
Abstract
Graft-versus-host disease (GVHD), an immunological disorder that arises from donor T cell activation through recognition of host alloantigens, is the major limitation in the application of allogeneic hematopoietic stem cell transplantation (allo-HSCT). Traditional immunosuppressive agents can relieve GVHD, but they induce serious side effects. It is highly required to explore alternative therapeutic strategy. Human amniotic epithelial stem cells (hAESCs) were recently considered as an ideal source for cell therapy with special immune regulatory property. In this study, we evaluated the therapeutic role of hAESCs in the treatment of GVHD, based on our previous developed cGMP-grade hAESCs product. Humanized mouse model of acute GVHD (aGVHD) was established by injection of huPBMCs via the tail vein. For prevention or treatment of aGVHD, hAESCs were injected to the mice on day -1 or on day 7 post-PBMC infusion, respectively. We showed that hAESCs infusion significantly alleviated the disease phenotype, increased the survival rate of aGVHD mice, and ameliorated pathological injuries in aGVHD target organs. We demonstrated that hAESCs directly induced CD4+ T cell polarization, in which Th1 and Th17 subsets were downregulated, and Treg subset was elevated. Correspondingly, the levels of a series of pro-inflammatory cytokines were reduced while the levels of the anti-inflammatory cytokines were upregulated in the presence of hAESCs. We found that hAESCs regulated CD4+ subset polarization in a paracrine mode, in which TGFβ and PGE2 were selectively secreted to mediate Treg elevation and Th1/Th17 inhibition, respectively. In addition, transplanted hAESCs preserved the graft-versus-leukemia (GVL) effect by inhibiting leukemia cell growth. More intriguingly, hAESCs infusion in HSCT patients displayed potential anti-GVHD effect with no safety concerns and confirmed the immunoregulatory mechanisms in the preclinical study. We conclude that hAESCs infusion is a promising therapeutic strategy for post-HSCT GVHD without compromising the GVL effect. The clinical trial was registered at www.clinicaltrials.gov as #NCT03764228.

Similar content being viewed by others


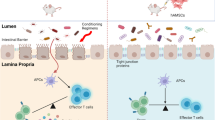
Introduction
Allogeneic hematopoietic stem cell transplantation (HSCT) is the most effective strategy to cure malignant blood disease in the clinic [1,2,3]. However, the incidence of graft-versus-host disease (GVHD) remains the major cause of morbidity and mortality after transplantation, as the common issue confronted by the mainstream HSCT protocols worldwide [3,4,5,6]. GVHD is initiated via recognition of host alloantigen by the donor T cells infused during HSCT [2, 3]. The activated donor T cells are involved in extensive tissue injuries in the recipient patients [7, 8]. Currently, high-dose corticosteroids administration is still the main first-line therapeutic medication for GVHD after HSCT in clinic and other immunosuppressive drugs are employed as preventive treatments [9,10,11]. Regrettably, more than 50% aGVHD patients exhibited steroid-resistant responses [12,13,14]. Moreover, as non-specific immunosuppressive agents, those drugs usually delay the reconstitution of immune system and therefore increase the opportunity for infections in patients receiving HSCT [14, 15]. On the other hand, graft versus leukemia (GVL), the immune response executed by graft lymphocytes to remove residual leukemia cells in HSCT recipients, was suppressed indiscriminately during the GVHD treatment, which raised the risk of leukemia relapse [16,17,18]. Therefore, alternative strategy against GVHD that retains GVL is urgently required to improve the therapeutic effect of HSCT.
Biologically, GVHD can be regarded as a severe inflammatory state in the HSCT recipients after breakdown of the host immune balance. Therefore, administration of immunomodulatory cells, which potentially regulates immune homeostasis with enduring effect, is accepted as a better intervention treatment of GVHD. To date, the regulatory T cells (Tregs) and mesenchymal stromal cells (MSCs) are most universal candidates in this category. Nevertheless, the low number of Tregs in both peripheral and umbilical cord blood, the major sources of Treg cellular products, limits the administration dose for patients [19, 20]. In addition, the function of Treg cells in suppressing antiviral immune responses may bring safety concern in GVHD patients [21, 22]. Furthermore, MSCs are the most extensively investigated cells for the GVHD therapy due to their immunosuppressive activities. However, clinical trials including ours yielded paradoxical results in the prevention and treatment of steroid-refractory GVHD, with positive and negative therapeutic effects [23, 24]. Impairing the GVL effect, higher relapse rate (RR) [24, 25] and more frequent occurrence of severe infection limit their further clinical applications [26].
