation and invasion and GSC self-renewal
We next examined the role of PLXNB2, a recently identified ANG receptor [7], in self-renewal of GSC and in proliferation, apoptosis and invasion of GBM cells. Knockdown of PLXNB2 by shRNA C1 and D2 in U87 and U251 cells (Fig. 5a) decreased their proliferation (Fig. 5b). Knockdown of PLXNB2 in GS6-22 cells reduced the number and size of spheres of GS6-22 (Fig. 5c) as compared to the NT control shRNA, indicating a role of PLXNB2 in GBM proliferation and GSC self-renewal. Moreover, colony formation of GBM cells was enhanced by PLXNB2 overexpression (PB2) and inhibited by PLXNB2 knockdown (C1, D2), accompanied by increased apoptosis (Fig. 5d). mAb17, a mAb developed against the ANG-binding site of PLXNB2 [7], inhibited U251 cell proliferation (Fig. 5e). We next examined the effect of PLXNB2 on GBM cell invasion. When equal numbers of GFP-labeled control shRNA-transfected cells (NT) and RFP-labeled PLXNB2 shRNA-transfected cells (D2) were mixed and examined in the 2-dimentional invasion assay, we found that both the number of invasive cells and the invading distance of these cells were significantly decreased in PLXNB2 knockdown cells (Fig. 5f). Together, these results demonstrate an essential role of PLXNB2 in proliferation, apoptosis and invasion of GBM cells, as well as self-renewal of GSCs.

a Immunoblot of PLXNB2 in U251 and U87 cells stably transfected with non-targeting (NT) control or PLXNB2-specific (C1, D2) shRNA. b Proliferation of control (NT) and PLXNB2 knockdown (C1, D1) U251 and U87 cells. Cell numbers were determined by a Coulter counter. c GSC spheres of control (NT) and PLXNB2 knockdown (D2) GS6-22 cells. The mRNA and protein levels of PLXNB2 in control and knockdown cells were shown on the left. Cells were seeded at various density ranging from 10 to 400 per well in 96-well plates and cultured in sphere medium for 7 days. The numbers of spheres with a diameter >30 µm were counted from the entire well. Sphere diameter shown were from the wells seeded with 200 cells. Bar = 500 μm. d Colonies of control (NT), PLXNB2 overexpression (PB2) and knockdown (C1, D2) U87 cells. AO and EB staining was used to visualise live and apoptotic cells in the colonies. PLXNB2 mRNA levels were determined by qRT-PCR, colony numbers were counted from 5 microscopic fields (×40), colony diameters were determined from all counted colonies, apoptotic cells were counted among 500 cells in the colony. Bar = 500 μm (phase contrast) or 200 μm (fluorescence). e Effect of anti-PLXNB2 IgG mAb17 on proliferation of U251 cells. Cells were cultured in the presence of 40 µg/ml non-immune isotype control IgG or various concentrations of mAb17 as indicated. Cell numbers were determined by a Coulter counter. f Effect of PLXNB2 knockdown on GBM spheroid invasion. Control (NT) and PLXNB2 knockdown (D2) U251 cells were transfected with vectors encoding GFP and RFP, respectively, mixed and plated in collagen/laminin gels. Phase contrast and fluorescent images were taken on days 0, 1 and 2. Bar = 200 μm. Numbers of invasive cells were counted and the invaded distance of each cell was measured on day 2. Data shown in the bar graphs are means ± SD of three replicates. *p < 0.05; **p < 0.01; ***p < 0.001.
PLXNB2 knockdown inhibits xenograft growth of human GBM cells in athymic mice
Next, we examined the effect of PLXNB2 knockdown on xenograft tumour growth in both ectopic and orthotopic athymic mouse models. In the ectopic model, tumour sizes derived from PLXNB2 knockdown U87 cells were 3-fold smaller than that derived from an equal number of control cells after 3 weeks of growth (Fig. 6a). In the orthotopic model, we transfected vector control and PLXNB2 knockdown U87 cells with a lentiviral vector-mediated luciferase gene and injected the cells into the SVZ region of athymic mice. Bioluminescence imaging showed that PLXNB2 knockdown markedly decreased orthotopic growth (16.7- and 22.6-fold difference in luminescence counts on day 16 and 21, respectively) (Fig. 6b) and prolonged animal survival (Fig. 6c). Postmortem examination showed that tumour lesions from the control group were more aggressive (Fig. 6d), proliferative (Fig. 6e), angiogenic (Fig. 6f) and invasive (Fig. 6g) as compared to PLXNB2 knockdown group.

