
Abstract
Background
There is no effective therapy for patients with malignant pleural mesothelioma (MPM) who progressed to platinum-based chemotherapy and immunotherapy.
Methods
We aimed to investigate the antitumor activity of CDK4/6 inhibitors using in vitro and in vivo preclinical models of MPM.
Results
Based on publicly available transcriptomic data of MPM, patients with CDK4 or CDK6 overexpression had shorter overall survival. Treatment with abemaciclib or palbociclib at 100 nM significantly decreased cell proliferation in all cell models evaluated. Both CDK4/6 inhibitors significantly induced G1 cell cycle arrest, thereby increasing cell senescence and increased the expression of interferon signalling pathway and tumour antigen presentation process in culture models of MPM. In vivo preclinical studies showed that palbociclib significantly reduced tumour growth and prolonged overall survival using distinct xenograft models of MPM implanted in athymic mice.
Conclusions
Treatment of MPM with CDK4/6 inhibitors decreased cell proliferation, mainly by promoting cell cycle arrest at G1 and by induction of cell senescence. Our preclinical studies provide evidence for evaluating CDK4/6 inhibitors in the clinic for the treatment of MPM.
Similar content being viewed by others


Introduction
Malignant pleural mesothelioma (MPM) is an aggressive, locally invasive and currently not curable malignancy of the pleura, which is associated with occupational and para-occupational exposure to asbestos [1]. Although asbestos use is banned in many countries, asbestos-insulated buildings are present throughout the world and some countries are still manufacturing and using large quantities of asbestos [2]. Germline mutations in BAP1 and in other cancer susceptibility genes such as PALB2, BRCA2, CHEK2 and MLH1 have been identified in about 10–15% of patients with MPM [3,4,5,6,7].
Treatment options are limited for patients with advanced MPM [8]. Cisplatin plus pemetrexed has been the standard treatment in patients with advanced MPM [9]. The addition of bevacizumab to chemotherapy modestly improved overall survival, but this treatment is not available in all countries [10]. Single-agent immunotherapy has demonstrated limited efficacy in the relapsed setting vs chemotherapy in the PROMISE-meso trial, while in the CONFIRM trial nivolumab has been superior to placebo [11, 12]. Recently, in the CheckMate-743 study, dual immune checkpoint inhibition with nivolumab plus ipilimumab has demonstrated superiority to platinum plus pemetrexed in the first-line setting and has been already approved by the Food and Drug Administration and recently got favourable opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use [13].
Comprehensive genomic analysis of MPM revealed that it is dominated by the inactivation of tumour suppressor genes by multiple mechanisms including single-nucleotide variants, copy number losses, gene fusions and splicing alterations [14, 15]. Commonly inactivated tumour suppressor genes include cyclin-dependent kinase inhibitor 2A (CDKN2A), BRCA1-associated protein 1 (BAP1) and neurofibromin 2 (NF2), large tumour suppressor kinase 2 (LATS2) and SET domain containing 2 (SETD2). MPM is characterised by chromosomal instability and extensive somatic copy number alterations with recurrent allelic losses in regions such as 1p, 3p21, 6q, 9p21, 15q11–15 and 22q [16].
CDKN2A deletions are found in 56–70% of MPM and are associated with shorter overall survival [17, 18]. The CDKN2A/ARF locus (9p21) encodes for two cell cycle regulatory proteins: p14ARF and p16INK4a, the latter being a negative regulator of cyclin-dependent kinase 4/6 (CDK4/6) [19, 20]. In a recent clinical trial of personalised therapy in advanced NSCLC, CDKN2A loss was associated with sensitivity to CDK4/6 inhibitors [19, 21]. Considering the high frequency of CDKN2A deletions in MPM and the fact that cell cycle deregulation is a hallmark of this disease, we postulated that CDK4/6 inhibitors might constitute a novel therapeutic approach in MPM. In the past decade, several selective CDK4/6 inhibitors, abemaciclib, ribociclib and palbociclib, have been approved for the treatment of metastatic breast cancer [22,23,24].
In the present work, we investigated the efficacy of CDK4/6 inhibitors in preclinical models of MPM to investigate their potential in the treatment of MPM. We also assessed the prognostic impact of CDK4 or CDK6 overexpression in primary tumours in patients with MPM using publicly available transcriptomic data.
Materials and methods
Cell culture and cell lines
Five human MPM cell lines, including H28, H2452, H2052, MSTO-211H and H226, were purchased from the American Type Culture Collection (Manassas, VA). Three additional primary cell lines (ICO_MPM1, ICO_MPM2 and ICO_MPM3) were derived from pleural effusions obtained from three patients with MPM. ICO_MPM1 and ICO_MPM3 were derived from two patients who progressed to standard chemotherapy with platinum and pemetrexed, while ICO_MPM2 was derived from a chemotherapy-naive patient. Primary cells were isolated and cultured as previously described [25]. All cell lines were incubated and maintained at 37 °C in a humidified chamber containing 5% CO2. All cells were tested routinely (after defrosting or every 4 months) for mycoplasma contamination by PCR. MSTO-211H and ICO_MPM3 cells were used for the in vivo experiments because they were able to form tumours in athymic mice.
Patient and tissue samples
Patients with confirmed histological diagnosis of MPM were scheduled for routine surgery involving extended pleurectomy decortication at the Glenfield Hospital (University of Leicester). Patients were approached 24 h prior to their operation and provided with patient information regarding the research. All patients signed informed consent prior to surgery. Seventy-nine patient MPM samples were obtained at the time of surgery. Following surgery, all patients were longitudinally tracked until disease progression with computed tomography monitoring and were monitored for survival.
Oncoscan analysis
DNA was extracted with the GeneRead DNA FFPE kit (Qiagen, Manchester, UK). Eighty nanograms of genomic DNA were analysed using the OncoScan FFPE Assay Kit (Affymetrix, Wooburn Green High Wycombe, UK). The BioDiscovery Nexus Express 10.0 for OncoScan software was used to determine copy number alterations and loss of heterozygosity (LOH).
Antibodies and drugs
Antibodies against total Rb (#9313), p-Rb (#8180), CDK4 (#12790), CDK6 (#13331), cyclin D1 (#2922), p16 (#80772) and β-actin (#4970) were purchased from Cell Signalling Technology (Danvers, MA and were used following the manufacturer’s instructions for Western blot.