Human amniotic epithelial stem cells (hAESCs), isolated from the amniotic epithelium layer that is closest to the fetus in the term placenta, have been drawing attention as an attractive candidate for cellular therapy in immune disease. This is based on the characterized features of hAESCs, in which the most attractive one is their immunoregulatory property. It has been demonstrated that hAESCs or their conditioned medium alone could dampen inflammatory reactions by suppressing the proliferation, inflammatory cytokine production, and cytotoxic activity in different immune cell subtypes in vitro and in vivo [27, 28]. The immunoregulatory mechanisms of hAESCs are attributed to the antibacterial and anti-inflammatory functions of amniotic membrane that help the fetus adapt the intricate preterm immune environment, although the detailed mechanisms have not been clearly illustrated. Furthermore, therapeutic effect of hAESCs administration were determined in different preclinical inflammatory disease models, including Crohn’s disease, fibrotic disease, and autoimmune disease, according to others and our previous studies [29,30,31,32,33,34]. Also, other characteristics of hAESCs appropriately meet the criteria of cellular product. Unlike the embryonic stem cells and induced pluripotent stem cells, hAESCs have no tumorigenicity due to the lack of telomerase, which is upregulated during oncogenesis. More intriguingly, immune privilege of hAESCs has been demonstrated after transplantation in mammalian animal species [35,36,37] and in human volunteers [38], mainly based on the absence of HLA class II molecules and the high expression of non-classical HLA class I molecules, corresponding to amniotic membrane serving as the barrier for maintaining feto-maternal tolerance during pregnancy. Moreover, sufficient supply (more than 100 million hAESCs harvested from a placenta) without ethical debates potentially fulfills hAESCs as an applicable source of biological product. Taken together, hAESCs have special and attractive potential as a superior candidate of cell therapy for GVHD in clinic. Aiming the clinical application, we have developed a current Good Manufacturing Practice (cGMP)-grade hAESCs cellular therapy product in a serum-free system, based on our previous study [35].
In the present study, the therapeutic effect of the hAESCs product on GVHD was investigated in a humanized mouse model and haploidentical related HSCT (HDR-HSCT) patients in a preliminary clinical study with small sample size. We demonstrated that hAESCs infusion can efficiently improve GVHD by repolarizing CD4+ T cells while maintaining the GVL effect in both preclinical and clinical studies.
Materials and methods
hAESCs collection, preparation, and release
The human amniotic epithelial stem cell (hAESC) injection is liquid cell suspension that is manufactured and provided under current Good Manufacturing Practice (cGMP) guidelines by Shanghai iCell Biotechnology Co., Ltd.
Briefly, the amnion was peeled away from the underlying chorion within the operating theater. After being rinsed with sterile saline, the amnion was immersed in tissue preservation solution and then transferred to the cell procedure center of Shanghai iCell Biotechnology Co., Ltd. There, all reagents, equipment, and procedures utilized for hAESCs manufacturing process were following cGMP and the guidance on cell-based therapy products. Proceeded hAESCs at passage 1 (15 mL) were packaged in the cryogenic cell storage bags (Miltenyi Biotec, Germany) and stored in vapor-phase liquid nitrogen.
Before being released for clinical settings the finial products should be proved that ≥90.0% of the cells were positive for the surface antigen CD324 (cat# 324120, Biolegend, CA, USA), and ≤2.0% were negative for CD146 (cat# 361036, Biolegend, CA, USA), CD34 (cat# 378602, Biolegend, CA, USA), and HLA-DR (cat# 307614, Biolegend, CA, USA). Furthermore, the final products were tested for cell viability, biological potency, impurity residue, sterility, mycoplasma, endotoxin, and viral pathogens.
On the day of cell intravenous infusion, the sealed bag of hAESCs was retrieved from vapor-phase liquid nitrogen and thawed in a 37–38 °C water bath for 1–2 min. Then 35 mL sterilized water for injection was aseptically added into the infusion bag for a final volume of 50 mL cell suspension. All patients intravenously received hAESCs injection consistent with the ethically permitted clinical study.
GVHD and GVL mouse models
All animal studies were approved by the Institutional Animal Care and Use Committee of Zhejiang University (ZJU20210127) and adhered to the NIH guidelines for the ethical treatment of animals. NCG mice (NOD-Prkdcem26Cd52Il2rgem26Cd22/Nju) were purchased from Model Animal Research Center of Nanjing University. The mice were acclimated to the room for 1 week after arrival and were maintained on a normal 12 h light-dark cycle. The mice were housed in conventional cages with free access to a standard pellet diet and water in specific pathogen-free conditions with a temperature of 24 ± 2 °C and 60%–70% relative humidity. Eight-week-old NCG mice were injected with 1 × 107 huPBMCs or 2 × 107 huPBMCs, respectively, via the tail vein to induce aGVHD. Mice receiving PBS alone were used as control group. In the aGVHD prevention group, 2 × 106 hAESCs were injected one day before PBMC. In the aGVHD treatment group, 2 × 106 hAESCs were injected on day 7 post-PBMC transfusion.
