
Abstract
Epstein-Barr virus (EBV) is a ubiquitous herpes virus that infects the majority of the population worldwide. The virus can establish a lifelong latent infection in host B-lymphocytes. In the setting of immunocompromise as is the case post transplantation, the virus can reactivate and cause one of the deadliest complications post hematopoietic stem cell transplantation (HSCT), post-lymphoproliferative disease (PTLD), the incidence of which has been increasing. Multiple risk factors have been associated with the onset of PTLD such as age, reduced intensity conditioning, EBV serology mismatch and cytomegalovirus (CMV) reactivation. The rarity of clinical trials involving PTLD and the lack of approved treatment modalities renders the management of PTLD challenging. While the first-line treatment involves weekly administration of rituximab, there is no consensus when treating rituximab-refractory PTLD. There is a handful of clinical trials that investigate the role of EBV-specific cytotoxic T-lymphocytes (CTLs) and novel agents, such as bortezomib, lenalidomide, everolimus, panobinostat, and brentuximab. This article aims to explore the entity of EBV-PTLD in HSCT recipients, expanding on clinical presentation, risk factors, modes of monitoring and treatment, and so highlighting the gaps in knowledge that are needed in order to build a treatment paradigm suitable for all patients at risk.
Similar content being viewed by others


Introduction
Epstein-Barr virus (EBV) is a ubiquitous herpes virus that establishes an infection in ~50% to 89% of children and over 90% of the adult population worldwide [1, 2]. It is infamous for causing one of the deadliest complications post hematopoietic stem cell transplantation (HSCT), post-transplant lymphoproliferative disease (PTLD), which plays a significant role in plummeting patient survival rates. Because of the low incidence of PTLD, there have been very few clinical trials that explore modalities to prevent, treat pre-emptively, and eradicate the disease once it develops. This article aims to explore the entity of EBV-PTLD in HSCT recipients, expanding on clinical presentation, risk factors, modes of monitoring, and treatment, and so highlighting the gaps in knowledge that are needed in order to build a treatment paradigm suitable for all patients at risk.
Pathogenesis
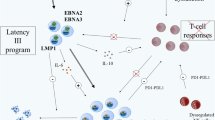
The virus preferentially infects B-lymphocytes via its target receptor CD21 whereby EBV exists in an asymptomatic latent state for the lifetime of the host but could, in the optimal setting of immune dysfunction, develop into EBV-related lymphomas. In an immunocompetent individual, after the primary infection, infectious mononucleosis, the virus is controlled by EBV-specific cytotoxic T-lymphocyte (CTL) activity [3]. Here, the virus circularizes its genome, limiting viral gene expression and establishing chronic latency [2, 4]. The panel of expressed viral latency proteins can drive naive B-cells into post-germinal center memory B-cells, allowing EBV to persist quiescently in peripheral blood and in the reticuloendothelial system [5]. This concept becomes important when considering antiviral agents such as ganciclovir that would usually target EBV during an active infection. Drugs such as ganciclovir require phosphorylation by viral kinases, which are usually not expressed during a latent infection thus rendering their use inadequate [6].
After establishing latency, the virus is capable of maintaining dormancy by hijacking and thus evading the host’s immune system. For instance, chronic exposure of an individual’s immune system to EBV latency proteins, such as latent-membrane protein (LMP), can activate the JAK/STAT pathway within infected B-cells resulting in increased expression of the co-inhibitory receptor, PD-L1 [7]. This results in faulty and exhausted immunity to EBV through T-cell anergy when bound to the PD-1 receptor [7]. In addition, the virus downgrades MHC class I and II proteins thus eluding immune destruction [8]. Even though HLA-class I downregulation allows EBV to evade the host’s immune system, it makes it more susceptible to natural killer (NK) cells [9]. One study explored this by studying the NK response of three donor peripheral blood mononuclear cells (PBMCs) to Ataka cells (Burkitt lymphoma cell line that is latently infected with EBV) compared to EBV-negative Ataka cells after incubation [10]. A significantly larger number of NK cells (p-value < 0.001) was evident after incubation of donor PBMCs with EBV-positive Ataka cells demonstrating the role of NK cells in immunity against EBV-positive cells [10].
While persistent latent EBV infection is common, even though rare, it is possible for an individual to develop EBV-related lymphomas. This is usually modulated by ethnic, geographic, genetic, immunologic, and infectious cofactors [11]. In immunocompromised individuals, compromised T-lymphocyte immunity allows EBV to cause EB viremia and fatal EBV-related lymphomas by extending the lifespan of virally infected B-lymphocytes and thus increasing their likelihood of gaining mutations such as alterations to BCL6, MYC, NF-kB, PI3K/AKT/mTOR, BCL2, and immunoglobulin switching that allows stepwise progression from early polymorphic lymphoma lesions to more advanced monoclonal ones [12,13,14,15,16,17]. Although >90% of EBV-PTLD cases in solid organ transplant (SOT) recipients are of host origin, the vast majority of EBV-PTLD post HSCT are of donor origin [18]. When immunocompromise is in the setting of HSCT, the transplant creates a unique immunity depleted environment paving the way for donor-derived EBV-infected B-lymphocytes to hijack the immunocompromised host and develop life-threatening complications such as PTLD [19].
While early post allogeneic transplantation PTLD is EBV-dependent, late onset PTLD is not. EBV-positive PTLD is usually reported within the first year post transplantation, with the majority of cases occurring within the first 6 months [20]. EBV-negative PTLD on the other hand, occurs >5 years post transplantation, with some reported cases as late as 10 years post transplantation. On a genomic level, there appears to be a significant difference between the two entities: EBV-negative cases share genomic features with diffuse large B-cell lymphoma in immunocompetent individuals whereby EBV-positive cases do not [7, 21]. This is reflected in the more numerous and more complex molecular aberrations in EBV-negative PTLD compared to EBV-positive PTLD [7, 21]. Thus, with fewer molecular/genetic changes, EBV-positive PTLD occurs in the early phase post transplantation during the nadir of immunocompromise. As such, EBV-negative PTLD is comparative to large B-cell lymphoma outside of the setting of transplantation. Irrespective of the clear differences between the two entities, they appear to respond to treatment similarly whereby EBV status does not appear to have a prognostic value [21].
EBV-PTLD epidemiology
The incidence of EBV-PTLD post allogeneic HSCT ranges from 0.5 to 17% [22,23,24,25]. The incidence rates of PTLD are witnessing an upward surge due to the exponentially increasing number of transplantations, relatively older donors and recipients, new immunosuppressive drugs, emergence of and increased demand for haploidentical HSCT, and increased awareness of the disorder [26]. This has however, led to improved diagnostic tools.
Clinical presentation and diagnosis
Clinically, PTLD presents with prolonged fever, lymphadenopathy, intermittent tonsillar enlargement, and progressive reduction in cellular lineages on complete blood count [27]. EBV can infect systems other than the hematopoietic system causing a myriad of events such as hepatitis, colitis, pneumonia, nephritis, and cerebritis [23, 28]. According to the 2016 World Health Organization (WHO) classification, PTLD can be classified as: plasmacytic hyperplasia, infectious mononucleosis, florid follicular hyperplasia, polymorphic, monomorphic, and classic Hodgkin lymphoma [29].
PTLD can develop at any time after receiving a transplant, even up to over 10 years post transplantation [30]. Nonetheless, the majority (60%) of cases develop within the first year of transplantation [31, 32]. This is in accordance with previous investigations, which demonstrated that the risk of developing EBV-PTLD is highest within the first year after HSCT with 83% of the patients presenting during this timeframe and with the majority presenting within the first 6 months post transplantation [20]. As mentioned earlier, while early PTLD tends to be EBV dependent, late PTLD does not [21].
PTLD, notorious for being one of the most fatal post-HSCT complications, can drastically affect survival. Developing PTLD alone decreases survival of patients from 62% to 20% [33]. Previously analyzed mortality rates among PTLD patients from >200 transplant centers found mortality due to PTLD to reach 84% [34]. Nonetheless, strict implementation of diagnostic and therapeutic strategies such as monitoring EBV DNAemia and pre-emptive therapy with novel drugs such as rituximab have managed to improve mortality due to PTLD from 84% before the year 2000 to 30% in 2013 [34,35,36].
To establish a diagnosis of EBV-PTLD, two of the following conditions must be met: (a) a lymphoproliferative process disrupting underlying cellular architecture, (b) presence of monoclonal or oligoclonal viral markers, and (c) established EBV infection with detectable viral nucleic acid or protein [29]. Once PTLD is suspected based on the clinical picture or rising peripheral blood EBV DNA levels, a tissue biopsy is required to confirm the diagnosis of PTLD and differentiate it from other entities [37]. The specimen is usually acquired via core needle or excisional biopsy from the most central or most fludeoxyglucose (FDG)-avid area of the tumor [37]. The specimen needs to be tested for clonal immunoglobulin heavy or light-chain, T-cell receptor gene rearrangements and EBV-LMP immunostaining [37]. It is important to stress that EBV nucleic acid in blood alone is not sufficient to establish a diagnosis of EBV-PTLD [35]. Once the diagnosis of EBV-PTLD is confirmed, a clinical evaluation becomes necessary to assess the burden of the disease through positron emission tomography/computed tomography (PET-CT), bone marrow biopsy, cerebrospinal fluid analysis, and magnetic resonance imagining of the brain and spine in the presence of neurological symptoms (Fig. 1) [37].