a Ectopic growth of not-targeting (NT, n = 8) control and PLXNB2 knockdown (D2, n = 8) U87 cells in athymic mice. Left panel, images of dissected tumours. Right panel, average tumour weights. b Orthotopic growth of control (NT, n = 4) and PLXNB2 knockdown (D2, n = 4) U87 cells. U87-NC and U87-D2 cells were stably transfected with a lentiviral vector encoding luciferase cDNA and intracranially injected into the SVZ region of the brain of athymic mice (1 × 105 cells in 2 μl) through a Hamilton syringe on a stereotactic fixation device: bregma, lateral 1.7 mm, posterior −0.5 mm, depth 2.5 mm. Left panel, representative bioluminescence images taken at 2 weeks post-inoculation. Right panel, weekly bioluminescence radiance (photons/s/cm2/cr). c Kaplan–Meier survival curves. d Gross histology of orthotopic tumour lesions. Yellow circles marked the tumour lesions, dotted white lines marked the midlines of the brains. e Cell proliferation examined by IHC of PCNA. The percentage of PCNA-positive cells in the core region and in the invasive front were determined from 500 cells in 5 microscopic fields (×400) and shown as means ± SD (n = 4). Bar = 50 μm. f Angiogenesis examined by IF of CD31. Vessels were counted in 5 microscopic fields (×400). Bar = 50 μm. g MMP9 level examined by IHC. Bar = 50 μm. *p < 0.05; **p < 0.01; ***p < 0.001.
Inhibitors of the ANG-PLXNB2 axis inhibit GBM growth
Finally, we assessed the potential of ANG inhibitors in GBM therapy in both transgenic and xenograft animal models. First, we examined the effect of neomycin, a small molecule that inhibits the binding of ANG to PLXNB2 [7], using paromomycin as a control. Paromomycin differs from neomycin only at position 6 of the glucose ring (–NH2 in neomycin and -OH in paromomycin) but does not inhibit ANG function in both in vitro and in vivo assays [46]. Treatment with neomycin increased the median survival of PDGF-induced pro-neural GBM mice from 51 days to 64 days, representing a 13 day or 25% increase in survival, as compared to the animals treated with paromomycin (Supplementary Fig. 6a). Histological and IHC analyses revealed that neomycin-treated mouse GBM tumours displayed a decrease in angiogenesis (Supplementary Fig. 6b), an increase in apoptosis (Supplementary Fig. 6c), a more homogenous ECM (Supplementary Fig. 6d) and a reduction in MMP9 expression (Supplementary Fig. 6e) as compared to paromomycin-treated tumours. These results demonstrate that blocking the binding of ANG to PLXNB2 prolonged the survival of PDGF-induced GBM mice, which was characterised by a reduction in angiogenesis and invasion and an increase in apoptosis. In order to determine if targeting ANG may have relevance to human GBM therapy, we examined the effects of ANG inhibitors on human GBM xenograft tumours in athymic mice. Treatment with human ANG-specific mAb 26-2 F (Supplementary Fig. 7a) or neomycin (Supplementary Fig. 7b) prevented the growth of U87 xenografts in 70% animals, accompanied by decreased expression of MMP9 and CD24 (Supplementary Fig. 7c) and reduced association of tumour cells with the vasculature (Supplementary Fig. 7d).