For GVL studies, 5 × 106 GFP-labeled HL60 cells and 107 huPBMCs were transplanted into the NCG mice via the tail vein to induce graft vs leukemia model. To determine the role of hAESCs in GVL, 2 × 106 hAESCs were co-transplanted into the GVL mice on day 0. Mice with an equivalent PBS injection were used as a control group. Mice from different groups were sacrificed 2 weeks post-transplantation to examine the HL60 in peripheral blood by flow cytometry or 3 weeks after cell transplantation to examine livers and spleens.
Histopathological evaluation of GVHD in mouse
The mouse GVHD assessment scale was according to the grading system established by Cooke KR et al. [39]. The body weight and clinical phenotypes, including weight loss, posture, activity, fur ruffling and diarrhea, as well as the survival status, were recorded every other day. The mice were sacrificed at day 10 after hAESCs injection. The aGVHD target organs were fixed in 4% paraformaldehyde and embedded in paraffin for histopathological analysis with H&E staining or anti-CD3 (cat# ab16669, Abcam, Cambridge, UK) immunohistochemistry staining. Histopathological scoring was assessed according to the system established by Polchert D et al. [40]. In vascular adhesion molecule staining, the arteries of target mice were harvested and embedded in optimal cutting temperature compound (OCT). The frozen sections were stained by anti-PECAM1 (cat# sc-376764, Santa Cruz, CA, USA), anti-ICAM1 (cat# 771504, Biolegend, CA, USA) or anti-VCAM1 (cat# MA134974, BD Pharmingen, Shanghai, China) followed by fluorescence-conjugated secondary antibodies. All experiments were repeated at least three times.
Flow cytometry and magnetic cell sorting
After washing with PBS, the hAESCs were incubated with the fluorescence-conjugated antibodies of anti-huCD34 (cat# 378602, Biolegend, CA, USA), anti-huCD45 (cat# 982322, Biolegend, CA, USA), anti-HLA-DR (cat# 307614, Biolegend, CA, USA) and anti-HLA-DQ (cat# 318104, Biolegend, CA, USA) and the HUVECs were stained with the fluorescence-conjugated monoclonal antibodies of anti-ICAM1 (cat# BMS313, Ebioscience, CA, USA) and anti-VCAM1 (cat# hzA547Ra, Ebioscience, CA, USA) for 30 min on ice. For detection of CD4 T cell subsets, PBMCs isolated from different groups of mice were incubated with fluorescence-conjugated antibodies of anti-CD4 (cat# 980804, Biolegend, CA, USA), anti-CD25 (cat# 985812, Biolegend, CA, USA), anti-HLA-DR (cat# 307614, Biolegend, CA, USA), anti-IFNγ (cat# 517901, Biolegend, CA, USA), anti-IL2 (cat# 503805, Biolegend, CA, USA), anti-IL17 (cat# 506907, Biolegend, CA, USA) and anti-FOXP3 (cat# 364702, Biolegend, CA, USA). Cells were permeated with a Fixation & Intracellular Permeabilization Kit (cat# 421002, Biolegend, CA, USA) for detection of intracellular proteins. Flow cytometry analysis was performed with a FC500 flow cytometer (Beckman-Coulter, CA, USA) and FlowJo software (Tree Star, Inc, OR, USA.). Isotype controls were used in each experiment. Magnetic cell sorting (MACS) assays were performed with the MojoSort isolation kit for human CD4+ T cells according to the manufacturer’s instructions. All experiments were repeated at least 3 times.
Immunocytofluorescence
After PBS wash, cells were fixed in 4% paraformaldehyde for 30 min on ice followed by treatment with 0.2% Triton X-100 for 30 min at room temperature. After blocking in 3% horse serum for 1 h, cells were incubated with primary antibody overnight at 4 °C. Primary antibodies against Pan-Cytokeratin (cat# ab7753, 1:200, Abcam, Cambridge, UK), E-cadherin (cat# ab233611, 1:200, Abcam, Cambridge, UK), OCT4 (cat# SC8826, 1:50, Santa Cruz, CA, USA), SSEA4 (cat# FCMAB116P, 1:100, Millipore, MA, USA) and NANOG (cat# AF1997, 1:40, R&D Systems, CA, USA) were diluted in blocking buffer. Fluorescent secondary antibodies were incubated for 1 h at room temperature. Cell nuclei were stained with DAPI (cat# D9542, Sigma-Aldrich, Shanghai, China).