Abemaciclib (LY2835219) was purchased from Selleckchem (Houston, TX). Palbociclib (PD0332991) was provided by Pfizer, Inc. (San Diego, CA). Cisplatin, pemetrexed and gemcitabine were obtained at the Catalan Institute of Oncology Pharmacy.
In vitro and in vivo drug experiments
For in vitro experiments, cell lines were plated into 6-well plates and treated with abemaciclib or palbociclib with 0 (control), 10, 100, 250 or 500 nM for 1, 3 or 15 days. Doses below micromolar range would be clinically well tolerated. For in vivo assays, mice were randomly treated with (i) vehicle, 200 μl of 0.05 N sodium lactate pH 4.0 by oral gavage 5 days a week; (ii) cisplatin alone, 3.5 mg/kg administered intraperitoneally once a week or combined with pemetrexed, 100 mg/kg administered intraperitoneally twice a week; (iii) gemcitabine, 75 mg/kg administered intraperitoneally twice a week; (iv) abemaciclib, 150 mg/kg by oral gavage 5 days over 7 days or (v) palbociclib, 150 mg/kg by oral gavage five days over 7 days. Mice with subcutaneous tumours were treated for 26 days, while mice harbouring orthotopic lung-derived MSTO-211H and ICO_MPM3 xenografts were treated during 40 and 52 days, respectively.
Western blot analysis
Total cell lysates and Western blotting were performed as previously described [26].
Cell viability, cell cycle and apoptosis analysis
Cell viability was evaluated by cell counting and colony formation assays as described elsewhere [27]. For colony formation tests, cells were grown for 15 days and the medium was renewed every 4 days. Cell cycle and apoptosis were analysed by flow cytometry and propidium iodide incorporation as described in [28]. A minimum of 1 × 104 cells was analysed per determination. All experiments were repeated at least three times with similar results: cell counting assay comprised three measurements in three biological replicates; cell cycle comprised three biological replicates; and apoptosis comprised multiple biological replicates: ten for MSTO-211H, four for H28 and three for ICO_MPM3. P values were adjusted using false discovery rate (FDR).
Measurement of cellular senescence
The evaluation of senescence-associated β-galactosidase expression was performed as previously described [29]. Experiments included three measurements in at least three biological replicates.
In vivo MPM subcutaneous preclinical drug assays in nude mice
To investigate the efficacy of palbociclib in the treatment of MPM, we used the MSTO-211H cell line, which was derived from a patient who had not received prior chemotherapy and was able to grow in athymic mice. For subcutaneous xenograft development, 4 × 106 MSTO-211H cells growing exponentially were suspended in 300 µl phosphate-buffered saline (PBS) and subcutaneously inoculated into the right flanks of 23 6-week-old male athymic nude (Hsd:Athymic Nude-Foxn1nu) mice (Envigo, Indianapolis, IA). Once the tumours reached a homogeneous average volume size of 300–400 mm3 as including criteria, mice (n = 21) were randomly assigned into three groups (n = 7 per group) and treated with (i) vehicle; (ii) cisplatin combined with pemetrexed or (iii) palbociclib as described above. To evaluate the efficacy, tumour volumes (V = π/6 × L × W2) were measured twice per week with callipers and the weight of each animal was measured every day. After 26 days of treatment, mice were euthanised by cervical dislocation and the tumours were excised, weighted and processed for histologic and RNA studies following standard protocols. The mean volume ± SD was calculated using R software v.3.5.0 [30]. Daily differences among treatments were analysed using Kruskal–Wallis tests, with FDR adjustment.
In vivo MPM orthotopic preclinical drug assays in tumours nude mice
To investigate the efficacy of CDK4/6 inhibitors in tumours after progression to standard first-line chemotherapy, we generated two different chemoresistant MPM orthotopic models by implanting (i) MSTO-211H subcutaneous xenografted tumours treated with cisplatin plus pemetrexed from the previous experiments or (ii) chemoresistant patient-derived subcutaneous xenografted tumours in the thoracic cavity of 6-week-old male athymic nude mice following our previously reported procedures [31]. As we described, mice were anesthetised with a continuous flow of 1–3% isoflurane/oxygen mixture (2 L/min) and subjected to a right thoracotomy. Mice were situated in the left lateral decubitus position, and a small transverse skin incision (5–8 mm) was made in the right chest wall. Chest muscles were separated by sharp dissection and costal and intercostal muscles were exposed. An intercostal incision of 2–4 mm on the third or fourth rib on the chest wall was made and a small tumour piece of 2–4 mm3 was introduced into the chest cavity and the tumour specimens were anchored to the lung surface with Prolene 7.0 suture. Next, the chest wall incision was closed with surgical staples, and finally, chest muscles and skin were closed. The waiting time between tumour implantation and the beginning of treatments was of 2 weeks based on our previous experience with orthotopic xenograft MPM models. For the orthotopic xenograft model, 33 mice were randomised into three groups (n = 11 per group) and treated with (i) vehicle; (ii) cisplatin alone or (iii) palbociclib as previously mentioned for 40 days. For the patient-derived orthotopic xenograft model, 31 mice were randomised into four groups (n = 7–8 per group) and treated with (i) vehicle; (ii) gemcitabine; (iii) abemaciclib or (iv) palbociclib as previously described for 52 days. In both orthotopic experiments, mice were weighed daily and monitored for the presence of breathing problems. After stopping treatments, all live mice remained untreated until the human endpoint (defined as presenting respiratory problems or excessive body weight loss). Orthotopic tumours were collected from euthanised mice and processed for histological studies. Survival curves for each cohort of mice were calculated using the Kaplan–Meier method and the differences between groups were compared using the Cox proportional-hazards model.
Inmunohistochemistry studies
Paraffin sections of subcutaneous MSTO-211H xenografted tumours harvested when the mice were euthanised 26 days post-treatment were used to assess macrophages and natural killer (NK) cells infiltration by immunohistochemistry. Antibodies used were anti-F4/80 (#70076 from Cell Signalling Technologies) as macrophage marker and recombinant anti-NCR1 (ab233558 from Abcam, Cambridge, UK) as NK cell marker. All slides were coded and examined in a blinded manner.
In silico analysis of publicly available RNA-sequencing (RNA-seq) data
Public data from RNA-seq cohorts published by Bueno et al. [14] and The Cancer Genome Atlas (TCGA-MESO) [15] were used to assess differences in survival. Gene expression (log2(TPM)) was stratified using the median, and Cox proportional-hazards models adjusted for sex, stage, age and histology were fitted to assess differences in survival using R software [30].