Proposed diagnostic and therapeutic approach to post-transplant lymphoproliferative disease (PTLD)
Multiple prognostic factors that affect overall survival negatively and influence clinician decision-making have been elucidated. Among them is older age, failure to respond to rituximab therapy, delayed immune reconstitution reflected by thrombocytopenia (<50 × 109/L) and leukopenia (<0.5 × 109/L) at the time of diagnosis, malignant disease at the time of PTLD diagnosis, presence of extranodal disease, and the presence of acute GVHD ≥ grade II [36, 38]. Elevated lactate dehydrogenase (LDH) levels have been previously identified as prognostic correlates in PTLD post SOT, but there are concerns regarding their applicability in the setting of PTLD post HSCT [39]. Nonetheless, it strongly correlates with the clinical manifestation of PTLD [30].
EBV-PTLD risk factors
Multiple risk factors that increase the likelihood of developing EBV-PTLD post HSCT have been elucidated. Patient baseline characteristics such as advanced age, especially above 50 years, and history of splenectomy pose significant risk for PTLD development, along with other transplant related factors described below. As a patient accumulates risk factors, the risk of developing PTLD also increases [33]. In one study, individuals with one risk factor had a 0.4% risk of developing PTLD, while individuals with two, three, four, and five risk factors had an increased risk of 3%, 10.4%, 26.5%, and 40%, respectively [33].
Reduced intensity conditioning
Reduced intensity conditioning (RIC) is usually used to decrease toxicity and early mortality following HSCT. Uhlin et al. [33] investigated 1021 patients, 4% of which developed PTLD, and found the use of RIC to be an independent risk factor for developing EBV-PTLD (hazard ratio (HR) 3.25; p-value 0.002). The influence of RIC on PTLD risk could be explained by EBV’s tropism to B-lymphocytes. After RIC, EBV-infected lymphocytes have a prolonged period of freedom to undergo transformation in the absence of normally restricting EBV-specific T-lymphocytes [33]. In addition, several studies have suggested that T-cell reconstitution is delayed after receiving RIC compared to myeloablative conditioning (MAC) [40, 41]. One study compared EBV-specific CTL reconstitution between HSCT patients who received RIC and those who received MAC, revealing that EBV-specific CTLs were detectable in only 1/9 patients at 6 months and 7/10 patients at 1 year in the RIC group compared to 6/9 and all patients, respectively, in the MAC group [40].
Degree of HLA-mismatch
Uhlin et al. [33] demonstrated an increased risk (HR 5.89, p-value < 0.001) of developing EBV-PTLD in recipients of HLA-mismatched grafts as opposed to recipients of HLA-identical grafts. Another study demonstrated that recipients of HSCT from a two or more HLA antigen-mismatched donor (relative risk (RR), 3.1) or unrelated donor (RR 4.2) had a higher risk of PTLD than patients receiving their transplant from an identical donor or donors with one HLA-mismatch (RR 1.8) [20].
Over the past decade, results from multiple studies regarding a possible association between EBV-PTLD and specific HLA-class I antigens have been conflicting. The inconsistency was perhaps influenced by different methodologies [42,43,44,45,46]. Jones et al. [47] tested HLA-class I associations with lymphoma (classical Hodgkin’s lymphoma or PTLD) in 263 patients, 97 of which had developed EBV-PTLD and no HLA-class I antigen associations with EBV-PTLD were found.
EBV donor/recipient serology mismatch
As expected, EBV serological mismatch between recipient and donor is another significant risk factor for reactivating EBV and developing PTLD, especially when the recipient is serologically EBV-negative and the donor is serologically EBV-positive [48, 49]. Such a combination has been shown to be a significant risk factor for developing PTLD in HSCT recipients with a HR of 4.97 (p-value < 0.001) [33]. The mechanism is not quite clear, with multiple studies agreeing with this and others failing to find a similar association [49, 50]. This could be explained by the total absence of EBV-specific CTLs in the recipient for the lack of previous exposure to EBV, and hence upon receiving a transplant from an EBV-positive donor, the donor’s EBV-infected B-lymphocytes are able to flourish without the slightest setback. As such when dealing with older patients who are conditioned with RIC, it is important for clinicians to factor in EBV donor/recipient serology when selecting a suitable donor especially with the availability of multiple donors.
Umbilical Cord Blood Transplantation (UCBT)
UCBT is an alternative used for patients who require an allogeneic HSCT but lack an HLA-matched donor with the advantage of immediate availability, low infection transmission risk, and lower than expected severe graft-versus-host disease (GVHD) rates [51,52,53]. Rates of EBV-related complications post UCBT have been found comparable to rates post bone marrow transplant (BMT) or peripheral blood stem cell (PBSC) infusion, whereby Barker et al.’s [54] multicenter retrospective analysis of 272 patients demonstrated a comparable rate of 2%. Nonetheless, the risk of developing PTLD post UCBT was significantly higher with the use of RIC instead of MAC regimens especially when combined with anti-thymocyte globulin (ATG). Brunstein et al. [55] and Peric et al. [56] demonstrated higher rates of 6–7% with the use of RIC regimens. When RIC was combined with ATG, Brunstein noted a 21% incidence of EBV-related complications whereby Peric noted a comparable 25% [55, 56]. This being said, the risk of EBV-PTLD associated with UCBT is in part due to the increased use of RIC and ATG in this setting.
Use of anti-thymocyte globulin (ATG) and T-cell depletion
T-cell depletion, which is increasingly used due to the increase in dependence on haploidentical transplantation, was found to increase the likelihood of reactivating EBV and as such, developing PTLD. T-cell depletion includes the in vivo and ex vivo depletion of T-cells from a graft in the hope of decreasing GVHD incidence and severity. Depleting T-cells depletes EBV-specific CTLs, which would compromise T-cellular immunity, the cornerstone of viral immunity, thus increasing the chance of EBV reactivation and PTLD development. Historically, total T-cell depletion was associated with worse outcomes mainly due to infections with CMV and EBV. Ninety-five percent of T-cells have been found to have the αβ receptor whereby the rest have the γδ receptor [57]. Selective depletion of αβ T-cells would decrease the risk of GVHD, all while allowing γδ T-cells to expand in frequency and function thus retaining cellular immunity against viral infections [58]. The role of γδ T-cells, specifically Vδ2-negative γδ T-cells, has already been demonstrated in CMV whereby significant expansions were noted with CMV reactivation and in CMV-seropositive patients [58]. The role of Vδ2-positive γδ T-cells in EBV immunity has been elegantly described, whereby an inverse relationship between Vδ2-positive γδ T-cells counts and EBV reactivation has been highlighted thus stressing their role in viral immunity [59].
Laberko et al. [60] described the outcome of EBV and CMV burden post TCR-α/β- and CD19- depleted grafts. The study demonstrated a 33% cumulative incidence of EBV viremia at 1 year with no relationship to the dose of α/β T-cells or CD19 cells infused [60]. The control of EBV was hypothesized to be due to simultaneous CD19 depletion and rituximab therapy [60]. Demonstrating similar results, Lang and colleagues [61] reported one EBV reactivation after a median follow-up of 367 days in their prospective multicenter, single-arm phase I/II clinical trial assessing the safety and feasibility of transplantation with TCR-α/β- and CD19- depleted haploidentical grafts in pediatric patients.
ATG is an in vivo mechanism to remove both recipient and donor T-cells to decrease the risk of GVHD in HSCT patients [20, 33, 62, 63]. One protocol that relies heavily on the use of ATG is the Beijing protocol, which involves transplanting an unmanipulated haploidentical graft with reliance on ATG for GVHD prophylaxis. There are currently multiple formulations of ATG that differ in terms of production and animal source. ATG is manufactured by immunizing animals (rabbits or horses) with human thymocytes (ATGAM and Thymoglobulin) or with Jurkat T-cells (ATG-F). Currently, there are very few studies that compare different ATG formulations in terms of immune reconstitution and risk of PTLD. One retrospective study compared different doses of ATG-G (thymoglobulin) to ATG-F in pediatric patients treated with allogeneic HSCT [64]. At the presently used doses (5–10 mg/kg ATG-G and 20–60 mg/kg ATG-F), no significant differences in immune reconstitution were observed, but PTLD rates were higher in patients treated with ATG-G [64]. In addition, the use of higher doses of ATG-G (20–40 mg/kg and 50–60 mg/kg) portrayed a dose-dependent effect on delaying immune reconstitution [64]. In addition, rabbit ATG appears to cause more profound lymphocytopenia when compared to its equine counterpart, even though given at higher doses, thus rendering the incidence of PTLD rather rare with the latter [65].
Use of alemtuzumab
Alemtuzumab (Campath) is a monoclonal antibody against CD52, a surface glycoprotein. Alemtuzumab, through poorly understood mechanisms, inhibits T- and B-lymphocytes, which is central to its use in GVHD prophylaxis. The T-cell compromise and delay in EBV-specific CTL reconstitution caused by alemtuzumab lowers viral immunity allowing EBV to grow rapidly. Studies have demonstrated alemtuzumab’s role as a risk factor for developing PTLD post HSCT [40, 66]. Nevertheless, there are opposing studies that have demonstrated a reduced incidence of PTLD with the use of alemtuzumab. This could be due to alemtuzumab’s equivalent effect on B-cells, which are the reservoir for EBV [67, 68].
PTLD risk is higher with some depletion procedures compared to others. For instance, broad lymphocyte depletion (such as the use of alemtuzumab and elutriation/density centrifugation) rather than other selective T-cell depletion procedures has been associated with a lower risk of developing PTLD (T-cell depletion RR 8.4–15.8; broad lymphocyte depletion RR 3.1) [20, 26, 69].
Post-transplantation cyclophosphamide
Another drug that has been increasingly employed in the setting of allogeneic transplantation as GVHD prophylaxis is post-transplant cyclophosphamide. Initially, like multiple other drugs that have proven effective in GVHD prophylaxis, cyclophosphamide was hypothesized to increase the incidence of EBV-PTLD post transplantation. A retrospective analysis of 785 allogeneic HSCT with post-transplant cyclophosphamide yielded zero cases of EBV-PTLD [70]. This was in concordance with many other centers that similarly reported the absence of EBV-PTLD in patients who received cyclophosphamide [71,72,73,74,75]. This could be due to destruction of donor and host EBV-infected cells with relative sparing of EBV-specific T-cells [70]. The use of post-transplant cyclophosphamide as opposed to ATG has been associated with a more rapid immune reconstitution of T-cells but not B-cells [76].