Quantitative real-time PCR
Tissue homogenates or cell lysates were prepared in TRK lysis buffer (cat# PR021, Omega, GA, USA). Total RNA was isolated using an E.N.Z.A. total RNA kit (cat# R6874-02, Omega, GA, USA) according to the manufacturer’s instructions. RNA was reverse transcribed into cDNA using the ReverTra Ace qPCR RT kit (cat# FSQ-101, Toyobo, Japan) according to the manufacturer’s instructions. Quantitative PCR was performed with SYBR FAST QPCR Kit (cat# SFUKB, KAPA Biosystems) using a Bio-Rad iCycler real-time PCR detection system (Bio-Rad, CA, USA). GAPDH was used as an internal control. The primer sequences are listed in Supplementary Table S1. All experiments were repeated at least 3 times.
Cytokine assay
The cytokines GM-CSF, IFNγ, IL-4, IL-8, IL-10, TGFβ, and TNFα in mouse serum were each quantified with the appropriate ELISA kits (all from Ebioscience, CA, USA) according to the manufacturer’s instructions. The analyses were performed with Bio-Plex 200 Systems (Bio-Rad, CA, USA). PGE2 were detected with an ELISA kit (Cayman, MI, USA) according to the manufacturer’s instructions and analyzed by Nanoquant (Tecan, Switzerland). All experiments were repeated at least 3 times.
Leukocyte-endothelial adhesion and transmigration assays
For adhesion assays, PHA-blasted human T cells were labeled with Calcein (cat# C3100MP, Invitrogen, CA, USA), and were then co-cultured with HUVECs in different conditional medium from hAESCs, PBMCs (with or without 5 μg/mL PHA) or co-cultured hAESCs+PBMCs for 30 min, followed by gently washing twice to remove non-adherent cells and microphotographs. Cell nuclei were stained with Hoechst. Transmigration was performed using 6.5 mm transwell filters with an 8 μm pore size (Costar, NY, USA). HUVECs were seeded on an insert coated with 0.1% gelatin (cat# 1288485, Sigma-Aldrich, Shanghai, China) and cultured until confluent. Calcein-labeled T cells were then added to the upper transwell chamber containing different conditional medium as described above for 12 h. Afterwards, the cells that transmigrated to the bottom compartment were imaged and quantified. All experiments were repeated at least 3 times.
Clinical trial design
The clinical trial was approved by the institutional review board of Peking University People’s Hospital (2018PHD006-01) and conducted under an investigational new stem cell application from the National Health Commission of China. All patients and donors provided written informed consent according to the Declaration of Helsinki. The trial was registered at https://www.clinicaltrials.gov/ as #NCT03764228. Patients with high risk for aGVHD were assessed for inclusion as follows: acute leukemia (≤CR2), myelodysplastic syndromes (MDSs, RA, RARS, RCMD, EB-1, EB-2), or chronic myeloid leukemia (CML) (CP or AP), underwent haploidentical hematopoietic stem cell transplantation with haplotype sibling female donor or with donor age >30 years old, adequate performance status, and organ function. Cryopreserved cGMP-grade hAESCs (Shanghai iCell Biotechnology, Shanghai, China) were I.V. infused into haploHSCT patients at days −1 and +7 of HSCT. Dose escalation was planned in cohorts of a minimum of 3 patients starting at 1 × 106 cells/kg per dose and escalating to 2 × 106 cells/kg per dose and up to 5 × 106 cells/kg per dose. The primary objectives were to evaluate the safety of hAESCs infusion and determine the maximum tolerated dose (MTD) of 3 doses of hAESCs cells administered in conjunction with haploHSCT (days −1 and +7 post-transplant). All involved subjects were monitored for 100 days after the first intravenous infusion of hAESCs. All untoward medial occurrences after the first hAESCs treatment were considered adverse events (AEs). The severity of AEs was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events 5.0. Monitoring for safety included vital signs (heart rate, respiration rate, temperature, and blood pressure), serial blood tests, and physical examination at the day 1, 2, 3, 4, 7, 10, 14, 21, 30, 60, and 100 after the first hAESC administration. To the purpose of dose finding, “toxicity” was defined as any of the events (1) death, (2) grade 3 − 4 infusion reaction, (3) grade 4 organ toxicity (not including mucositis or myelosuppression) within 28 days of the first hAESCs cell infusion. Secondary objectives were to determine the efficacy of hAESCs infusion on GVHD prophylaxis, estimate the proportion of patients with engraftment/graft failure, 100-day nonrelapse mortality (NRM), cumulative incidence (CI) of aGVHD, RR, overall survival (OS), and disease-free survival (DFS).