Whole-exome sequencing (WES) and RNA-seq analysis of patient-derived cell lines
Paired-end RNA-seq was performed on an Illumina HiSeq 2500, with 100 bp long reads. Genomic DNA and total RNA were submitted to the Centro National de Análisis Genómico (Barcelona, Spain), for WES and RNA-seq library preparation and sequencing. All statistical analyses were done using R software v.3.5.0 [30]. Sequence data have been deposited at the European Genome-phenome Archive (EGA), which is hosted by the EBI and the CRG, under accession number EGAS00001005352.
Statistics
Cell proliferation assay was assessed using Wilcoxon’s signed-rank tests comparing each treatment with vehicle condition and adjusted using FDR correction. Differences among treatment and vehicle conditions in the cell death experiment were evaluated using Mann–Whitney U test for each comparison and Kruskal–Wallis test if three conditions were simultaneously tested and adjusted afterward using FDR. Cell cycle and senescence experiments were analysed using proportion tests taking into consideration all the cells counted in the abovementioned experiments. P values were adjusted using FDR. For in vivo experiments, the analysis of differences in body weight for orthotopic xenografts was computed using a Mann–Whitney U test comparing the first and last day of treatment in each treatment and adjusted with FDR. For subcutaneous xenografts, a longitudinal analysis testing for differences in treatment slopes was done using an analysis of covariance. All tests were two-sided, and assumptions were verified for all tests that required it. Homoscedasticity was also verified using a Levene test. Regarding survival analysis, the sample size was not calculated since we used publicly available data Bueno et al. [14]. and TCGA [15]. Cox proportional-hazards models adjusted for sex, stage, age, and histology were fitted to assess the differences between gene expression (categorised using the median log2TPM value for each gene). For in vivo experiments, a Cox proportional-hazards model was fitted to assess differences among groups. Survival curves were plotted using Kaplan–Meier curves. P value <0.05 was considered statistically significant. All statistical analyses were done using R software v.3.5.0 [30].
For additional information about methodology, see Supplementary material.
Results
Genomic characterisation of patient-derived MPM models and baseline expression of genes involved in the cell cycle in MPM cell lines
Clinicopathological characteristics and main genomic alterations are shown in Table 1. In all three patient-derived cell lines, CDKN2A/p16 was deleted and NF2 was wild type, while BAP1 was mutated in ICO_MPM1 (p.K651Yfs*1) and ICO_MPM2 (p.R60X). Additional information about their mutational profile is provided in Supplementary Table S1.
We examined by Western blot the expression levels of CDK4, CDK6, cyclin D1, CDKN2A/p16 and RB proteins in five commercial and in three patient-derived MPM cell lines (Fig. 1a). p16 expression was not detected in any cell line, while all cell lines showed some degree of cyclin D1 expression and retained RB expression. CDK4 was expressed at a relatively high level in most MPM cells, whereas CDK6 expression appeared to be comparatively lower. Additional information about their mutational profile is provided in Supplementary Table S2.

a Baseline protein expression levels by Western blot of CDK4, CDK6, cyclin D1, Rb, phosphor-RB and p16. b Number of viable cells was determined in vitro by cell counting in the panel of cells after 3 days of treatment with increasing concentrations (0, 10, 100 and 500 nM) of abemaciclib or palbociclib. Bar plots represent the means ± SD of three measurements in three biological replicates. Adjusted p values were calculated with Wilcoxon’s signed-rank tests. In the graph, the p values are reported with respect to 0 nM (*p < 0.05; **p < 0.01). c Colony formation assay displaying treatment response to abemaciclib and palbociclib. A representative image from three biologically independent replicates is displayed.
Antiproliferative effect of CDK4/6 inhibitors on human MPM cell lines
All MPM cell lines treated with increasing concentrations of abemaciclib or palbociclib for 72 h showed a decrease in cell number (Fig. 1b and Supplementary Fig. S1). Treatment with abemaciclib and palbociclib at 100 or 500 nM significantly reduced cell number in comparison to control in all cell lines tested (p < 0.05). At 10 nM, the cell number was significantly reduced in six out of eight cell lines after treatment with abemaciclib (p < 0.01), while it was significantly reduced in all cell lines after palbociclib treatment (p < 0.05).
The reduction in cell number after exposure to CDK4/6 inhibitors at 100 nM was nearly 50% (mean 54.5% ± 5.5 with abemaciclib and mean 53.4% ± 4.9 with palbociclib). At 500 nM, a reduction of 64.3% and 64.1% were observed with abemaciclib and palbociclib, respectively. All primary cell lines were sensitive to CDK4/6 inhibitors regardless of whether they had been derived from a patient who was chemotherapy-naive or who had received prior chemotherapy. ICO_MPM2, which was derived from a chemotherapy-naive patient, was the most sensitive primary cell line to palbociclib with a cell number reduction of 45.3% ± 5.2 at 100 nM (Fig. 1b). These antiproliferative effects were confirmed by cell colony formation assay and crystal violet staining (Supplementary Fig. S2A). The ability to form colonies was prevented when H226, H2052, ICO_MPM1 and ICO_MPM3 were treated with abemaciclib and palbociclib at 250 and 500 nM (Fig. 1c).
Effect of CDK4/6 inhibitors on cell cycle in human MPM cell lines
Three cell lines selected for expressing high levels of CDK4 and CDK6 proteins (MSTO-211H, H28 and ICO_MPM3) were evaluated for alterations in cell cycle progression after 24h treatment with 250 or 500 nM abemaciclib or palbociclib. Compared with control, all cells treated with abemaciclib or palbociclib at 500 nM were arrested at the G1 phase (p < 0.001, Fig. 2a). In addition, a significant decrease in cell percentage in the G2/M phase (p < 0.001) and in the S phase (p < 0.001) was observed in the three cell lines after treatments at 250 and 500 nM compared to non-treated cells (Supplementary Fig. S2B). All changes in the percentage of cells in each phase of the cell cycle after abemaciclib and palbociclib treatment are summarised in Fig. 2b.