Acute graft-versus-host disease (GVHD)
GVHD is another independent risk factor for developing EBV-PTLD post HSCT (HR 2.65; p-value 0.006) [33]. Other studies have also agreed with this result [60, 77]. This could be due to impaired specific immunity due to pro-inflammatory cytokines released in GVHD in addition to immunosuppressive treatments given for prophylaxis [34].
Cytomegalovirus (CMV) reactivation
Immunosuppression post HSCT puts the recipient at risk of infection by various microbes, including bacteria, viruses, fungi and parasites. One particular infectious agent is CMV, another herpes virus. CMV is linked to multiple complications, including bone marrow suppression, increased risk of GVHD, and increased likelihood of EBV reactivation. Multiple studies have shown an increased incidence of EBV reactivation in HSCT recipients who have also reactivated CMV [78, 79].
Splenectomy
The spleen is integral to immunity since it is the site of T-cell, B-cell, and immunoglobulin production [80, 81]. As such, it is hypothesized that splenectomy impairs the function of CD5+ B-cells thus leading to EBV growth and reactivation [33]. The independent effect of splenectomy as a risk factor for developing PTLD and EBV reactivation has been demonstrated [33].
EBV monitoring and surveillance
EBV surveillance is standard clinical practice for early detection of a possible viral reactivation in patients undergoing transplantation [82, 83]. It is usually maintained by quantifying EBV-DNA in peripheral blood as a surrogate for EBV-related malignancies [84,85,86,87]. Viral load is monitored using quantitative polymerase chain reaction (PCR) but there is no consensus regarding the EBV threshold before initiating further work-up and pre-emptive treatment [37]. Importantly, EBV-DNA is readily detected in peripheral blood mononuclear cells of healthy seropositive individuals, which reflects circulating latently infected EBV B-lymphocytes [11]. On the other hand, free EBV-DNA in plasma or serum is not typically detected in a healthy individual and is usually derived from dying EBV-infected tumor cells, dying latently infected B-lymphocytes, or virions [11]. As such and as previously discussed, the detection of EBV-DNA alone is not enough to establish a diagnosis of PTLD in an immunocompromised individual at risk of reactivation [11]. Currently, there is no data that supports preference for whole blood, plasma or serum during monitoring or testing [35, 88, 89]. As such, according to ECIL-6 (European Conference on Infections in Leukemia) guidelines, there are no current recommendations [83].
Even though solely insufficient to diagnose PTLD, EBV-DNA levels are of prognostic value when measured pre-treatment. They also serve as a means to follow-up disease progression and relapse post treatment initiation. In some lymphomas, high pre-treatment EBV-DNA correlates with lower response rates to treatment, higher International Prognostic Index scores and inferior survival outcomes [85,86,87, 90, 91].
As patients differ inherently in terms of risk factors and comorbidities and thus receive different conditioning cocktails, it becomes important to tailor EBV monitoring duration and frequency to each patient. The latest ECIL-6 (European Conference on Infections in Leukemia) guidelines advise starting EBV monitoring no later than 4 weeks post HSCT and at least weekly until reconstitution of cellular immunity (~4 months after HSCT) [83]. Longer EBV monitoring is recommended in patients considered to have poor T-cell reconstitution: currently treated for GVHD, post haplo-transplantation, prior T-cell depletion, received ATG/Alemtuzumab or having experienced early EBV reactivation in the post-transplant course (Fig. 1) [83].
Treatment
Treating EBV-PTLD is rather problematic since there is no approved treatment in neither the United States, nor Europe. Obstacles in management are due to variability in patient response to treatment, limitations and toxicities associated with current therapies, absence of EMA and FDA approved therapies, lack of a global standard of care and most importantly, lack of randomized trials [26, 92].
There are currently two approaches to treatment: 1) pre-biopsy treatment of “suspected PTLD”, which includes treating EB viremia and pre-emptive therapy in high-risk patients and 2) post-biopsy treatment whereby disease is confirmed with biopsy before initiating therapy.
An elevation in or continuously high blood EBV-DNA levels should trigger pre-emptive treatment [37]. This includes reduction of immunosuppression (RIS), rituximab, or cellular immunotherapy all of which will be discussed in this section [37].
Reduction of immunosuppression (RIS)
RIS remains the first-line therapy for pre-emptive treatment, as well as for patients who develop PTLD where reducing immunosuppression allows recovery of the host’s immune system, thereby enabling EBV-specific T-lymphocytes to proliferate and control the disease [26]. Responses to RIS are highly variable ranging from 0% to 73%, but are not sustainable in time, since durable responses are maintained in < 10–20% of the cases [92,93,94,95,96,97,98,99,100].
There are two specific concerns when dealing with HSCT patients, (a) relatively long median time to response after RIS of ~3–5 weeks and (b) increased risk of graft failure/rejection upon RIS [32]. The median time to response after RIS is considerably affected by procedures such as T-cell depletion and administration of ATG or alemtuzumab [11]. Even if immune reconstitution starts shortly after initiation of RIS, prior exposure to T-cell depletion strongly affects T-lymphocyte reconstruction thus delaying the development of the cornerstone of viral immunity and in this case, delaying elimination of EBV-PTLD.
The possibility of developing graft rejection or GVHD after RIS is detrimental. One retrospective study demonstrated that 15% of patients receiving treatment for PTLD developed allograft rejection, 95% of which had been treated with RIS [101]. A retrospective study incorporated RIS in a sequential treatment protocol for PTLD in solid organ transplant (SOT) recipients. This study demonstrated that 37% of the patients developed acute graft rejection after immunosuppression was reduced [102]. Even though this study examined the impact of RIS among SOT recipients, the results could probably be extrapolated to the more immunosuppressed HSCT recipients thus rendering the use of RIS questionable.
Multiple factors have been pinpointed as possible predictors of poor outcome in patients receiving RIS. These include increased lactate dehydrogenase levels, multi-organ involvement/dysfunction, late onset of disease, age > 50 years, and B-symptoms (fever, night sweats, weight loss, lymphadenopathy) [32].
A probable alternative to RIS is the incorporation of agents with anti-tumor and antiviral properties thus allowing for the treatment of PTLD while maintaining the level of immunosuppression necessary to prevent graft rejection and GVHD [11]. Immunosuppressive agents such as sirolimus (mTOR inhibitor) have been found to counteract EBV due to the important role of the mTOR pathway in B-cell proliferation and early EBV-PTLD lesions [103, 104]. In recipients of SOT, remissions of EBV-PTLD have been noted upon the replacement of a calcineurin inhibitor with an mTOR inhibitor [105, 106]. Whether this could be extrapolated to HSCT recipients remains to be identified by future studies.
As such, we conclude that RIS maintains an integral role in treating EBV-PTLD but there appears to be a fine balance between reducing immunosuppression just enough to allow the host’s immune system to clear EBV-PTLD but not so much that it allows the grave possibility of developing rejection and GVHD.
Rituximab
Rituximab is a monoclonal, anti-CD20 antibody that has become standard in patients with non-destructive PTLD, polymorphic PTLD, or monomorphic diffuse large B-cell lymphoma-like PTLD, which is resistant to RIS [26]. Rituximab is effective in EBV-PTLD not only due to it targeting the CD20 + tumor but also due to its effect on B-cell depletion, which shifts the ratio of EBV-infected B-lymphocytes to EBV-specific T-lymphocytes in favor of an antiviral/anti-tumor response [107].
As frontline monotherapy, rituximab has reported response rates of about 60–65% and up to 80% [107,108,109]. The 20% chance of treatment failure renders the use of rituximab as monotherapy, insufficient. As such, rituximab is often combined with RIS or cellular immunotherapy such as EBV-CTLs. Approximately 44–79% of patients respond to rituximab therapy with RIS, with complete remission rates of 20–55% [109,110,111,112,113,114,115]. The obstacle is in treating patients with relapsed or rituximab-resistant PTLD due to the lack of consensus and approved therapies. As such, survival remains dismal in patients with relapsed or rituximab-refractory disease with only 28% of HSCT patients and 36% of SOT patients expected to be alive at 1 year [33, 114].
Pre-emptively, rituximab can be used before the diagnosis of EBV-PTLD is established due to rising EBV viral load in peripheral blood in the absence of clinical symptoms as recommended by American and European guidelines [82, 83]. As previously mentioned, there is no consensus on threshold EBV-DNA levels after which pre-emptive treatment should be initiated. In the absence of standard laboratory assays, some scientists employ a threshold of 1000 EBV copies/mL, others use thresholds of 10,000 EBV copies/mL or 40,000 EBV copies/mL [37, 83]. As such, there are currently no specific guidelines regarding a specific cut-off before which pre-emptive therapy is to be initiated. There are thus center-specific cutoff values depending on the correlation between clinical and laboratory data [83]. Rituximab when used in this setting is administered once weekly (375 mg/m2) until EBV-DNA negativity (usually 1–4 doses) [83]. It becomes important to stress the role of pre-emptive rituximab in the setting of allogeneic HSCT post T-cell depletion (an independent risk factor of PTLD). One prospective study demonstrated significantly reduced rates of PTLD (18% vs. 49%) and annulled PTLD-mortality (0% vs. 26%) with pre-emptive rituximab in patients receiving T-cell depleted grafts [116]. A recent retrospective study of 16 patients who were treated pre-emptively with a lower dose of rituximab (100 mg/m2 per dose) demonstrated a 93.4% success rate with only 1/16 developing PTLD despite negative viremia [117]. As such, we highlight rituximab’s role in the pre-emptive setting, especially in the setting of T-cell depletion, whereby it demonstrated high response rates with a significant drop in PTLD incidence.