Haplo-HSCT procedure
All patients received a myeloablative conditioning regimen (MAC) without in vitro T-cell depletion. All patients received haploidentical allo-HSCT, the conditioning regimen was modified as BUCY + ATG (thymoglobulin) consisting of cytarabine 4 g/m2 per day intravenously on days −10 to −9; busulfan (3.2 mg/kg per day, intravenously on days −8 to −6); cyclophosphamide (CY, 1.8 g/m2 per day) intravenously on days −5 to −4; Me-CCNU (250 mg/m2), orally once on day −3; and thymoglobulin (ATG, Sang Stat, Lyon, France; 2.5 mg/kg per day) intravenously for 4 consecutive days from days −5 to −2. All subjects received fresh granulocyte colony-stimulating factor (G-CSF) mobilized peripheral blood cells. Ganciclovir was administered during conditioning (through day −2) and acyclovir (400 mg twice a day) was given until the discontinuation of all immunosuppressive agents. Patients also received prophylactic drugs to prevent infection by fungi.
Definitions and endpoints
hAESCs engraftment was defined as over 0.08% using Chimerism analyses (Shanghai Tissuebank Diagnostics) [41]. White blood cell engraftment was defined as an ANC count of 0.5 × 109 cells/L or more for 3 consecutive days. Platelet engraftment was defined as a platelet count of 20 × 109 cells/L or more for 7 consecutive days without transfusion. Acute and chronic GVHD were diagnosed and graded according to established criteria [42, 43]. CMV infection was defined as a plasma PCR result above 1000 copies/mL. EBV infection was defined as a plasma PCR result above 1000 copies/mL. OS times were measured as the date of HSCT until death from any cause. DFS was defined as the time from transplantation to relapse or death from any cause. Surviving patients were censored on the date of their last follow-up. Non-relapse mortality (NRM) was defined as death after allo-HSCT without disease progression or relapse. Relapse was defined by the appearance of morphological evidence of the disease obtained from testing samples from the peripheral blood, bone marrow, or extramedullary sites or else by the recurrence and sustained presence of pre-transplantation chromosomal abnormalities.
Clinical blood sample preparation and immunoassays
Blood samples of the 10 subjects underwent hAESCs trials were obtained at day 7, 14, 30, and 90 after HSCT and were then analyzed by flow cytometry for detecting the reconstitution of T, B, NK cells and monocytes. The plasma sample at day +7 were collected and screened the plasma cytokine levels with Luminex assay (Luminex, TX, USA) or ELISA kit (R&D Systems, CA, USA).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software). The data are presented as the mean ± SEM. Comparisons between two groups were by unpaired t-test, between more than two groups by one-way ANOVA followed by Bonferroni’s post-hoc test or by two-way ANOVA. P < 0.05 was considered to reach statistically significance.
Results
hAESCs isolation and identification in a serum-free system
To exclude the serum-derived contamination, we set up a specific serum-free isolating and culture system for the cGMP-grade hAESCs [35]. Morphological observation of primary-cultured hAESCs showed the normal cobblestone-like monolayer cells (Fig. 1a). Negative expression of the hematopoietic marker CD45 and CD34 (Fig. 1b), and the full expression of signature epithelial marker pan-Cytokeratin and E-cadherin (Fig. 1c) were detected by flow cytometry, indicating the purity of hAESCs without contamination of blood cells. Moreover, compared to human umbilical cord MSCs (hUC-MSCs), low level of classical mesenchymal marker group, and abundant expressions of classical epithelial marker group and epithelial-associated protein group were detected in hAESCs by proteomic analysis (Fig. 1d). The extensive expression of pluripotent marker OCT4, NANOG and SSEA4 proved the homeostatic and low-differentiated status of hAESCs (Fig. 1e). On the other hand, little expression of HLA class II molecules HLA-DR and HLA-DQ was detected in hAESCs (Fig. 1f). Taken together, these data demonstrate that hAESCs, different from MSCs, are a certain kind of epithelial stem cells; the cGMP-grade hAESCs show decent homogeneity and maintain their biological characteristics.