Effects of cell line treatments with CDK4/6 inhibitors abemaciclib or palbociclib at 0, 250 or 500 nM doses to induce (a, b) cell cycle arrest and (c) senescence. a MSTO-211H, H28 and ICO_MPM3 cells were untreated and treated with both inhibitors for 24 h and DNA content was analysed by flow cytometry. Cell cycle arrest at the G1 phase was induced by both CDK4/6 inhibitors in the cell lines. b Percentage of cells in each phase of the cell cycle phase in response to abemaciclib and palbociclib treatments at doses of 0, 250 or 500 nM for 24 h. Cell cycle phase distribution analysis was done using the FlowJo software. Each value represents the mean ± SD of three replicates. Adjusted p values were considered significant when the mean differed from control within each phase of the cell cycle (*p < 0.05; ***p < 0.001). c A significant increase in the number of SA-β-gal-positive cells was detected in MSTO-211H, H28 and ICO_MPM2 treated. Data are expressed as a percentage of senescent cells obtained from the mean value ± SD of three replicates. Adjusted p values <0.05 were considered significant.
Effect of CDK4/6 inhibitors on cell death and senescence in human MPM cell lines
As a next step, we investigated whether treatment with CDK4/6 inhibitors could induce cell death in MPM cells. MSTO-211H, H28 and ICO_MPM3 cells were treated with different concentrations of abemaciclib and palbociclib, as single agents or in combination with the apoptosis inhibitor QVD for 72 h and cell death was quantified by FACS (Supplementary Fig. S3). Neither inhibitor was able to significantly increase the levels of apoptosis in MSTO-211H, H28 and ICO_MPM3 cells at any of the doses tested. At the highest dose (500 nM), the percentage of apoptotic cells reached 12% with abemaciclib and 8% with palbociclib in MSTO-211H cells, 2% after abemaciclib and 3% after palbociclib in H28 cells, and around 6% after either treatment in ICO_MPM3 cells.
To investigate whether CDK4/6 inhibitors promote senescence, both treated and control MSTO-211H, H28 and ICO_MPM2 cells were stained using β-galactosidase. A significant increase in the percentage of senescent cells was detected in all cell lines treated with different concentrations of abemaciclib or palbociclib (p < 0.001, Fig. 2c). Specifically, the proportion of senescent SA-β-gal-positive MSTO-211H cells increased from 18 to 54% with 250 nM abemaciclib and to 61% with 500 nM abemaciclib and to 52% with 250 nM palbociclib and to 59% with 500 nM palbociclib. Likewise, an increase of senescent cells was also observed in H28 cells treated with 250 or 500 nM of either inhibitor. In ICO_MPM2 cells, the percentage of SA-β-gal-positive cells increased from 12% in control cells to 41% after abemaciclib and to 46% after palbociclib treatments at 250 and 500 nM, respectively.
Palbociclib reduced tumour growth and improved overall survival in mice bearing MPM tumours
The effect of palbociclib in vivo was examined by implanting subcutaneously MSTO-211H cells into the right flanks of athymic mice. After 26 days of treatment, the mean volume of tumours implanted subcutaneously in vehicle-treated mice was 1816 ± 795.2 mm3 and in cisplatin plus pemetrexed-treated mice it was 1647.1 ± 733.8 mm3, whereas in palbociclib-treated mice it was 524.2 ± 236.6 mm3 (Fig. 3a and Supplementary Fig. S4A). Differences among palbociclib and the two other cohorts were already statistically significant at day 16 (p = 0.043, Fig. 3b). At mice sacrifice, 26 days post-treatment, a significant decrease in the tumour weight was observed for palbociclib-treated mice with respect to the vehicle and combined chemotherapy-treated mice (0.35 vs 1.1 and 1.13 g; p = 0.01 and 0.007, respectively, Fig. 3c, d). No differences were observed at the histological level (Supplementary Fig. S5). The body weight of the mice was monitored to evaluate the potential side effects of treatments (Fig. 3e). In those mice treated with palbociclib, no body weight loss was observed during the experiment, suggesting that palbociclib did not exert significant systemic toxicity at the doses used in this study. After 26 days of treatment, we quantified the macrophages and NK cell infiltration into the subcutaneous MPM tumours xenografts (Fig. 3f). A statistically significant increase in the tumour-associated macrophages (F4/80+ cells) was observed for palbociclib-treated tumours with respect to vehicle and combined chemotherapy-treated tumours (0.09 vs 0.048 and 0.048; p = 0.041, respectively, Fig. 3g). No significant differences in the percentage of tumour-infiltrated NK cells (NCR1+ cells) were found between vehicle, cisplatin plus pemetrexed and palbociclib-treated tumours (0.04 vs 0.004 and 0.10; p = 0.58, respectively, Fig. 3h).

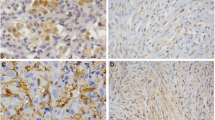
A xenografted subcutaneous tumour model was established by inoculation of MSTO-211H cells into the flanks of athymic nude mice (n = 7 per group). Tumours’ volume (a) was monitored by calliper measure every 4 days (at each time point, a SD bar is shown). b Differences in tumour volume for each day were represented using boxplots and compared using Kruskal–Wallis test, adjusted by FDR and were significantly different at day 16. At the end of the experiment, mice were sacrificed and (c, d) the tumours were removed, weighted and photographed. Asterisks indicated the absence of apparent macroscopic tumour at sacrifice, while residual cells were identified by H&E analysis. e Summary of the body weight values among first and last day of treatment from all mice in in vivo subcutaneous tumour xenograft growth experiment. Palbociclib did not exert any substantial change in the mice body weight. Differences were evaluated by Mann–Whitney test and adjusted for FDR. f Representative IHC images for (F.A) mouse macrophages that express F4/80 (brown staining) and (F.B) mouse NK cells that express NCR1 (brown staining) infiltrated in the subcutaneous MPM tumours xenografted in athymic nude mice after 26 days of treatment with vehicle, platinum plus pemetrexed or palbociclib. Inset photos contain the digital whole slide image showing the infiltrated area of tumour. Scale bar = 50 µm. Dot plots showing (g) the mean F4/80+ intensity per pixel (x-axis) or (h) the percentage of NCR1+ cells over total cells infiltrated in the subcutaneous MPM tumour xenografted in athymic nude mice after 26 days of treatment with vehicle, platinum plus pemetrexed or palbocilib (n = 5–7 per group). Data are expressed as single data values (dots) + the mean. ANOVA test was used to detect statistical differences between treatments (*p < 0.05).