Rituximab has also been employed for use in the prophylactic setting whereby it is administered with the transplant, before or shortly after before the onset of EBV DNAemia. A retrospective study highlighted the significant role of rituximab in both, the pre-emptive setting and prophylactic setting after alemtuzumab-conditioned allogeneic transplantation, whereby 92% of patients with EBV reactivation exhibited complete response after 4 weekly rituximab dosages [66]. In addition, the association between pre-transplant use of rituximab and lack of EBV reactivation (HR 0.34, p-value 0.001) was highlighted [66]. A recently published prospective study compared cord transplant patients conditioned with thymoglobulin, both documented risk factors for EBV-PTLD development, on the basis of prophylactic rituximab [118]. Patients who received rituximab prior to transplantation where less likely to reactive EBV (18% vs. 2%) or develop PTLD (12% vs. 0%) thereafter when compared to their counterparts who did not receive rituximab [118]. Nonetheless, it is important to consider the differences between the two population groups of patients, especially in terms of disease severity, whereby patients who did not receive prophylactic rituximab appeared to have higher indices of disease severity [118].
The routine pre-emptive use of rituximab, even though central to decreasing PTLD rates, allows for the emergence of CD20-negative lymphomas refractory to rituximab. Hiraga et al. [119] reported that 5 out of 19 cases of relapsed B-cell lymphomas previously treated with rituximab, downregulated CD20. Tsai et al. [120] demonstrated decreased promoter activity of the CD20 gene and decreased CD20 expression in rituximab-resistant cell lines after repeated rituximab exposure. Although there are multiple mechanisms proposed to explain rituximab-refractoriness, the absence of CD20 is the most important determinant.
The use of rituximab comes at a cost with several possible side effects including delayed humoral immune reconstitution, prolonged hypogammaglobulinemia, prolonged neutropenia and a magnified risk of infections [121, 122].
Second and third generation CD20 monoclonal antibodies have been developed such as the fully human anti-CD20, ofatumumab (OFA), and the humanized anti-CD20, obinutuzumab (OBZ). Ofatumumab, similar to rituximab, is a type I anti-CD20 whereby they both lead to effective complement lytic cytotoxicity [123]. Obinutuzumab, on the other hand, is a type II antibody and has limited cytotoxicity [124]. There are limited studies that investigate the added benefit of OBZ compared to rituximab. Upfront, comparing the addition of OBZ or rituximab to chemotherapy regimens such as CHOP in the treatment of DLBCL, there appears to be limited differences in disease control and overall survival [125, 126]. In the setting of relapsed, rituximab-refractory disease after receiving prior rituximab-containing regimens, the addition of OBZ appears to play a more fundamental role [127, 128]. Ofatumumab combined with chemotherapy in the treatment of rituximab-refractory DLBCL also showed promising results [129]. When compared to rituximab in the setting of relapsed/refractory DLBCL, there appeared to be no difference between the two salvage therapies [130]. Owing to the absence of trials that investigate the role of OBZ and OFA in the setting untreated or rituximab-refractory PTLD, we are limited to the potential extrapolation of the previously mentioned results.
Adoptive cellular immunotherapy
Adoptive T-cell therapy using EBV-specific CTLs has been incorporated in the treatment and prevention of EBV-PTLD long since 1995 with proven efficacy and safety even in the face of refractory or relapsed disease [131, 132]. Patients generally show a good response to treatment or prophylaxis with CTLs and among 101 high-risk HSCT recipients who received prophylactic EBV-CTLs, none developed EBV-PTLD [131]. The same group demonstrated EBV-CTLs’ efficacy in treating EBV-PTLD where 11 out of 13 patients with established EBV-PTLD achieved complete remission after treatment with EBV-CTLs [131]. If the donor source of EBV-CTLs is not available or the donor is EBV negative, third-party HLA-matched EBV-CTLs can be employed [133]. A trial that compared outcomes between patients treated with donor EBV-CTLs and third-party EBV-CTLs retrieved from a donor bank demonstrated similar results of ~60% long remissions. This makes treatment with EBV-CTLs feasible in case the donor is EBV-negative [134].
With T-lymphocytes being involved in the pathogenesis of GVHD, there was concern regarding possible increased rates of GVHD upon infusion of EBV-CTLs. In one series, >two-thirds of HSCT patients who developed EBV-PTLD had a durable response upon receiving EBV-CTLs with no cases of GVHD [135]. This is compared to 17% GVHD rates in patients who received HLA-matched donor lymphocyte infusion (DLI) for EBV-PTLD [135]. Owing to the rising concern that CTLs might induce GVHD, cellular products have been engineered with “suicide genes” so that in the event that these infused cells lead to GVHD, the cells may be “turned off” pharmacologically [136, 137].
Currently, there are multiple trials that are looking into the use of both, donor-derived and third-party EBV-CTLs in the setting of treatment-refractory EBV-PTLD of which the phase III MATCH trial is one.
Chimeric antigen receptor T-cell (CAR-T cell) therapy is a novel adoptive immunotherapy whereby lymphocytes are engineered with chimeric receptors allowing infused CAR T-cells to recognize and eliminate cancer cells. Among the earliest CAR T-cells is the anti-CD19 CAR T-cells. Second and third generation anti-CD19 CAR T-cells (with additional targets such as CD28 and 4-1BB) have demonstrated promising results in B-cell lymphomas with CR rates >50% [138]. There are currently no trials investigating CAR-T cells for treating PTLD but with the many trials that have proved effectiveness of CAR-T cells in B-cell lymphomas, one can hypothesize that similar results could be possible in the setting of PTLD.
Chemotherapy
There appears to be no role for chemotherapy in the treatment of EBV-PTLD post HSCT, whereby it is reserved as last resort salvage therapy in this patient group [107]. A retrospective study by Fox et al. [139] portrayed the limited efficacy of chemotherapy in patients who failed rituximab whereby none of the patients attained complete remission. In SOT recipients, on the other hand, chemotherapy is less limited with efficacy established with some regimens offered to rituximab non-responders, most classically R-CHOP (rituximab, cyclophosphamide, hydroxydaunorubicin, oncovin and prednisone) [26, 114, 139, 140]. Nonetheless, even though active against PTLD in this population, mortality is greatly affected by the regimen’s side effects. R-CHOP in the first-line setting has been associated with 50% mortality, dropping to 27% when sequentially used after rituximab [26, 115, 140].
Trials of chemotherapy regimens such as R-CHOP in HSCT recipients are underway.
Antiviral therapy
During the primary infection, EBV encodes thymidine kinase enzyme that can convert nucleoside analogs to their monophosphate forms, which are later converted by cellular enzymes to triphosphates targeting viral DNA polymerase, inhibiting viral replication, and thus inducing apoptosis. As such, antivirals such as ganciclovir have an inhibitory effect on EBV-infected cells during a lytic viral infection in vitro. EBV-infected cells are transformed B-cells without a lytic viral infection and so are not susceptible to antiviral therapy in vitro [141]. In addition and as previously mentioned, during a latent infection, EBV circularizes and inhibits the expression of particular proteins such as thymidine kinase [142]. This renders EBV not susceptible to antivirals during latency. A meta-analysis investigated the effect of prophylactic antivirals on PTLD incidence in high-risk EBV serologically mismatched patients after SOT [142]. There was no effect irrespective of the type of transplant, type of antiviral, duration of prophylaxis or patient age group [142]. Such results could be extrapolated to the setting post HSCT transplantation since proliferating EBV-infected B-cells will behave similarly whether EBV is of donor or recipient origin.
Future prospects
As previously highlighted, the biggest setback in treating EBV-PTLD is the lack of clinical trials due to the rarity of the disease. Currently, there is a handful of trials aiming at elucidating treatment regimens for post HSCT patients who develop EBV-PTLD (Table 1).
Interestingly, immunomodulatory drugs that have not been known to contribute to the treatment of EBV-PTLD are being investigated in clinical trials. One such drug is lenalidomide with its known propensity to augment cellular immunity. It has been shown to demonstrate durable efficacy when used in treating relapsed PTLD in two case reports [143, 144]. Currently, one trial is investigating the role of lenalidomide combined with everolimus, a derivative of sirolimus, which inhibits mTOR, in treating lymphoproliferative disease.
Another novel agent under investigation is the anti-CD30 monoclonal antibody, brentuximab vedotin (BV) for CD30-expressing PTLD [145]. One study combined BV with rituximab as induction and maintenance therapies for EBV-associated CD20 + non-Hodgkin lymphomas that co-express CD30 [146]. The preliminary results revealed a complete response rate that exceeds 70% [146].
The role of the proteasome inhibitor, bortezomib is also being investigated in a clinical trial combined with rituximab. The recently completed phase II trial demonstrated an overall response rate of 42.9% and a 14.29% all-cause mortality for participants in the study [147]. All patients developed adverse events to treatment, including leucocytosis, cardiac arrest, GI distress, infections, metabolite disturbances, cytopenias, or peripheral neuropathy with 28.6% developing serious adverse events [147].
The role of panobinostat, a histone deacetylase inhibitor is also being investigated in clinical trials both, as single agent and in combination with everolimus.
Ibrutinib, a Bruton tyrosine kinase inhibitor (BTK), has established its role in the setting of mantle cell lymphoma and CLL/SLL [148, 149]. BTK plays an important role in signaling pathways involved in B-cell proliferation and apoptosis and thus inhibiting it would alleviate the proliferative, anti-apoptotic properties of B-cell malignancies. Whether or not it will be of benefit in treating PTLD is yet to be investigated.