a Morphology of hAESCs in light field; scale bar = 50 μm. b Expression of blood cell markers CD45 and CD34 (blue) in hAESCs by flow cytometry; isotype antibodies were used as control (light red). c Immunocytochemistry for indication of epithelium marker Pan-cytokeratin (green) and E-cadherin (green) expression in hAESCs. Nuclei were stained with DAPI (blue); scale bar = 50 μm. d Heatmap representing color-coded log2 transformed relative abundance ratio from proteomics analysis of signature genes in hUC-MSCs and hAESCs. e Immunocytochemistry for indicating the pluripotent marker OCT4 (green), Nanog (red), and SSEA4 (green) expression in hAESCs. Nuclei were stained with DAPI (blue); scale bar = 50 μm. f Flow cytometry detection of the human MHC class II cell surface markers HLA-DR, DQ (blue) in hAESCs; isotype antibodies were used as the control (light red).
hAESCs inhibit the development of aGVHD in humanized mice model
NCG mice, a novel immune-deficient mouse strain lacking T, B, and NK cells, were employed to establish an aGVHD humanized mice model. As reported in NOG mice, we also found that total body irradiation was not necessary to induce aGVHD in NCG mice. To investigate the therapeutic effect of hAESCs in aGVHD, NCG mice were divided into four groups: aGVHD group (human PBMCs transplantation only), aGVHD prevention group (hAESCs injection 1 day before PBMCs transplantation), aGVHD treatment group (hAESCs injection on day 7 after PBMCs transplantation) and control group (PBS injection only) (Fig. 2a). High chimerism of transplanted cells of human origin (huCD45+) was detected in the peripheral blood of recipient NCG mice at day 14 after PBMCs transplantation (Fig. 2b). Most transfused huCD45+ cells were found as human CD3 positive (huCD3+), suggesting a robust expansion of human T cells in recipient mice (Fig. 2b). The engraftment rates were comparable in mice with or without hAESCs injection.

a Illustration of the experimental design. The human PBMCs (2 × 107 cells/mouse) and hAESCs (2 × 106 cells/mouse) were injected via caudal vein; the mice injected with PBS were used as controls. b Two weeks after human PBMCs transplantation, the engraftment rate of each group was determined by detection of huCD45 and huCD3 immune cells. c Representative images of disease phenotype in each group on day 21 after human PBMCs transplantation. d The body weight change of each group was recorded every other day (n = 10). e The aGVHD clinical scores of each group were recorded every other day (n = 10). f The survival rates of each group were recorded every day (n = 10). Data are presented as the mean ± SEM, ns no significance; ***P < 0.001; ****P < 0.0001.
The aGVHD disease phenotypes began to be observed around the second week after PBMCs transfusion in the aGVHD group, including body weight loss, mobility decrease, hunching, diarrhea, and ruffled hair (Fig. 2c–e). By contrast, in the hAESCs-injected groups, particularly the aGVHD prevention group, the aGVHD disease phenotypes were postponed and relieved significantly (Fig. 2c–e). Moreover, the survival durations were prolonged and survival rates were increased in hAESCs-injected groups compared with the aGVHD group; the anti-aGVHD effect of hAESCs was better in the prevention group than the treatment group (Fig. 2f).
To highlight the advantages of hAESCs in the prevention and treatment of GVHD, we included human umbilical cord mesenchymal stem cells (hUCMSCs) and two widely utilized immunosuppressants, Cyclosporine A and Mycophenolate Mofetil, as control treatment groups. Similar preventive effects were observed in both the immunosuppressant groups and the aGVHD prevention group (Supplementary Fig. S1a–d). However, while the aGVHD treatment group did not demonstrate superior therapeutic effects compared to the immunosuppressant groups, it exhibited superiority over the hUCMSCs group.
hAESCs ameliorate the pathological injuries of aGVHD
To determine the effect of hAESCs on pathological injuries of aGVHD, histological analysis was conducted on the major GVHD target organs from all mouse groups. In the aGVHD group, pathological progression resulted in significant regions of inflammation around the hepatic ducts in the livers, perivascular cuffing, and infiltration in alveolar in the lungs, dropsy in the renal tubulars and blunting of the villi in the intestines. In the presence of hAESCs, these pathological injuries were reduced markedly, with a more pronounced rescue effect was observed in the prevention group compared to the treatment group (Fig. 3a, b). Similarly, consistent with the aforementioned phenotypic descriptions, the aGVHD prevention group exhibited superior mitigation of pathological injuries, resembling the outcomes observed in the immunosuppressant groups. However, the aGVHD treatment group did not manifest superior therapeutic effects compared to the groups treated with immunosuppressants but exhibited superiority over the hUCMSCs group (Supplementary Fig. S1e, f).