To preclinically investigate the efficacy of palbociclib as second-line treatment in MPM tumours refractory to conventional chemotherapy, we re-implanted orthotopically in the thoracic space small fragments from one of the subcutaneous tumours derived from MSTO-211H cells previously treated with cisplatin plus pemetrexed. After 40 days of treatment, no vehicle-treated mice were alive (0/11; 0%); only three platinum-treated mice were alive (3/11; 27.3%), while seven palbociclib-treated mice were still alive (7/11; 63.6%) (Fig. 4a). Animals (93.9%) were sacrificed due to dyspnoea or excessive weight loss (31/33). Overall survival analysis showed a significant reduction in the risk of death for palbociclib-treated mice compared with vehicle (hazard ratio (HR) = 0.04 [95% confidence interval (CI) 0.01–0.17]) or with cisplatin (HR = 0.11 [95% CI 0.03–0.41]).

Two advanced orthotopic models were generated by implantation in the pleural space of mice small solid fragments (2–3 mm3) of previously generated (a–c) MSTO-211H subcutaneous cisplatin plus pemetrexed-resistant tumour xenograft or (d–f) chemoresistant patient-derived tumour xenograft. a Kaplan–Meier curves showing survival of MSTO-211H orthotopic tumour-bearing mice (n = 33). b Representative MSTO-211H images of orthotopic tumours dissected from each group of treatment and c histological characterisation on H&E sections (scale bars = 100 µm). d Kaplan–Meier curves showing survival of ICO_MPM3 orthotopic tumour-bearing mice (n = 29). e Representative images of patient-derived orthotopic tumours dissected from each group of treatment and f histological characterisation on H&E sections (scale bars = 100 µm). Both orthotopic models accurately reproduce human MPM disease characteristics as tumours grown from the site of implantation to all the pleural space. Tumour mass area is delimited by a white line.
Those mice that were alive after 40 days of treatment (n = 10) were maintained without treatment and followed up until the endpoint. After 6 days of stopping treatments, all the remaining platinum-treated mice (n = 3) were dead, whereas two out of seven palbociclib-treated mice were still alive after 2 months without receiving any treatment. Palbociclib treatment did not exert any substantial change in body weight between the first and the last day of treatment (Supplementary Fig. S4B). Representative pictures of MSTO-211H orthotopic tumours from each group of treatment are shown in Fig. 4b and Supplementary Fig. S6. Histopathological analysis of the MPM tumour xenografts grown orthotopically in mice accurately reproduced the natural history of mesothelioma (Fig. 4c and Supplementary Fig. S7).
Next, we generated an additional orthotopic model by implanting into the pleural space of nude mice small tumour fragments harvested from a xenograft generated by injecting subcutaneously the patient-derived cell line ICO_MPM3. Orthotopically implanted mice (n = 31) were allocated in the different treatment groups and treatment was started 15 days after implantation, and mice were treated continuously for 52 days. Then, mice were maintained alive without treatment and followed up until the human endpoint. (Fig. 4d). Overall survival analysis shows a significant reduction in the risk of death for palbociclib-treated mice compared with the vehicle group (HR = 0.23 [95% CI 0.06–0.85]; p = 0.027). No significant differences were observed in the risk of death for gemcitabine-treated mice (HR = 0.50 [95% CI 0.17–1.40]; p = 0.19) and abemaciclib-treated mice (HR = 0.42 [95% CI 0.14–1.23]; p = 0.12) compared to vehicle. Representative images of orthotopic tumours from each treatment group are shown in Fig. 4e, f.
Gene expression profiling in cell lines and xenografts treated with CDK4/6 inhibitors
To determine the functional consequences of CDK4/6 inhibitor treatment, we performed a transcriptomic analysis of MSTO-211H cells treated with abemaciclib or palbociclib at 250 nM for 72 h. In addition, tumour xenografts treated with palbociclib were also evaluated. After treatment with either CDK4/6 inhibitor, a significant downregulation in the expression levels was observed in the MSTO-211H cell line for genes related to cell cycle, such as regulation of transcription genes involved in G1–S transition of the mitotic cell cycle, nucleus organisation and mitotic spindle assembly and organisation (Supplementary Fig. S8 and Supplementary Table S3). On the other hand, there was a significant upregulation of genes related to interferon signalling pathways, lymphocyte migration and chemotaxis, complement activation and antigen presentation pathways, such as major histocompatibility complex protein. Furthermore, the transcriptomic analysis of palbociclib-treated tumours in xenografted mice showed similar results (Supplementary Fig. S9).
CDK4 and CDK6 overexpression are associated with poor prognosis in patients with MPM
Based on these results, we evaluated the prognostic value of CDK4 and CDK6 overexpression using publicly available transcriptomic data from two cohorts of MPM patients. Patients with CDK4 overexpression (i.e. above the median) had significantly shorter overall survival in both cohorts (12.6 and 13.3 months, respectively) compared with patients with lower expression (23.5 and 25.9 months, respectively). CDK4 overexpression remained statistically significant after adjusting by age, gender, tumour stage and histologic subtype in each dataset, as well as in the combined analysis of both series (HR = 2.10 [95% CI 1.53–2.88]; p = 4.2e–06; Fig. 5a). Patients with CDK6 overexpression (i.e. above the median) had significantly shorter overall survival (12.6 months) compared with patients with lower expression (20.3 months; p = 0.00026) in the Bueno cohort and there was a trend toward shorter overall survival in the TCGA dataset (15 vs 23.6 months; p = 0.060). Nevertheless, CDK6 overexpression remained statistically significant in the combined analysis after adjusting by age, gender, tumour stage and histologic subtype in the Bueno cohort, and also in the combined cohort (HR = 1.74 [95% CI 1.32–2.29]; p = 5.4e–05; Fig. 5b).

Kaplan–Meier plots of overall survival (OS) in MPM patients according to a CDK4 and b CDK6 gene expression levels based on data obtained from Bueno et al. (left column) and Hmeljak et al. (middle column) cohorts or the combination of both (right column). High levels of CDK4 or CDK6 (red line) were significantly associated with poor OS in patients with MPM. In each cohort, the high and low expression levels were defined based upon the median. P values and hazard ratios (HRs) were calculated by likelihood ratio test and multivariate Cox regression analysis, respectively.
As previously reported [9, 10], low expression of CDKN2A was associated with shorter overall survival in both cohorts and was independently associated with worse prognosis in the combined cohort including Bueno and TCGA (HR = 0.49 [95% CI 0.36–0.66]; p = 3.4e–06; Supplementary Fig. S10A). In the TCGA cohort, only two tumours harboured an RB1 homologous deletion, while CDKN2A/p16 deletion was a common event present in 34 out of 74 cases (46%).