Conclusion
EBV-PTLD is one of the detrimental complications that HSCT recipients are at risk of developing when severely immunocompromised. Despite the entity markedly affecting survival, there is a lack of comparative data evaluating potential therapeutic strategies and no clear consensus for their use in the management of PTLD in transplant recipients due to the few clinical trials and the rarity of the disease. Studies have determined risk factors for developing PTLD such as HLA-mismatch, EBV-serology mismatch, development of GVHD, RIC instead of MAC, and T-cell depletion. Bearing in mind these risk factors and the characteristics of individual patients allows the physician to highlight a subset of patients that are at high risk of developing the disease. Currently, guidelines encourage weekly monitoring of EBV-DNA and pre-emptive treatment with rituximab weekly (1–4 doses), alone or combined with RIS or CTLs when feasible. A comprehensive treatment paradigm awaits the results of clinical trials that are looking into novel tools or agents such as ‘off-the-shelf’’ CTLs, bortezomib, lenalidomide, everolimus, brentuximab and panobinostat.
References
Balfour HH Jr., Dunmire SK, Hogquist KA. Infectious mononucleosis. Clin Transl Immunology. 2015;4:e33.
Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000;343:481–92.
Comoli P, Basso S, Zecca M, Pagliara D, Baldanti F, Bernardo ME, et al. Preemptive therapy of EBV-related lymphoproliferative disease after pediatric haploidentical stem cell transplantation. Am J Transplant. 2007;7:1648–55.
Yates J, Warren N, Reisman D, Sugden B. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc Natl Acad Sci USA. 1984;81:3806–10.
Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-Barr virus-associated lymphomas. Philos Trans R Soc Lond B Biol Sci. 2017;372:1732.
Kalinova L, Indrakova J, Bachleda P. Post-transplant lymphoproliferative disorder. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2009;153:251–7.
Ferreiro JF, Morscio J, Dierickx D, Vandenberghe P, Gheysens O, Verhoef G, et al. EBV-positive and EBV-negative posttransplant diffuse large B cell lymphomas have distinct genomic and transcriptomic features. Am J Transplant. 2016;16:414–25.
Quinn LL, Williams LR, White C, Forrest C, Zuo J, Rowe M. The missing link in epstein-barr virus immune evasion: the BDLF3 gene induces ubiquitination and downregulation of major histocompatibility complex class I (MHC-I) and MHC-II. J Virol. 2016;90:356–67.
Ressing ME, Keating SE, van Leeuwen D, Koppers-Lalic D, Pappworth IY, Wiertz EJ, et al. Impaired transporter associated with antigen processing-dependent peptide transport during productive EBV infection. J Immunol. 2005;174:6829–38.
Djaoud Z, Guethlein LA, Horowitz A, Azzi T, Nemat-Gorgani N, Olive D, et al. Two alternate strategies for innate immunity to Epstein-Barr virus: One using NK cells and the other NK cells and gammadelta T cells. J Exp Med. 2017;214:1827–41.
Kanakry JA, Ambinder RF. EBV-related lymphomas: new approaches to treatment. Curr Treat Options Oncol. 2013;14:224–36.
Henderson S, Rowe M, Gregory C, Croom-Carter D, Wang F, Longnecker R, et al. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell. 1991;65:1107–15.
Capello D, Rossi D, Gaidano G. Post-transplant lymphoproliferative disorders: molecular basis of disease histogenesis and pathogenesis. Hematol Oncol. 2005;23:61–7.
Krams SM, Martinez OM. Epstein-Barr virus, rapamycin, and host immune responses. Curr Opin Organ Transplant. 2008;13:563–8.
Vereide DT, Sugden B. Lymphomas differ in their dependence on Epstein-Barr virus. Blood. 2011;117:1977–85.
Burns DM, Tierney R, Shannon-Lowe C, Croudace J, Inman C, Abbotts B, et al. Memory B-cell reconstitution following allogeneic hematopoietic stem cell transplantation is an EBV-associated transformation event. Blood. 2015;126:2665–75.
Friedberg JW, Swinnen L. Post-transplant lymphoproliferative disease in the lymphomas. 2nd ed. Philadelphia, PA, USA: Elsevier; 2006.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H et al. WHO classification of tumors of haemotopoietic and lymphoid tissue. 4th ed. Lyon: IARC; 2008.
Ruf S, Moser O, Wossmann W, Kreyenberg H, Wagner HJ. Examining the origin of posttransplant lymphoproliferative disorder in a patient after a second allogeneic hematopoeitic stem cell transplantation for relapsed BCR-ABL positive acute lymphoblastic leukemia. J Pedia Hematol Oncol. 2011;33:50–4.
Landgren O, Gilbert ES, Rizzo JD, Socie G, Banks PM, Sobocinski KA, et al. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood. 2009;113:4992–5001.
Luskin MR, Heil DS, Tan KS, Choi S, Stadtmauer EA, Schuster SJ, et al. The impact of EBV status on characteristics and outcomes of posttransplantation lymphoproliferative disorder. Am J Transplant. 2015;15:2665–73.
Johansson JE, Remberger M, Lazarevic V, Hallbook H, Wahlin A, Kimby E, et al. Allogeneic haematopoietic stem-cell transplantation with reduced intensity conditioning for advanced stage Hodgkin’s lymphoma in Sweden: high incidence of post transplant lymphoproliferative disorder. Bone Marrow Transplant. 2011;46:870–5.
Hou HA, Yao M, Tang JL, Chen YK, Ko BS, Huang SY, et al. Poor outcome in post transplant lymphoproliferative disorder with pulmonary involvement after allogeneic hematopoietic SCT: 13 years’ experience in a single institute. Bone Marrow Transplant. 2009;43:315–21.
Buyck HC, Ball S, Junagade P, Marsh J, Chakrabarti S. Prior immunosuppressive therapy with antithymocyte globulin increases the risk of EBV-related lymphoproliferative disorder following allo-SCT for acquired aplastic anemia. Bone Marrow Transplant. 2009;43:813–6.
Ocheni S, Kroeger N, Zabelina T, Sobottka I, Ayuk F, Wolschke C, et al. EBV reactivation and post transplant lymphoproliferative disorders following allogeneic SCT. Bone Marrow Transplant. 2008;42:181–6.
Dierickx D, Habermann TM. Post-transplantation lymphoproliferative disorders in adults. N Engl J Med. 2018;378:549–62.
Gu B, Chen GH, Wu DP. [Recent advances on diagnosis and therapy of lymphoproliferative disorders after allo-HSCT]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2014;22:538–42.
Deeg HJ, Socie G. Malignancies after hematopoietic stem cell transplantation: many questions, some answers. Blood. 1998;91:1833–44.
Tamaru JI. 2016 revision of the WHO classification of lymphoid neoplasms. Rinsho Ketsueki. 2017;58:2188–93.
Rasche L, Kapp M, Einsele H, Mielke S. EBV-induced post transplant lymphoproliferative disorders: a persisting challenge in allogeneic hematopoetic SCT. Bone Marrow Transplant. 2014;49:163–7.
Zimmermann H, Trappe RU. Therapeutic options in post-transplant lymphoproliferative disorders. Ther Adv Hematol. 2011;2:393–407.
Al-Mansour Z, Nelson BP, Evens AM. Post-transplant lymphoproliferative disease (PTLD): risk factors, diagnosis, and current treatment strategies. Curr Hematol Malig Rep. 2013;8:173–83.
Uhlin M, Wikell H, Sundin M, Blennow O, Maeurer M, Ringden O, et al. Risk factors for Epstein-Barr virus-related post-transplant lymphoproliferative disease after allogeneic hematopoietic stem cell transplantation. Haematologica. 2014;99:346–52.
Curtis RE, Travis LB, Rowlings PA, Socie G, Kingma DW, Banks PM, et al. Risk of lymphoproliferative disorders after bone marrow transplantation: a multi-institutional study. Blood. 1999;94:2208–16.
Styczynski J, Reusser P, Einsele H, de la Camara R, Cordonnier C, Ward KN, et al. Management of HSV, VZV and EBV infections in patients with hematological malignancies and after SCT: guidelines from the Second European Conference on Infections in Leukemia. Bone Marrow Transplant. 2009;43:757–70.
Styczynski J, Gil L, Tridello G, Ljungman P, Donnelly JP, van der Velden W, et al. Response to rituximab-based therapy and risk factor analysis in Epstein Barr Virus-related lymphoproliferative disorder after hematopoietic stem cell transplant in children and adults: a study from the Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Clin Infect Dis. 2013;57:794–802.
DeStefano CB, Desai SH, Shenoy AG, Catlett JP Management of post-transplant lymphoproliferative disorders. Br J Haematol. 2018;182:330–343.
Garcia-Cadenas I, Yanez L, Jarque I, Martino R, Perez-Simon JA, Valcarcel D, et al. Frequency, characteristics and outcome of PTLD after allo-SCT: a multicenter study from the Spanish group of blood and marrow transplantation (GETH). Eur J Haematol. 2019;00:1–7.
Evens AM, David KA, Helenowski I, Nelson B, Kaufman D, Kircher SM, et al. Multicenter analysis of 80 solid organ transplantation recipients with post-transplantation lymphoproliferative disease: outcomes and prognostic factors in the modern era. J Clin Oncol. 2010;28:1038–46.
Chakrabarti S, Milligan DW, Pillay D, Mackinnon S, Holder K, Kaur N, et al. Reconstitution of the Epstein-Barr virus-specific cytotoxic T-lymphocyte response following T-cell-depleted myeloablative and nonmyeloablative allogeneic stem cell transplantation. Blood. 2003;102:839–42.