a H&E staining of the major aGVHD target organs, including the liver, intestine, lung, and kidney from each mouse group. The mice were sacrificed on day 12 after human PBMCs transplantation. b The pathological score of the major aGVHD target organs from each mouse group (n = 5). c Immunohistochemistry detection of infiltrated donor cells in sections of target organs from each group with huCD3 staining. The mice were sacrificed on day 12 after human PBMCs transplantation. d Masson staining of lung sections from each group. The mice were sacrificed on day 12 after human PBMCs transplantation. e hAESCs were labeled with EGFP via lentivirus infection for 48 h. The EGFP expression (bottom) and light field (upper) of hAESCs are shown. f Localization of hAESCs in the main organs when transplanted with or without PBMCs. Mice were sacrificed on day 5 after hAESCs injection and the EGFP expression was detected by RT-PCR. Data are presented as the mean ± SEM, ns no significance; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Corresponding to the trend of pathological injuries, immunohistochemical staining for human CD3 showed remarkable T cell infiltration into these GVHD target organs in the aGVHD group mice, whereas decreased infiltration of T cells as observed in these organs in the aGVHD treatment mice, and further inhibition in the prevention group (Fig. 3c). Intriguingly, fibrosis in the lungs, identified as a manifestation of chronic GVHD, was also observed in our mouse model. In the aGVHD group, Masson’s staining exhibited collagen accumulation, which was ameliorated in both mouse groups with hAESCs injection (Fig. 3d). To identify the residence of hAESCs in the GVHD target organs and whether PBMCs would affect hAESCs distribution, transduced EGFP in hAESCs (Fig. 3e) were checked by RT-PCR in the main target organs. Strong EGFP signal was detected in the lungs, liver, and kidney, while moderate signal was detected in the intestine, spleen, and lymph nodes, with or without PBMC transplantation (Fig. 3f).
hAESCs modulate cytokine production and CD4+ T cell polarization in mouse aGVHD model
Based on hAESCs modulation of tissue injury and immune cell infiltration during aGVHD, we asked whether infused hAESCs directly modulated the immune system. To this end, cytokine level was first examined in mouse serum of different groups. Compared with the aGVHD group, the pro-inflammatory cytokines TNFα, IFNγ, GM-CSF and IL8 decreased and the anti-inflammatory cytokines TGFβ and IL10 increased in both aGVHD prevention and aGVHD treatment groups (Fig. 4a–f), while no change in the IL4 level among all mouse groups (Fig. 4g). Notably, the expression of the alloantigen HLA-DR, indicative of T cell activation and cytokines generation in GVHD, was highly inhibited in the hAESCs prevention group and partially inhibited in the hAESCs treatment group (Fig. 4h, i). These results implied that the hAESCs modulation targeted the T cell population in aGVHD. There is accumulated evidence that the composition of CD4+ T cell subsets is a significant factor to evoke aGVHD [21, 22, 44, 45]. In the present study, Th1 and Th17 subsets were found to decline in CD4+ T cells in mice of hAESCs-injected groups, especially in the aGVHD prevention group, compared with the aGVHD group (Fig. 4j, k and Supplementary Fig. S2a, b), while the proportion of Th2 subset was not affected by hAESCs infusion (Fig. 4l and Supplementary Fig. S2c). However, the Treg subset was specifically elevated in the aGVHD prevention group (Fig. 4m, n).

a–g On day 10 after human PBMCs transplantation, the peripheral blood was collected for cytokine concentration evaluation by ELISA. h–n Two weeks after human PBMCs transplantation, the mice were sacrificed and spleens were harvested for CD4+ subset analysis. The activation marker, HLA-DR, in CD4+ lymphocytes (h, i), Th1 (CD4+IFNγ+) in CD4+ lymphocytes (j), Th17 (CD4+IL17+) in CD4+ lymphocytes (k), Th2 (CD4+IL4+) in CD4+ lymphocytes (l) and Treg (CD4+CD25hiFOXP3+) in CD4+ lymphocytes (m, n) of each group were detected by flow cytometry. Data are presented as the mean ± SEM, ns no significance; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Pathologically, the pro-inflammatory cytokines induce endothelial dysfunction for organ immune infiltration. To examine whether hAESCs impacted the failure of the first barrier against aGVHD, the endothelial activation and consequent dysfunction were examined. hAESCs infusion reduced expression of endothelial ICAM1 and VCAM1 in vivo (Supplementary Fig. S3a), indicating the inhibition on inflammatory endothelial activation, which was confirmed in human umbilical vein endothelial cells (HUVECs) treated with conditional medium from hAESCs, PBMCs or co-cultured PBMCs-hAESCs (Supplementary Fig. S3b, c). Correspondingly, hAESCs effectively sequestered the T cell-endothelial adhesion and transmigration in the same in vitro system (Supplementary Fig. S3d, e).