CDKN2A copy number was assessed in an independent cohort of 79 MPM acquired at radical surgery involving extended pleurectomy decortication. Patient clinicopathological characteristics are outlined in Supplementary Table S4. Homozygous loss of 9p21.3 encompassing CDKN2A was observed in 40 samples (50.6%), while copy number loss/LOH was observed in 18 (22.7%). CDKN2A homozygous loss was associated with shorter median overall survival (10.98 months) compared to euploid CDKN2A (45.8 months; HR = 0.37 [95% CI 0.22–0.62]; p = 0.0002; Supplementary Fig. S10B). CDKN2A copy number loss/LOH was associated with shorter median overall survival (8.52 months) compared to wild-type CDKN2A (45.8 months; HR = 0.18 [95% CI 0.08–0.40]; p = 0.0001). There were no statistically significant differences in overall survival among patients harbouring CDKN2A homozygous deletion compared to those with CDKN2A copy number loss/LOH (HR = 0.89 [95% CI 0.49–1.59]; p = 0.158).
Discussion
MPM is a rapidly fatal neoplastic disease in which therapeutic options are limited. We investigated the role of CDK4/6 inhibition in MPM because cell cycle deregulation is a relevant hallmark in this disease and CDKN2A/p16 deletion is a common genomic event associated with worse clinical outcomes in MPM [17, 18].
The efficacy of palbociclib has been previously studied in in vitro models of MPM [29]. However, the antitumour activity of CDK4/6 inhibitors has not yet been evaluated using primary patient-derived cell models of MPM neither in vivo models of MPM. In our work, we assessed the efficacy of two CDK4/6 inhibitors, abemaciclib and palbociclib, in a subset of five commercial and three primary patient-derived cell culture models obtained from pleural effusions of patients with MPM (one chemotherapy-naive and two after progression to standard first-line chemotherapy). Furthermore, we performed not only in vivo basic subcutaneous drug response studies in xenografts derived from one chemotherapy-naive MPM cell line but also advanced studies by means of orthotopic implanted xenografts from a cell line-derived tumour previously treated with cisplatin plus pemetrexed and from a chemoresistant primary cell line-derived tumour. Remarkably all the cell lines were sensitive to palbociclib and to abemaciclib. Treatment with abemaciclib or palbociclib significantly reduced cell proliferation, as evaluated by cell number counting or by colony formation ability, in all the cell lines including in the primary ones derived from pleural liquid from patients resistant to chemotherapy. In vitro experiments underscore two important points: (i) no substantial differences were found in the antiproliferative effect of both inhibitors; and (ii) the sensitivity to CDK4/6 inhibitors was not correlated with the endogenous expression levels of CDK4 or CDK6. We consider that there is still an unmet need for biomarkers able to predict clinical benefit to CDK4/6 inhibitors. In our work, we confirmed that CDKN2A/p16 deletion is associated with shorter overall survival in a cohort of MPM patients with resectable disease. These results are consistent with previous publications where CDKN2A/p16 loss predicted worse clinical outcomes in patients with resected and advanced epithelioid MPM [18].
By analysing publicly available transcriptomic data of MPM, we found that overexpression of CDK4 or CDK6 is also associated with shorter overall survival. Together, these findings suggest that cell cycle deregulation may confer an aggressive biological behaviour in mesothelioma and reinforce our hypothesis about further investigating CDK4/6 inhibitors in MPM patients.
We investigated the impact of abemaciclib and palbociclib treatment on cell cycle progression, cellular senescence and apoptosis induction. These experiments were performed using three cell lines that were sensitive to both drugs, including a primary cell line derived from a patient resistant to chemotherapy. As expected, the treatment with abemaciclib and palbociclib caused cell cycle arrest at the G1 phase but also promoted cellular senescence. However, neither abemaciclib nor palbociclib activated programmed cell death or apoptosis as indicated by negligible sub-G1 accumulation or propidium iodide incorporation. The observed increase in cellular senescence induced by both drugs could be linked to apoptosis resistance mechanisms [23, 32]. As proposed by other groups [29, 33, 34], our results reinforce the cytostatic mechanism of action of CDK4/6 inhibitors and underscore that these should be given sequentially after completing chemotherapy treatment [35]. However, the low cell death observed in vitro does not eliminate the possibility that palbociclib, by inducing senescence in a few cells, may promote in vivo cytotoxicity mediated by NK cells. This phenomenon has been described in lung cancer, in which NKs participate in tumour reduction upon treatment with MEK inhibitors and palbociclib [36, 37].
To explore potential activation of compensatory pathways, we performed gene set enrichment analysis of the transcriptome of MSTO-211H cells treated with abemaciclib or palbociclib in vitro or subcutaneously implanted in mice. This experiment showed downregulation of genes involved in G1–S transition of the mitotic cell cycle, nucleus organisation and mitotic spindle assembly and organisation. In concordance with studies conducted in other tumour types, genes encoding interferon signalling and antigen presentation pathways were upregulated after CDK4/6 pharmacological inhibition [34]. In melanoma, CDK4/6 inhibition activates p53 by lowering PRMT5, which leads to altered MDM4 splicing and significantly reduced protein expression [38, 39]. Other studies performed in breast cancer cell lines and transgenic mice models have shown that abemaciclib treatment increased the expression of antigen processing and presentation and even suppressed the proliferation of regulatory T cells [24, 34]. In the present study, we have observed that palbociclib treatment increase the number of tumour-associated macrophages in subcutaneous MPM tumour xenografts and there was a trend toward higher NK cell infiltration, but further studies evaluating the functional consequences of the treatment with CDK4/6 inhibitors on the tumour immune contexture are warranted in mesothelioma.
To the best of our knowledge, this is the first time that the effectiveness of abemaciclib and palbociclib was evaluated using preclinical in vivo subcutaneous and orthotopic MPM tumour models. Among the available cell line models, the MSTO-211H cell line was selected for the in vivo subcutaneous and orthotopic experiments because (i) it expresses CDK4 and CDK6; (ii) it is the most sensitive cell line to palbociclib at 500 nM; and (iii) it was tumorigenic in athymic mice. Our results showed that palbociclib reduced tumour size in a subcutaneous mouse model of chemo-naïve MSTO-211H cells compared with standard chemotherapy (cisplatin plus pemetrexed). An increased expression of anti-apoptotic proteins after long-term chemotherapy treatment could explain the chemoresistance in this model [40]. We tried to replicate a situation representing treatment after progression to platinum-based chemotherapy by generating an orthotopic tumour mouse model and implanting in the pleura small solid fragments of MSTO-211H xenografted tumours previously treated with chemotherapy. In this advanced model of MPM, palbociclib significantly increased the overall survival of mice compared with cisplatin-based chemotherapy or vehicle; the benefits from treatment persisted even after stopping the treatment. Our results reinforce the potential use of palbociclib as a second-line treatment for patients with MPM that is resistant or has relapsed after standard chemotherapy doublet treatment. Finally, we also used ICO_MPM3, a patient-derived cell line also tumorigenic in athymic mice to test the efficacy of CDK4/6 inhibitors in vivo.