Saito T, Kanda Y, Nakai K, Kim SW, Arima F, Kami M, et al. Immune reconstitution following reduced-intensity transplantation with cladribine, busulfan, and antithymocyte globulin: serial comparison with conventional myeloablative transplantation. Bone Marrow Transplant. 2003;32:601–8.
Reshef R, Luskin MR, Kamoun M, Vardhanabhuti S, Tomaszewski JE, Stadtmauer EA, et al. Association of HLA polymorphisms with post-transplant lymphoproliferative disorder in solid-organ transplant recipients. Am J Transplant. 2011;11:817–25.
Pourfarziani V, Einollahi B, Taheri S, Nemati E, Nafar M, Kalantar E. Associations of Human Leukocyte Antigen (HLA) haplotypes with risk of developing lymphoproliferative disorders after renal transplantation. Ann Transplant. 2007;12:16–22.
Subklewe M, Marquis R, Choquet S, Leblond V, Garnier JL, Hetzer R, et al. Association of human leukocyte antigen haplotypes with posttransplant lymphoproliferative disease after solid organ transplantation. Transplantation. 2006;82:1093–100.
Wheless SA, Gulley ML, Raab-Traub N, McNeillie P, Neuringer IP, Ford HJ, et al. Post-transplantation lymphoproliferative disease: Epstein-Barr virus DNA levels, HLA-A3, and survival. Am J Respir Crit Care Med. 2008;178:1060–5.
Lustberg ME, Pelletier RP, Porcu P, Martin SI, Quinion CD, Geyer SM, et al. Human leukocyte antigen type and posttransplant lymphoproliferative disorder. Transplantation. 2015;99:1220–5.
Jones K, Wockner L, Thornton A, Gottlieb D, Ritchie DS, Seymour JF, et al. HLA class I associations with EBV+post-transplant lymphoproliferative disorder. Transpl Immunol. 2015;32:126–30.
Walker RC, Marshall WF, Strickler JG, Wiesner RH, Velosa JA, Habermann TM, et al. Pretransplantation assessment of the risk of lymphoproliferative disorder. Clin Infect Dis. 1995;20:1346–53.
Sundin M, Le Blanc K, Ringden O, Barkholt L, Omazic B, Lergin C, et al. The role of HLA mismatch, splenectomy and recipient Epstein-Barr virus seronegativity as risk factors in post-transplant lymphoproliferative disorder following allogeneic hematopoietic stem cell transplantation. Haematologica. 2006;91:1059–67.
Hoegh-Petersen M, Goodyear D, Geddes MN, Liu S, Ugarte-Torres A, Liu Y, et al. High incidence of post transplant lymphoproliferative disorder after antithymocyte globulin-based conditioning and ineffective prediction by day 28 EBV-specific T lymphocyte counts. Bone Marrow Transplant. 2011;46:1104–12.
Kelly SS, Parmar S, De Lima M, Robinson S, Shpall E. Overcoming the barriers to umbilical cord blood transplantation. Cytotherapy. 2010;12:121–30.
Barker JN, Krepski TP, DeFor TE, Davies SM, Wagner JE, Weisdorf DJ. Searching for unrelated donor hematopoietic stem cells: availability and speed of umbilical cord blood versus bone marrow. Biol Blood Marrow Transplant. 2002;8:257–60.
Grewal SS, Barker JN, Davies SM, Wagner JE. Unrelated donor hematopoietic cell transplantation: marrow or umbilical cord blood? Blood. 2003;101:4233–44.
Barker JN, Martin PL, Coad JE, DeFor T, Trigg ME, Kurtzberg J, et al. Low incidence of Epstein-Barr virus-associated posttransplantation lymphoproliferative disorders in 272 unrelated-donor umbilical cord blood transplant recipients. Biol Blood Marrow Transplant. 2001;7:395–9.
Brunstein CG, Weisdorf DJ, DeFor T, Barker JN, Tolar J, van Burik JA, et al. Marked increased risk of Epstein-Barr virus-related complications with the addition of antithymocyte globulin to a nonmyeloablative conditioning prior to unrelated umbilical cord blood transplantation. Blood. 2006;108:2874–80.
Peric Z, Cahu X, Chevallier P, Brissot E, Malard F, Guillaume T, et al. Features of EBV reactivation after reduced intensity conditioning unrelated umbilical cord blood transplantation. Bone Marrow Transplant. 2012;47:251–7.
Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13:88–100.
Knight A, Madrigal AJ, Grace S, Sivakumaran J, Kottaridis P, Mackinnon S, et al. The role of Vdelta2-negative gammadelta T cells during cytomegalovirus reactivation in recipients of allogeneic stem cell transplantation. Blood. 2010;116:2164–72.
Liu J, Bian Z, Wang X, Xu LP, Fu Q, Wang C, et al. Inverse correlation of Vdelta2(+) T-cell recovery with EBV reactivation after haematopoietic stem cell transplantation. Br J Haematol. 2018;180:276–85.
Laberko A, Bogoyavlenskaya A, Shelikhova L, Shekhovtsova Z, Balashov D, Voronin K, et al. Risk factors for and the clinical impact of cytomegalovirus and Epstein-Barr virus infections in pediatric recipients of TCR-alpha/beta- and CD19-depleted grafts. Biol Blood Marrow Transplant. 2017;23:483–90.
Lang PJ, Schlegel PG, Roland M, Schulz AS, Greil J, Bader P, et al. Safety and efficacy of Tcralpha/beta and CD19 depleted haploidentical stem cell transplantation following reduced intensity conditioning in children: Results of a prospective multicenter phase I/II clinical trial. Blood. 2017;130(Suppl 1):214
Rooney CM, Loftin SK, Holladay MS, Brenner MK, Krance RA, Heslop HE. Early identification of Epstein-Barr virus-associated post-transplantation lymphoproliferative disease. Br J Haematol. 1995;89:98–103.
van Esser JW, van der Holt B, Meijer E, Niesters HG, Trenschel R, Thijsen SF, et al. Epstein-Barr virus (EBV) reactivation is a frequent event after allogeneic stem cell transplantation (SCT) and quantitatively predicts EBV-lymphoproliferative disease following T-cell-depleted SCT. Blood. 2001;98:972–8.
Mensen A, Na IK, Hafer R, Meerbach A, Schlecht M, Pietschmann ML, et al. Comparison of different rabbit ATG preparation effects on early lymphocyte subset recovery after allogeneic HSCT and its association with EBV-mediated PTLD. J Cancer Res Clin Oncol. 2014;140:1971–80.
Scheinberg P, Nunez O, Weinstein B, Scheinberg P, Biancotto A, Wu CO, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365:430–8.
Burns DM, Rana S, Martin E, Nagra S, Ward J, Osman H, et al. Greatly reduced risk of EBV reactivation in rituximab-experienced recipients of alemtuzumab-conditioned allogeneic HSCT. Bone Marrow Transplant. 2016;51:825–32.
Sica S, Metafuni E, Bellesi S, Chiusolo P. Epstein-barr virus related lymphoproliferations after stem cell transplantation. Mediterr J Hematol Infect Dis. 2009;1:e2009019.
Scheinberg P, Fischer SH, Li L, Nunez O, Wu CO, Sloand EM, et al. Distinct EBV and CMV reactivation patterns following antibody-based immunosuppressive regimens in patients with severe aplastic anemia. Blood. 2007;109:3219–24.
Nijland ML, Kersten MJ, Pals ST, Bemelman FJ, Ten Berge IJ. Epstein-Barr virus-positive posttransplant lymphoproliferative disease after solid organ transplantation: pathogenesis, clinical manifestations, diagnosis, and management. Transpl Direct. 2016;2:e48.
Kanakry JA, Kasamon YL, Bolanos-Meade J, Borrello IM, Brodsky RA, Fuchs EJ, et al. Absence of post-transplantation lymphoproliferative disorder after allogeneic blood or marrow transplantation using post-transplantation cyclophosphamide as graft-versus-host disease prophylaxis. Biol Blood Marrow Transplant. 2013;19:1514–7.
Ciurea SO, Mulanovich V, Saliba RM, Bayraktar UD, Jiang Y, Bassett R, et al. Improved early outcomes using a T cell replete graft compared with T cell depleted haploidentical hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2012;18:1835–44.
Raiola AM, Dominietto A, Ghiso A, Di Grazia C, Lamparelli T, Gualandi F, et al. Unmanipulated haploidentical bone marrow transplantation and posttransplantation cyclophosphamide for hematologic malignancies after myeloablative conditioning. Biol Blood Marrow Transplant. 2013;19:117–22.
Solomon SR, Sizemore CA, Sanacore M, Zhang X, Brown S, Holland HK, et al. Haploidentical transplantation using T cell replete peripheral blood stem cells and myeloablative conditioning in patients with high-risk hematologic malignancies who lack conventional donors is well tolerated and produces excellent relapse-free survival: results of a prospective phase II trial. Biol Blood Marrow Transplant. 2012;18:1859–66.
Bilmon IA, Kwan J, Gottlieb D, Kerridge I, McGurgan M, Huang G, et al. Haploidentical bone marrow transplants for haematological malignancies using non-myeloablative conditioning therapy and post-transplant immunosuppression with cyclophosphamide: results from a single Australian centre. Intern Med J. 2013;43:191–6.
Bashey A, Zhang X, Sizemore CA, Manion K, Brown S, Holland HK, et al. T-cell-replete HLA-haploidentical hematopoietic transplantation for hematologic malignancies using post-transplantation cyclophosphamide results in outcomes equivalent to those of contemporaneous HLA-matched related and unrelated donor transplantation. J Clin Oncol. 2013;31:1310–6.
Retiere C, Willem C, Guillaume T, Vie H, Gautreau-Rolland L, Scotet E, et al. Impact on early outcomes and immune reconstitution of high-dose post-transplant cyclophosphamide vs anti-thymocyte globulin after reduced intensity conditioning peripheral blood stem cell allogeneic transplantation. Oncotarget. 2018;9:11451–64.