Discussion
We have identified ANG and its receptor PLXNB2 as a pair of novel regulators that promote GBM progression by enhancing proliferation, angiogenesis, invasion and vascular association in both genetic and xenograft GBM mouse models, and in cultured GSC and GBM cells. Knockout of Ang1 gene slowed the progression of PDGF-induced pro-neural GBM in a genetic mouse model in the Ink4a/Arf−/−;Pten−/− background and extended the median survival of these mice from 38 days to 48 days (Fig. 1a). A 10-day extension of survival is significant in these mice considering the relatively short median survival of 38 days. A beneficial effect of ANG inhibition on GBM survival has also been shown by a 13-day life extension in the same genetic mouse GBM model upon treatment of a small molecule inhibitor that blocks the binding of ANG to its receptor PLXNB2 (Supplementary Fig. 6a). Pathological examination and histological analyses revealed that ANG deficiency or inhibition resulted in a decrease in angiogenesis and cell proliferation, and an increase in apoptosis in Ang1−/− GBM or in WT GBM after treatment with ANG inhibitor. These findings can be attributed to the properties of ANG as an angiogenic molecule and to the known function of ANG in regulating cell proliferation and survival. More significantly, we noticed a dramatic reduction in invasion and vascular association of tumour cells in Ang1−/− GBM, suggesting that ANG mediates tumour invasion and vascular association, the two essential components in perivascular invasion of GBM, the primary mode of brain tumour spread. Brain ECM lacks rigid collagen protein and is enriched with less rigid proteoglycan that slows down cell migration. Additionally, the carbohydrate component of proteoglycan is resistant to protease degradation. Therefore, brain ECM poses a barrier to tumour invasion even after extensive remodelling by MMPs. Primary brain tumours and brain metastasis all show widespread perivascular invasion as a unique way to infiltrate into brain tissues [47]. Perivascular invasion can be advantageous to GBM infiltration because there is considerably less physical resistance in the perivascular space [48]. It was reported that GBM cells with perivascular invasion migrated 3-fold faster than those without perivascular invasion [49]. The perivascular invasion of GBM cells involves two main steps, that is, invasion and vascular association [48]. We demonstrated that ANG functions in both steps by upregulating MMP9 and CD24 that mediates invasion and vascular association and of GBM cells, respectively, both of which are important for perivascular invasion.
MMP9 expression has been shown to be correlated with a higher glioma grade and invasiveness [40], and to be localised at the perivascular niche in GBM [41]. MMP9 inhibition by RNAi significantly reduced GBM tumour invasion, tumour growth and angiogenesis [50]. MMP9 has also been reported as pro-invasive factor of GSC [51]. Although there was no experimental data presented in this study that ANG also upregulates MMP9 expression in GSCs, it would be interesting to examine the role of ANG in GSC invasion in future studies.
We also showed that ANG regulates the expression of CD24, a cell surface molecule that has been shown to be correlated with a poor GBM prognosis [44]. CD24 overexpression increased GBM tumour invasiveness in vivo but not in vitro [43], suggesting that CD24-mediated GBM invasion is dependent on the in vivo microenvironment.
The finding is that ANG and PLXNB2 regulate sphere formation of patient-derived GSCs is of interest. As cancer stem cells are thought to be a major reason for chemo-resistance and disease relapse, inhibitors of the ANG-PLXNB2 axis might thus have therapeutic implications.
Data were presented to show that inhibition of ANG-PLXNB2 inhibits GBM growth in both an immunocompetent genetic mouse model and an immunodeficient xenograft animal model, suggesting a therapeutic potential of ANG-PLXNB2 inhibitors in GBM treatment. The potential role of ANG in the immune escape of GBM would be an interesting area for further investigation as ANG has indeed been reported to have an immune-regulatory function. ANG is expressed in the paneth cells of mouse intestine where it regulates innate immunity [4]. It is an acute-phase protein [52, 53] and is upregulated by IL-6 [53], TNF-α and IL-1β [54] and in inflammatory bowel disease [55]. An anti-inflammatory function of ANG has also been proposed [56]. ANG has been shown to suppress TNF-α- and LPS-induced expression of pro-inflammatory cytokines including IL-1β, IL-6, IL-8, TNFR1, TNFR2, MCP-1, MCP-2 and to promote the expression of anti-inflammatory cytokines including IL-4 and IL-10 in corneal fibroblasts [57, 58]. The anti-inflammatory and immune-regulatory functions of ANG in the eyes have been confirmed in both mouse [57] and rat [59] models.
In summary, we have identified a novel pathway of invasive growth of GBM in which the ANG-PLXNB2 axis enhances GSC self-renewal, stimulates GBM invasion and vascular association as well as angiogenesis and cell proliferation, and decreases apoptosis. In particular, we showed that ANG upregulates MMP9 and CD24 expression leading to enhanced ECM degradation and tumour-vessel interaction. We demonstrated the role of ANG-PLXNB2 in GBM pathogenesis as well as the potential therapeutic activity of ANG inhibitors against GBM in both the immunocompetent genetic GBM model and the immunodeficient xenograft model.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
No datasets were generated or analysed during the current study. All data generated or analysed during this study are included in this published article and in its supplementary information files.