Some limitations of our study are the absence of a wide range of available commercial MPM cell lines and the need for preclinical in vivo models representing the heterogeneity of the disease, including the adaptive immune system. However, in our study, we have combined commercial and primary patient-derived lines as well as orthotopic models where mesothelioma grows in its corresponding microenvironment and can recapitulate the disease behaviour.
A phase II clinical study of abemaciclib in patients harbouring p16ink4a-deficient, relapsed MPM has recently completed accrual (NCT03654833). CDK4/6 inhibition in this cohort has been associated radiological responses; however, the underlying molecular correlates of response are under investigation. Accordingly, WES of the trial cohort is planned to uncover genomic determinants of response.
In conclusion, our data support that treatment with CDK4/6 inhibitors, abemaciclib or palbociclib, can reduce cell proliferation and induce cellular senescence in MPM cell lines and palbociclib can increase overall survival of mice with orthotopically implanted MPM cells. A remarkable and sustained response to palbociclib was observed in xenografts of MPM tumour resistant to cisplatin and pemetrexed, which was then implanted orthotopically in the pleural space of mice. Transcriptomic analysis of cell lines and xenografted tumours treated with CDK4/6 inhibitors showed an increased expression of interferon signalling pathway and antigen-presenting processes, suggesting that CDK4/6 inhibitors may favour potential response to immunotherapy. Our results warrant further evaluation of CDK4/6 inhibitors as a second-line treatment in patients with advanced MPM that has failed standard platinum-based chemotherapy.
Data availability
All data generated or analysed during this study will be stored in EGA and available on reasonable request.
References
Delgermaa V, Takahashi K, Park EK, Le GV, Hara T, Sorahan T. Global mesothelioma deaths reported to the World Health Organization between 1994 and 2008. Bull World Health Organ. 2011;89:716–24, 24A–24C.
Chen T, Sun XM, Wu L. High time for complete ban on asbestos use in developing countries. JAMA Oncol. 2019;5:779–80.
Hassan R, Morrow B, Thomas A, Walsh T, Lee MK, Gulsuner S, et al. Inherited predisposition to malignant mesothelioma and overall survival following platinum chemotherapy. Proc Natl Acad Sci USA. 2019;116:9008–13.
Panou V, Gadiraju M, Wolin A, Weipert CM, Skarda E, Husain AN, et al. Frequency of germline mutations in cancer susceptibility genes in malignant mesothelioma. J Clin Oncol. 2018;36:2863–71.
Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43:1022–5.
Guo R, DuBoff M, Jayakumaran G, Kris MG, Ladanyi M, Robson ME, et al. Novel germline mutations in DNA damage repair in patients with malignant pleural mesotheliomas. J Thorac Oncol. 2020;15:655–60.
Zauderer MG, Jayakumaran G, DuBoff M, Zhang L, Francis JH, Abramson DH, et al. Prevalence and preliminary validation of screening criteria to identify carriers of germline BAP1 mutations. J Thorac Oncol. 2019;14:1989–94.
Woolhouse I, Bishop L, Darlison L, de Fonseka D, Edey A, Edwards J, et al. BTS guideline for the investigation and management of malignant pleural mesothelioma. BMJ Open Respir Res. 2018;5:e000266.
Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21:2636–44.
Zalcman G, Mazieres J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet. 2016;387:1405–14.
Popat S, Curioni-Fontecedro A, Dafni U, Shah R, O’Brien M, Pope A, et al. A multicentre randomised phase III trial comparing pembrolizumab versus single-agent chemotherapy for advanced pre-treated malignant pleural mesothelioma: the European Thoracic Oncology Platform (ETOP 9-15) PROMISE-meso trial. Ann Oncol. 2020;31:1734–45.
Fennel DO, Ottensmeier C, Califano R, Hanna GG, Ewings S, Hill K, et al. Nivolumab versus placebo in relapsed malignant mesothelioma: preliminary results from the CONFIRM phase 3 trial. In: Presented at: International Association for the Study of Lung Cancer 2020 World Conference on Lung Cancer_Abstract PS01.11. 2020.
Baas P, Scherpereel A, Nowak AK, Fujimoto N, Peters S, Tsao AS, et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet. 2021;397:375–86.
Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48:407–16.
Hmeljak J, Sanchez-Vega F, Hoadley KA, Shih J, Stewart C, Heiman D, et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 2018;8:1548–65.
Björkqvist AM, Tammilehto L, Anttila S, Mattson K, Knuutila S. Recurrent DNA copy number changes in 1q, 4q, 6q, 9p, 13q, 14q and 22q detected by comparative genomic hybridization in malignant mesothelioma. Br J Cancer. 1997;75:523–7.
López-Ríos F, Chuai S, Flores R, Shimizu S, Ohno T, Wakahara K, et al. Global gene expression profiling of pleural mesotheliomas: overexpression of aurora kinases and P16/CDKN2A deletion as prognostic factors and critical evaluation of microarray-based prognostic prediction. Cancer Res. 2006;66:2970–9.
Dacic S, Kothmaier H, Land S, Shuai Y, Halbwedl I, Morbini P, et al. Prognostic significance of p16/cdkn2a loss in pleural malignant mesotheliomas. Virchows Arch. 2008;453:627–35.
Hylebos M, Van Camp G, van Meerbeeck JP, Op de Beeck K. The genetic landscape of malignant pleural mesothelioma: results from massively parallel sequencing. J Thorac Oncol. 2016;11:1615–26.
Frizelle SP, Grim J, Zhou J, Gupta P, Curiel DT, Geradts J, et al. Re-expression of p16INK4a in mesothelioma cells results in cell cycle arrest, cell death, tumor suppression and tumor regression. Oncogene. 1998;16:3087–95.
Middleton G, Fletcher P, Popat S, Savage J, Summers Y, Greystoke A, et al. The National Lung Matrix Trial of personalized therapy in lung cancer. Nature. 2020;583:807–12.
Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6:353–67.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35.
Turner NC, Huang Bartlett C, Cristofanilli M. Palbociclib in hormone-receptor-positive advanced breast cancer. N. Engl J Med. 2015;373:1672–3.
Oie HK, Russell EK, Carney DN, Gazdar AF. Cell culture methods for the establishment of the NCI series of lung cancer cell lines. J Cell Biochem Suppl. 1996;24:24–31.
Nadal E, Chen G, Gallegos M, Lin L, Ferrer-Torres D, Truini A. Epigenetic inactivation of microRNA-34b/c predicts poor disease-free survival in early-stage lung adenocarcinoma. Clin Cancer Res. 2013;19:6842–52.
Bollard J, Miguela V, Ruiz de Galarreta M, Venkatesh A, Bian CB, Roberto MP, et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut. 2017;66:1286–96.
Püschel F, Muñoz-Pinedo C. Measuring the activation of cell death pathways upon inhibition of metabolism. Methods Mol Biol. 2019;1862:163–72.
Bonelli MA, Digiacomo G, Fumarola C, Alfieri R, Quaini F, Falco A. et al. Combined inhibition of CDK4/6 and PI3K/AKT/mTOR pathways induces a synergistic anti-tumor effect in malignant pleural mesothelioma cells. Neoplasia. 2017;19:637–48.
Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2020.
Ambrogio C, Carmona FJ, Vidal A, Falcone M, Nieto P, Romero OA, et al. Modeling lung cancer evolution and preclinical response by orthotopic mouse allografts. Cancer Res. 2014;74:5978–88.
Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40.
Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77.
Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548:471–5.
Salvador-Barbero B, Alvarez-Fernández M, Zapatero-Solana E, El Bakkali A, Menéndez MDC, López-Casas PP, et al. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell. 2020;38:584.
Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science. 2018;362:1416–22.
Wagner V, Gil J. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene. 2020;39:5165–76.
Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, et al. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013;27:1903–16.
AbuHammad S, Cullinane C, Martin C, Bacolas Z, Ward T, Chen H, et al. Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proc Natl Acad Sci USA. 2019;116:17990–8000.
Varin E, Denoyelle C, Brotin E, Meryet-Figuière M, Giffard F, Abeilard E, et al. Downregulation of Bcl-xL and Mcl-1 is sufficient to induce cell death in mesothelioma cells highly refractory to conventional chemotherapy. Carcinogenesis. 2010;31:984–93.
Acknowledgements
We thank CERCA Programme/Generalitat de Catalunya for their institutional support and grant 2017SGR448.
Funding
EN received support from the SLT006/17/00127 grant, funded by the Department of Health of the Generalitat de Catalunya by the call “Acció instrumental d’intensificació de professionals de la salut”. This study was supported by grants from Instituto de Salud Carlos III (grants PI14/01109 and PI18/00920) (co-funded by European Regional Development Fund. ERDF, a way to build Europe), Spanish Society of Medical Oncology grant for emerging research groups and from Pfizer (WI244174) to EN. MH-M was supported by a Marie Skłodowska-Curie grant, agreement no. 766214.
Author information
Authors and Affiliations
Contributions
EA: conceptualisation, methodology, validation, formal analysis, investigation, resources, data curation, writing—original draft, writing—review and editing, visualisation. AA: conceptualisation, software, validation, formal analysis, resources, data curation, writing—review and editing, visualisation. MM-I: methodology, investigation, resources, data curation. MH-M: methodology, validation, investigation, data curation. DC: conceptualisation, software, validation, formal analysis, data curation. MG: conceptualisation, resources, writing—review and editing. EP: resources, writing—review and editing. MS: resources, writing—review and editing. SB: software, validation, data curation. AJS: resources, writing—review and editing. AD: resources, writing—review and editing. RP: resources, data curation, writing—review and editing. SP: resources, writing—review and editing. SA: resources, writing—review and editing. IE: resources, writing—review and editing. RR: resources, writing—review and editing. RL: resources, validation, data curation. AV: resources, validation, data curation. ED: data curation. MV: resources, validation, data curation. MS-C: resources, writing—review and editing. DF: resources, validation, formal analysis, investigation, data curation, writing—review and editing. CM-P: conceptualisation, methodology, validation, formal analysis, investigation, data curation, writing—review and editing, visualisation. AV: conceptualisation, methodology, validation, formal analysis, investigation, data curation, writing—review and editing, visualisation. XS: conceptualisation, software, formal analysis, data curation, writing—review and editing, visualisation. EN: conceptualisation, methodology, validation, formal analysis, investigation, resources, data curation, writing—original draft, writing—review and editing, visualisation, supervision, project administration, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
EN received research support from Roche, Pfizer, Bristol Myers Squibb and Merck Serono and participated in advisory boards from Bristol Myers Squibb, Merck Serono, Merck Sharpe & Dohme, Lilly, Roche, Pfizer, Takeda, Boehringer Ingelheim, Bayer, Amgen and Astra Zeneca. RP has participated in advisory boards from Bristol Myers Squibb, Merck Sharpe & Dohme, Roche, Pfizer, Lilly, Boehringer Ingelheim and Astra Zeneca. DF has received research support from Astex Therapeutics, Astra Zeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Clovis Oncology, Merck Sharpe & Dohme, Lilly Oncology, Roche and participated in advisory boards from Atara Therapeutics, Bayer, Boehringer Ingelheim and Inventiva. The other authors declare no competing interests.
Ethics approval and consent to participate
The retrospective cohort was approved by a National Ethical Committee, under the references 4/LO/1527 (a translational research platform entitled Predicting Drug and Radiation Sensitivity in Thoracic Cancers—also approved by University Hospitals of Leicester NHS Trust under the reference IRAS131283) and 14/EM/1159 (retrospective cohort). Pleural effusions samples were obtained after patients signed the informed consent approved by the Hospital de Bellvitge Ethical Committee (PR152/14). All the animal experiments were performed in accordance with protocols approved by Animal Research Ethics Committee at IDIBELL. This study was performed in accordance with the principles outlined in the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aliagas, E., Alay, A., Martínez-Iniesta, M. et al. Efficacy of CDK4/6 inhibitors in preclinical models of malignant pleural mesothelioma. Br J Cancer 125, 1365–1376 (2021). https://doi.org/10.1038/s41416-021-01547-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01547-y
- Springer Nature Limited