Zhang Q, Zou BH, Lou X, Liu H, Zhang B, Chen H. [An analysis of risk factors and prognosis of Epstein-Barr virus infection after allogeneic hematopoietic stem cell transplantation]. Zhonghua Nei Ke Za Zhi. 2016;55:619–23.
Zallio F, Primon V, Tamiazzo S, Pini M, Baraldi A, Corsetti MT, et al. Epstein-Barr virus reactivation in allogeneic stem cell transplantation is highly related to cytomegalovirus reactivation. Clin Transplant. 2013;27:E491–7.
Bao X, Zhu Q, Qiu H, Chen F, Xue S, Ma X, et al. [Clinical risks analysis of EBV infection in patients with allogeneic hematopoietic stem cell transplantation]. Zhonghua Xue Ye Xue Za Zhi. 2016;37:138–43.
Abedi MR, Linde A, Christensson B, Mackett M, Hammarstrom L, Smith CI. Preventive effect of IgG from EBV-seropositive donors on the development of human lympho-proliferative disease in SCID mice. Int J Cancer. 1997;71:624–9.
Gary-Gouy H, Harriague J, Bismuth G, Platzer C, Schmitt C, Dalloul AH. Human CD5 promotes B-cell survival through stimulation of autocrine IL-10 production. Blood . 2002;100:4537–43.
Tomblyn M, Chiller T, Einsele H, Gress R, Sepkowitz K, Storek J, et al. Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant. 2009;15:1143–238.
Styczynski J, van der Velden W, Fox CP, Engelhard D, de la Camara R, Cordonnier C, et al. Management of Epstein-Barr Virus infections and post-transplant lymphoproliferative disorders in patients after allogeneic hematopoietic stem cell transplantation: Sixth European Conference on Infections in Leukemia (ECIL-6) guidelines. Haematologica. 2016;101:803–11.
Jones K, Nourse JP, Keane C, Crooks P, Gottlieb D, Ritchie DS, et al. Tumor-specific but not nonspecific cell-free circulating DNA can be used to monitor disease response in lymphoma. Am J Hematol. 2012;87:258–65.
Wang ZY, Liu QF, Wang H, Jin J, Wang WH, Wang SL, et al. Clinical implications of plasma Epstein-Barr virus DNA in early-stage extranodal nasal-type NK/T-cell lymphoma patients receiving primary radiotherapy. Blood. 2012;120:2003–10.
Gandhi MK, Lambley E, Burrows J, Dua U, Elliott S, Shaw PJ, et al. Plasma Epstein-Barr virus (EBV) DNA is a biomarker for EBV-positive Hodgkin’s lymphoma. Clin Cancer Res. 2006;12:460–4.
Hohaus S, Santangelo R, Giachelia M, Vannata B, Massini G, Cuccaro A, et al. The viral load of Epstein-Barr virus (EBV) DNA in peripheral blood predicts for biological and clinical characteristics in Hodgkin lymphoma. Clin Cancer Res. 2011;17:2885–92.
Hakim H, Gibson C, Pan J, Srivastava K, Gu Z, Bankowski MJ, et al. Comparison of various blood compartments and reporting units for the detection and quantification of Epstein-Barr virus in peripheral blood. J Clin Microbiol. 2007;45:2151–5.
Ruf S, Behnke-Hall K, Gruhn B, Bauer J, Horn M, Beck J, et al. Comparison of six different specimen types for Epstein-Barr viral load quantification in peripheral blood of pediatric patients after heart transplantation or after allogeneic hematopoietic stem cell transplantation. J Clin Virol. 2012;53:186–94.
Ito Y, Kimura H, Maeda Y, Hashimoto C, Ishida F, Izutsu K, et al. Pretreatment EBV-DNA copy number is predictive of response and toxicities to SMILE chemotherapy for extranodal NK/T-cell lymphoma, nasal type. Clin Cancer Res. 2012;18:4183–90.
Kanakry JA, Li H, Gellert LL, Lemas MV, Hsieh WS, Hong F, et al. Plasma Epstein-Barr virus DNA predicts outcome in advanced Hodgkin lymphoma: correlative analysis from a large North American cooperative group trial. Blood. 2013;121:3547–53.
Elstrom RL, Andreadis C, Aqui NA, Ahya VN, Bloom RD, Brozena SC, et al. Treatment of PTLD with rituximab or chemotherapy. Am J Transplant. 2006;6:569–76.
Ghobrial IM, Habermann TM, Maurer MJ, Geyer SM, Ristow KM, Larson TS, et al. Prognostic analysis for survival in adult solid organ transplant recipients with post-transplantation lymphoproliferative disorders. J Clin Oncol. 2005;23:7574–82.
Ghobrial IM, Habermann TM, Ristow KM, Ansell SM, Macon W, Geyer SM, et al. Prognostic factors in patients with post-transplant lymphoproliferative disorders (PTLD) in the rituximab era. Leuk Lymphoma. 2005;46:191–6.
Swinnen LJ, Mullen GM, Carr TJ, Costanzo MR, Fisher RI. Aggressive treatment for postcardiac transplant lymphoproliferation. Blood. 1995;86:3333–40.
Knight JS, Tsodikov A, Cibrik DM, Ross CW, Kaminski MS, Blayney DW. Lymphoma after solid organ transplantation: risk, response to therapy, and survival at a transplantation center. J Clin Oncol. 2009;27:3354–62.
Jagadeesh D, Woda BA, Draper J, Evens AM. Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options Oncol. 2012;13:122–36.
Oton AB, Wang H, Leleu X, Melhem MF, George D, Lacasce A, et al. Clinical and pathological prognostic markers for survival in adult patients with post-transplant lymphoproliferative disorders in solid transplant. Leuk Lymphoma. 2008;49:1738–44.
Caillard S, Dharnidharka V, Agodoa L, Bohen E, Abbott K. Posttransplant lymphoproliferative disorders after renal transplantation in the United States in era of modern immunosuppression. Transplantation. 2005;80:1233–43.
Maecker B, Jack T, Zimmermann M, Abdul-Khaliq H, Burdelski M, Fuchs A, et al. CNS or bone marrow involvement as risk factors for poor survival in post-transplantation lymphoproliferative disorders in children after solid organ transplantation. J Clin Oncol. 2007;25:4902–8.
Franke AJ, Bishnoi R, Bajwa R, Skelton WP, Patel N, Slayton WB, et al. Association of allograft rejection with reduction of immunosuppression for post-transplant lymphoproliferative disorder: Analysis of a 20-year single-institutional experience. J Clin Oncol. 2017;35(15suppl):19047. Abstract
Swinnen LJ, LeBlanc M, Grogan TM, Gordon LI, Stiff PJ, Miller AM, et al. Prospective study of sequential reduction in immunosuppression, interferon alpha-2B, and chemotherapy for posttransplantation lymphoproliferative disorder. Transplantation. 2008;86:215–22.
Nelson BP, Wolniak KL, Evens A, Chenn A, Maddalozzo J, Proytcheva M. Early posttransplant lymphoproliferative disease: clinicopathologic features and correlation with mTOR signaling pathway activation. Am J Clin Pathol. 2012;138:568–78.
El-Salem M, Raghunath PN, Marzec M, Wlodarski P, Tsai D, Hsi E, et al. Constitutive activation of mTOR signaling pathway in post-transplant lymphoproliferative disorders. Lab Invest. 2007;87:29–39.
Garcia VD, Bonamigo Filho JL, Neumann J, Fogliatto L, Geiger AM, Garcia CD, et al. Rituximab in association with rapamycin for post-transplant lymphoproliferative disease treatment. Transpl Int. 2003;16:202–6.
Gibelli NE, Tannuri U, Pinho-Apezzato ML, Tannuri AC, Maksoud-Filho JG, Andrade WC, et al. Sirolimus in pediatric liver transplantation: a single-center experience. Transpl Proc. 2009;41:901–3.
Styczynski J, Einsele H, Gil L, Ljungman P. Outcome of treatment of Epstein-Barr virus-related post-transplant lymphoproliferative disorder in hematopoietic stem cell recipients: a comprehensive review of reported cases. Transpl Infect Dis. 2009;11:383–92.
Xu LP, Zhang CL, Mo XD, Zhang XH, Chen H, Han W, et al. Epstein-Barr virus-related post-transplantation lymphoproliferative disorder after unmanipulated human leukocyte antigen haploidentical hematopoietic stem cell transplantation: Incidence, risk factors, treatment, and clinical outcomes. Biol Blood Marrow Transplant. 2015;21:2185–91.
Gonzalez-Barca E, Domingo-Domenech E, Capote FJ, Gomez-Codina J, Salar A, Bailen A, et al. Prospective phase II trial of extended treatment with rituximab in patients with B-cell post-transplant lymphoproliferative disease. Haematologica. 2007;92:1489–94.
Trappe RU, Dierickx D, Zimmermann H, Morschhauser F, Mollee P, Zaucha JM, et al. Response to rituximab induction is a predictive marker in B-cell post-transplant lymphoproliferative disorder and allows successful stratification into rituximab or R-CHOP consolidation in an international, prospective, multicenter phase II trial. J Clin Oncol. 2017;35:536–43.
Oertel SH, Verschuuren E, Reinke P, Zeidler K, Papp-Vary M, Babel N, et al. Effect of anti-CD 20 antibody rituximab in patients with post-transplant lymphoproliferative disorder (PTLD). Am J Transplant. 2005;5:2901–6.
Blaes AH, Peterson BA, Bartlett N, Dunn DL, Morrison VA. Rituximab therapy is effective for posttransplant lymphoproliferative disorders after solid organ transplantation: results of a phase II trial. Cancer. 2005;104:1661–7.