References
Marx JL. The 23-million-dollar quest pays off. Science. 1985;230:161.
Fett JW, Strydom DJ, Lobb RR, Alderman EM, Bethune JL, Riordan JF, et al. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry. 1985;24:5480–6.
Subramanian V, Feng Y. A new role for angiogenin in neurite growth and pathfinding: implications for amyotrophic lateral sclerosis. Hum Mol Genet. 2007;16:1445–53.
Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4:269–73.
Silberstein L, Goncalves KA, Kharchenko PV, Turcotte R, Kfoury Y, Mercier F, et al. Proximity-based differential single-cell analysis of the niche to identify stem/progenitor cell regulators. Cell Stem Cell. 2016;19:530–43.
Goncalves KA, Silberstein L, Li S, Severe N, Hu MG, Yang H, et al. Angiogenin promotes hematopoietic regeneration by dichotomously regulating quiescence of stem and progenitor cells. Cell. 2016;166:894–906.
Yu W, Goncalves KA, Li S, Kishikawa H, Sun G, Yang H, et al. Plexin-B2 mediates physiologic and pathologic functions of angiogenin. Cell. 2017;171:849–64 e825.
Weiner HL, Weiner LH, Swain JL. Tissue distribution and developmental expression of the messenger RNA encoding angiogenin. Science. 1987;237:280–2.
Hu JL, Luo WJ, Wang H. Angiogenin upregulation independently predicts unfavorable overall survival in proneural subtype of glioblastoma. Technol Cancer Res Treat. 2019;18:1533033819846636.
Eberle K, Oberpichler A, Trantakis C, Krupp W, Knupfer M, Tschesche H, et al. The expression of angiogenin in tissue samples of different brain tumours and cultured glioma cells. Anticancer Res. 2000;20:1679–84.
van Es MA, Schelhaas HJ, van Vught PW, Ticozzi N, Andersen PM, Groen EJ, et al. Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann Neurol. 2011;70:964–73.
Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–76.
Shinoura N, Shamraj OI, Hugenholz H, Zhu JG, McBlack P, Warnick R, et al. Identification and partial sequence of a cDNA that is differentially expressed in human brain tumors. Cancer Lett. 1995;89:215–21.
Saha B, Ypsilanti AR, Boutin C, Cremer H, Chedotal A. Plexin-B2 regulates the proliferation and migration of neuroblasts in the postnatal and adult subventricular zone. J Neurosci. 2012;32:16892–905.
Le AP, Huang Y, Pingle SC, Kesari S, Wang H, Yong RL, et al. Plexin-B2 promotes invasive growth of malignant glioma. Oncotarget. 2015;6:7293–304.
Huang Y, Tejero R, Lee VK, Brusco C, Hannah T, Bertucci TB, et al. Plexin-B2 facilitates glioblastoma infiltration by modulating cell biomechanics. Commun Biol. 2021;4:145.
Towner RA, Jensen RL, Vaillant B, Colman H, Saunders D, Giles CB, et al. Experimental validation of 5 in-silico predicted glioma biomarkers. Neuro Oncol. 2013;15:1625–34.
Montana V, Sontheimer H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J Neurosci. 2011;31:4858–67.
Fett JW, Olson KA, Rybak SM. A monoclonal antibody to human angiogenin. Inhibition of ribonucleolytic and angiogenic activities and localization of the antigenic epitope. Biochemistry. 1994;33:5421–7.
Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 2009;27:2383–92.
Li S, Yu W, Kishikawa H, Hu GF. Angiogenin prevents serum withdrawal-induced apoptosis of P19 embryonal carcinoma cells. FEBS J. 2010;277:3575–87.
Ibaragi S, Yoshioka N, Kishikawa H, Hu JK, Sadow PM, Li M, et al. Angiogenin-stimulated Ribosomal RNA transcription is essential for initiation and survival of AKT-induced prostate intraepithelial neoplasia. Mol Cancer Res. 2009;7:415–24.
Sidaway P. CNS cancer: glioblastoma subtypes revisited. Nat Rev Clin Oncol. 2017;14:587.
Honma K, Miyata T, Ochiya T. Type I collagen gene suppresses tumor growth and invasion of malignant human glioma cells. Cancer Cell Int. 2007;7:12.
Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling adult gliomas using RCAS/t-va technology. Transl Oncol. 2009;2:89–95.
Sheng J, Yu W, Gao X, Xu Z, Hu GF. Angiogenin stimulates ribosomal RNA transcription by epigenetic activation of the ribosomal DNA promoter. J Cell Physiol. 2014;229:521–9.
Xu ZP, Tsuji T, Riordan JF, Hu GF. Identification and characterization of an angiogenin-binding DNA sequence that stimulates luciferase reporter gene expression. Biochemistry. 2003;42:121–8.
Kishimoto K, Liu S, Tsuji T, Olson KA, Hu GF. Endogenous angiogenin in endothelial cells is a general requirement for cell proliferation and angiogenesis. Oncogene. 2005;24:445–56.
Tsuji T, Sun Y, Kishimoto K, Olson KA, Liu S, Hirukawa S, et al. Angiogenin is translocated to the nucleus of HeLa cells and is involved in ribosomal RNA transcription and cell proliferation. Cancer Res. 2005;65:1352–60.
Li S, Hu MG, Sun Y, Yoshioka N, Ibaragi S, Sheng J, et al. Angiogenin mediates androgen-stimulated prostate cancer growth and enables castration resistance. Mol Cancer Res. 2013;11:1203–14.
Hirukawa S, Olson KA, Tsuji T, Hu GF. Neamine inhibits xenografic human tumor growth and angiogenesis in athymic mice. Clin Cancer Res. 2005;11:8745–52.
Ibaragi S, Yoshioka N, Li S, Hu MG, Hirukawa S, Sadow PM, et al. Neamine inhibits prostate cancer growth by suppressing angiogenin-mediated ribosomal RNA transcription. Clin Cancer Res. 2009;15:1981–8.
Yoshioka N, Wang L, Kishimoto K, Tsuji T, Hu GF. A therapeutic target for prostate cancer based on angiogenin-stimulated angiogenesis and cancer cell proliferation. Proc Natl Acad Sci USA. 2006;103:14519–24.
Liu S, Yu D, Xu ZP, Riordan JF, Hu GF. Angiogenin activates Erk1/2 in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2001;287:305–10.
Kim HM, Kang DK, Kim HY, Kang SS, Chang SI. Angiogenin-induced protein kinase B/Akt activation is necessary for angiogenesis but is independent of nuclear translocation of angiogenin in HUVE cells. Biochem Biophys Res Commun. 2007;352:509–13.
Trouillon R, Kang DK, Park H, Chang SI, O’Hare D. Angiogenin induces nitric oxide synthesis in endothelial cells through PI-3 and Akt kinases. Biochemistry. 2010;49:3282–8.
Kieran D, Sebastia J, Greenway MJ, King MA, Connaughton D, Concannon CG, et al. Control of motoneuron survival by angiogenin. J Neurosci. 2008;28:14056–61.
Steidinger TU, Slone SR, Ding H, Standaert DG, Yacoubian TA. Angiogenin in Parkinson disease models: role of Akt phosphorylation and evaluation of AAV-mediated angiogenin expression in MPTP treated mice. PLoS ONE. 2013;8:e56092.
Steidinger TU, Standaert DG, Yacoubian TA. A neuroprotective role for angiogenin in models of Parkinson’s disease. J Neurochem. 2011;116:334–41.
Zhao Y, Xiao A, diPierro CG, Carpenter JE, Abdel-Fattah R, Redpath GT, et al. An extensive invasive intracranial human glioblastoma xenograft model: role of high level matrix metalloproteinase 9. Am J Pathol. 2010;176:3032–49.
Forsyth PA, Wong H, Laing TD, Rewcastle NB, Morris DG, Muzik H, et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br J Cancer. 1999;79:1828–35.
Bagci T, Wu JK, Pfannl R, Ilag LL, Jay DG. Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene. 2009;28:3537–50.
Senner V, Sturm A, Baur I, Schrell UH, Distel L, Paulus W. CD24 promotes invasion of glioma cells in vivo. J Neuropathol Exp Neurol. 1999;58:795–802.
Deng J, Gao G, Wang L, Wang T, Yu J, Zhao Z. CD24 expression as a marker for predicting clinical outcome in human gliomas. J Biomed Biotechnol. 2012;2012:517172.
Seano G, Jain RK. Vessel co-option in glioblastoma: emerging insights and opportunities. Angiogenesis. 2020;23:9–16.
Hu GF. Neomycin inhibits angiogenin-induced angiogenesis. Proc Natl Acad Sci USA. 1998;95:9791–5.
Baker GJ, Yadav VN, Motsch S, Koschmann C, Calinescu AA, Mineharu Y, et al. Mechanisms of glioma formation: iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia. 2014;16:543–61.
Cuddapah VA, Robel S, Watkins S, Sontheimer H. A neurocentric perspective on glioma invasion. Nat Rev Neurosci. 2014;15:455–65.
Winkler F, Kienast Y, Fuhrmann M, Von Baumgarten L, Burgold S, Mitteregger G, et al. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia. 2009;57:1306–15.
Jadhav U, Chigurupati S, Lakka SS, Mohanam S. Inhibition of matrix metalloproteinase-9 reduces in vitro invasion and angiogenesis in human microvascular endothelial cells. Int J Oncol. 2004;25:1407–14.
Pietras A, Katz AM, Ekstrom EJ, Wee B, Halliday JJ, Pitter KL, et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14:357–69.
Olson KA, Verselis SJ, Fett JW. Angiogenin is regulated in vivo as an acute phase protein. Biochem Biophys Res Commun. 1998;242:480–3.
Verselis SJ, Olson KA, Fett JW. Regulation of angiogenin expression in human HepG2 hepatoma cells by mediators of the acute-phase response. Biochem Biophys Res Commun. 1999;259:178–84.
Etoh T, Shibuta K, Barnard GF, Kitano S, Mori M. Angiogenin expression in human colorectal cancer: the role of focal macrophage infiltration. Clin Cancer Res. 2000;6:3545–51.
Oikonomou KA, Kapsoritakis AN, Kapsoritaki AI, Manolakis AC, Tiaka EK, Tsiopoulos FD, et al. Angiogenin, angiopoietin-1, angiopoietin-2, and endostatin serum levels in inflammatory bowel disease. Inflamm Bowel Dis. 2011;17:963–70.
Bai R, Sun D, Chen M, Shi X, Luo L, Yao Z, et al. Myeloid cells protect intestinal epithelial barrier integrity through the angiogenin/plexin-B2 axis. EMBO J. 2020;39:e103325.
Lee SH, Kim KW, Joo K, Kim JC. Angiogenin ameliorates corneal opacity and neovascularization via regulating immune response in corneal fibroblasts. BMC Ophthalmol. 2016;16:57.
Lee SH, Kim KW, Min KM, Kim KW, Chang SI, Kim JC. Angiogenin reduces immune inflammation via inhibition of TANK-binding kinase 1 expression in human corneal fibroblast cells. Mediators Inflamm. 2014;2014:861435.
Park J, Kim JT, Lee SJ, Kim JC. The anti-inflammatory effects of angiogenin in an endotoxin induced uveitis in rats. Int J Mol Sci. 2020;21:413.
Acknowledgements
We thank late Dr. Bert L. Vallee of Harvard Medical Scholl for support and guidance. We thank Dr. James F. Riordan of Harvard Medical Scholl for advice and discussion.
Funding
This work was supported in part by NIH grants R01CA105241, R01NS065237, R01HL135160 (G-FH), and Vertex Pharmaceuticals Inc.
Author information
Authors and Affiliations
Contributions
HY and G-FH developed the hypothesis, designed experiments, analysed the data and wrote the manuscript. HY, LY, SI, SL, NV, KAG, WY, HK, YJ, AJH and G-FH performed experiments. RS contributed to design and generation of Ang1 KO mice. HY, DJ, BC, EH and G-FH interpreted data. G-FH supervised the project. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yang, H., Yuan, L., Ibaragi, S. et al. Angiogenin and plexin-B2 axis promotes glioblastoma progression by enhancing invasion, vascular association, proliferation and survival. Br J Cancer 127, 422–435 (2022). https://doi.org/10.1038/s41416-022-01814-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-022-01814-6
- Springer Nature Limited
This article is cited by
-
Expression analysis, clinical significance and potential function of PLXNB2 in acute myeloid leukaemia
Molecular Biology Reports (2023)
-
Muscone Can Improve Spinal Cord Injury by Activating the Angiogenin/Plexin-B2 Axis
Molecular Neurobiology (2022)