Choquet S, Leblond V, Herbrecht R, Socie G, Stoppa AM, Vandenberghe P, et al. Efficacy and safety of rituximab in B-cell post-transplantation lymphoproliferative disorders: results of a prospective multicenter phase 2 study. Blood. 2006;107:3053–7.
Choquet S, Oertel S, LeBlond V, Riess H, Varoqueaux N, Dorken B, et al. Rituximab in the management of post-transplantation lymphoproliferative disorder after solid organ transplantation: proceed with caution. Ann Hematol. 2007;86:599–607.
Trappe R, Oertel S, Leblond V, Mollee P, Sender M, Reinke P, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell post-transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol. 2012;13:196–206.
van Esser JW, Niesters HG, van der Holt B, Meijer E, Osterhaus AD, Gratama JW, et al. Prevention of Epstein-Barr virus-lymphoproliferative disease by molecular monitoring and preemptive rituximab in high-risk patients after allogeneic stem cell transplantation. Blood . 2002;99:4364–9.
Delapierre B, Reman O, Dina J, Breuil C, Bellal M, Johnson-Ansah H, et al. Low dose rituximab for pre-emptive treatment of Epstein Barr virus reactivation after allogenic hematopoietic stem cell transplantation. Curr Res Transl Med. 2019. https://doi.org/10.1016/j.retram.2019.03.001.
Van Besien K, Bachier-Rodriguez L, Satlin M, Brown MA, Gergis U, Guarneri D, et al. Prophylactic rituximab prevents EBV PTLD in haplo-cord transplant recipients at high risk. Leuk Lymphoma. 2019. https://doi.org/10.1080/10428194.2018.1543877.
Hiraga J, Tomita A, Sugimoto T, Shimada K, Ito M, Nakamura S, et al. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood. 2009;113:4885–93.
Tsai PC, Hernandez-Ilizaliturri FJ, Bangia N, Olejniczak SH, Czuczman MS. Regulation of CD20 in rituximab-resistant cell lines and B-cell non-Hodgkin lymphoma. Clin Cancer Res. 2012;18:1039–50.
McIver Z, Stephens N, Grim A, Barrett AJ. Rituximab administration within 6 months of T cell-depleted allogeneic SCT is associated with prolonged life-threatening cytopenias. Biol Blood Marrow Transplant. 2010;16:1549–56.
Petropoulou AD, Porcher R, Peffault de Latour R, Xhaard A, Weisdorf D, Ribaud P, et al. Increased infection rate after preemptive rituximab treatment for Epstein-Barr virus reactivation after allogeneic hematopoietic stem-cell transplantation. Transplantation. 2012;94:879–83.
Teeling JL, Mackus WJ, Wiegman LJ, van den Brakel JH, Beers SA, French RR, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol. 2006;177:362–71.
Herter S, Herting F, Mundigl O, Waldhauer I, Weinzierl T, Fauti T, et al. Preclinical activity of the type II CD20 antibody GA101 (obinutuzumab) compared with rituximab and ofatumumab in vitro and in xenograft models. Mol Cancer Ther. 2013;12:2031–42.
Vitolo U, Trneny M, Belada D, Burke JM, Carella AM, Chua N, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol. 2017;35:3529–37.
Marcus R, Davies A, Ando K, Klapper W, Opat S, Owen C, et al. Obinutuzumab for the first-line treatment of follicular lymphoma. N Engl J Med. 2017;377:1331–44.
Sehn LH, Chua N, Mayer J, Dueck G, Trneny M, Bouabdallah K, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17:1081–93.
Sehn LH, Goy A, Offner FC, Martinelli G, Caballero MD, Gadeberg O, et al. Randomized phase II trial comparing obinutuzumab (GA101) with rituximab in patients with relapsed CD20+indolent B-cell non-Hodgkin lymphoma: Final analysis of the GAUSS study. J Clin Oncol. 2015;33:3467–74.
Tang T, Lim C, Tao M, Quek R, Farid M, Kim WS, et al . A multi-center, non-randomized phase 2 study of ofatumumab in combination with ICE-chemotherapy (O-ICE) in patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) [abstract]. Blood. 2014;124:5645.
van Imhoff GW, McMillan A, Matasar MJ, Radford J, Ardeshna KM, Kuliczkowski K, et al. Ofatumumab versus rituximab salvage chemoimmunotherapy in relapsed or refractory diffuse large B-cell lymphoma: The ORCHARRD Study. J Clin Oncol. 2017;35:544–51.
Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–35.
Rooney CM, Smith CA, Ng CY, Loftin S, Li C, Krance RA, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet. 1995;345:9–13.
Vickers MA, Wilkie GM, Robinson N, Rivera N, Haque T, Crawford DH, et al. Establishment and operation of a Good Manufacturing Practice-compliant allogeneic Epstein-Barr virus (EBV)-specific cytotoxic cell bank for the treatment of EBV-associated lymphoproliferative disease. Br J Haematol. 2014;167:402–10.
Prockop SE, Doubrovina E, Baroudy K, Boulad F, Khalaf R, Papadopoulose EB, et al. Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes (EBV-CTLs) for treatment of rituximab-refractory EBV-associated lymphoproliferative disorder (EBV-LPD). Presented at: 2015 AACR Annual Meeting; April 18–22, Philadelphia, PA: 2015. Abstract 8841.
Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+lymphomas after allogeneic hematopoietic cell transplantation. Blood. 2012;119:2644–56.
Quintarelli C, Savoldo B, Dotti G. Gene therapy to improve function of T cells for adoptive immunotherapy. Methods Mol Biol. 2010;651:119–30.
Marin V, Cribioli E, Philip B, Tettamanti S, Pizzitola I, Biondi A, et al. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum Gene Ther Methods. 2012;23:376–86.
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–9.
Fox CP, Burns D, Parker AN, Peggs KS, Harvey CM, Natarajan S, et al. EBV-associated post-transplant lymphoproliferative disorder following in vivo T-cell-depleted allogeneic transplantation: clinical features, viral load correlates and prognostic factors in the rituximab era. Bone Marrow Transplant. 2014;49:280–6.
Dotti G, Fiocchi R, Motta T, Mammana C, Gotti E, Riva S, et al. Lymphomas occurring late after solid-organ transplantation: influence of treatment on the clinical outcome. Transplantation. 2002;74:1095–102.
Gershburg E, Marschall M, Hong K, Pagano JS. Expression and localization of the Epstein-Barr virus-encoded protein kinase. J Virol. 2004;78:12140–6.
AlDabbagh MA, Gitman MR, Kumar D, Humar A, Rotstein C, Husain S. The role of antiviral prophylaxis for the prevention of Epstein-Barr virus-associated posttransplant lymphoproliferative disease in solid organ transplant recipients: A systematic review. Am J Transplant. 2017;17:770–81.
Portell C, Nand S. Single agent lenalidomide induces a response in refractory T-cell posttransplantation lymphoproliferative disorder. Blood. 2008;111:4416–7.
Laubli H, Tzankov A, Juskevicius D, Degen L, Rochlitz C, Stenner-Liewen F. Lenalidomide monotherapy leads to a complete remission in refractory B-cell post-transplant lymphoproliferative disorder. Leuk Lymphoma. 2016;57:945–8.
Haque T, Chaggar T, Schafers J, Atkinson C, McAulay KA, Crawford DH. Soluble CD30: a serum marker for Epstein-Barr virus-associated lymphoproliferative diseases. J Med Virol. 2011;83:311–6.
Gandhi M, Ma S, Smith SM, Nabhan C, Evens AM, Winter JN et al. Brentuximab vedotin (BV) plus rituximab (R) as frontline therapy for patients (Pts) with Epstein Barr Virus (EBV)+and/or CD30+lymphoma: phase I results of an ongoing phase I-II study. Blood. 2014;124:3096.
Massachusetts General Hospital, Dana-Farber Cancer Institute, Brigham and Women's Hospital, Beth Israel Deaconess Medical Center, Millennium Pharmaceuticals, Inc. Bortezomib plus rituximab for EBV+PTLD. https://ClinicalTrials.gov/show/NCT01058239; 2011.
Noy A, de Vos S, Thieblemont C, Martin P, Flowers CR, Morschhauser F, et al. Targeting Bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood. 2017;129:2224–32.
Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Al Hamed, R., Bazarbachi, A. & Mohty, M. Epstein-Barr virus-related post-transplant lymphoproliferative disease (EBV-PTLD) in the setting of allogeneic stem cell transplantation: a comprehensive review from pathogenesis to forthcoming treatment modalities. Bone Marrow Transplant 55, 25–39 (2020). https://doi.org/10.1038/s41409-019-0548-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41409-019-0548-7
- Springer Nature Limited
This article is cited by
-
Prediction model for EBV infection following HLA haploidentical matched hematopoietic stem cell transplantation
Journal of Translational Medicine (2024)
-
Rituximab for posttransplant lymphoproliferative disorder – therapeutic, preemptive, or prophylactic?
Bone Marrow Transplantation (2024)
-
Molecular monitoring of viral infections in immunocompromised patients in a large university hospital in Italy: reflections after thirteen years of real-life activity
European Journal of Clinical Microbiology & Infectious Diseases (2024)
-
Applying Rituximab During the Conditioning Regimen Prevents Epstein–Barr Virus Infection Following Allogeneic Hematopoietic Stem Cell Transplant in a Children’s Cohort: A Retrospective Case–Control Study
Infectious Diseases and Therapy (2023)
-
Expert Consensus on the Characteristics of Patients with Epstein–Barr Virus-Positive Post-Transplant Lymphoproliferative Disease (EBV+ PTLD) for Whom Standard-Dose Chemotherapy May be Inappropriate: A Modified Delphi Study
Advances in Therapy (2023)