hAESCs polarize the CD4+ T cell subsets by producing TGFβ and PGE2
To further investigate the mechanism of CD4+ subset polarization regulated by hAESCs in the pathological environment of GVHD, a hAESCs-PBMCs co-culture system was employed. Consistent with the result in vivo, the proportion of Treg subset in CD4+ cells increased (Fig. 5a and Supplementary Fig. S4a), concomitant with up-regulation of FOXP3, the key transcription factor of Treg cells (Fig. 5b). A decrease of the pro-inflammatory cytokines TNFα and IFNγ and increase of anti-inflammatory factors IL-10 was detected in PBMC co-cultured with hAESCs, when compared with PBMC cultured alone (Fig. 5c). To further determine the regulation of hAESCs in CD4+ subset polarization, CD4+ cells were sorted and co-cultured with hAESCs. Again, the presence of hAESCs enhanced Treg proportion as expected (Fig. 5d and Supplementary Fig. S4b). However, the hAESCs-induced Treg elevation was not affected by IFNγ pretreatment (Fig. 5d and Supplementary Fig. S4b) or Toll-like receptors (TLRs) induction (Fig. 5a, b, d and Supplementary Fig. S4a–f). This is different from the case in MSCs, in which IFNγ stimulation and TLRs activation were considered to be essential to upregulate Treg [46].

a, b hAESCs, with or without LPS (5 μg/mL) pretreatment for 1 h, were co-cultured with human PBMCs (hAESCs:PBMC = 1:10). After 5 days, the PBMCs were collected and the Treg in CD4+ cells were analyzed by flow cytometry (a) and the relative mRNA level of FOXP3, the key transcription factor of Treg, was determined by real-time RT-PCR (b). c The relative mRNA level of TNFα, IFNγ and IL10 in human PBMCs or PBMCs co-cultured with hAESCs were determined by real-time RT-PCR. d Human CD4+ T cells were isolated from human PBMCs by magnetic cell sorting (MACS) and treated with IFNγ (500 U) or LPS (5 μg/mL) before co-culturing with hAESCs (hAESCs:CD4+ PBMC = 1:10). After 5 days, Treg cells in the CD4+ cells were analyzed by flow cytometry. Human CD4+ cells were isolated from human PBMCs by MACS and co-cultured with hAESCs (hAESCs:CD4+ PBMC = 1:10) in contact, separated by transwell or treated with hAESCs conditional medium. After 5 days, the CD4+ cells were collected and the Treg (e), Th1 (f), Th17 (g) and Th2 (h) in CD4+ cells were analyzed by flow cytometry and the relative mRNA level of corresponding lineage-specific transcription factors Foxp3, T-BET, RORc and GATA3 were determined by real-time RT-PCR, respectively (i). j–l Human PBMCs were co-cultured with hAESCs (hAESCs:PBMC = 1:10) with the TGFβ neutralizing antibody, an IDO inhibitor (1-MT), and a PGE2 inhibitor (indomethacin), respectively. Five days later, the percentages of Treg (j), Th1 (k), and Th17 (l) were analyzed by flow cytometry. m The culture media of hAESCs, PBMCs or hAESCs-PBMCs were collected for PGE2 analysis by ELISA.
Since TLR-mediated Treg elevation is dependent on cell contact as reported, we hypothesized that the hAESCs-regulated CD4+ T cell polarization could be in a cell non-contact pattern. Thereby, hAESCs and CD4+ cells were co-cultured in different chambers in a transwell system. Flow cytometry showed that the polarization profile of CD4+ subsets in the non-contact transwell group was similar to the contacted co-culture group (Fig. 5e–i and Supplementary Fig. S5a–d). Similar results were also observed in the hAESCs conditional medium-treated CD4+ cells (Fig. 5e–i and Supplementary Fig. S5a–d). In addition, expression of the lineage-specific transcription factors FOXP3 (Treg), T-BET (Th1), RORc (Th17) and GATA3 (Th2) demonstrated comparable switch trends in all the groups with hAESCs co-culture (Fig. 5i), correlating to the results of immunophenotype shifts. Taken together, these data indicated that hAESCs mediated CD4+ subset polarization by cytokine secretion but were independent of cell contact.
Thereby, our study further focused on the potential roles of critical soluble cytokines in hAESCs regulation. In the transwell co-culture system, the administration of the TGFβ neutralizing antibody abolished the effect of hAESCs on Treg up-regulation but did not affect the regulation of Th1 and Th17 subsets (Fig. 5j–l and Supplementary Fig. S5e–g). On the other hand, the prostaglandin E2 (PGE2) inhibitor, indomethacin, disabled the suppression of both Th1 and Th17 subsets by hAESCs, but had no effect on hAESCs-induced Treg elevation (Fig. 5j–l and Supplementary Fig. S5e–g). Indeed, high PGE2 concentration was detected in the hAESCs culture medium, particularly after being co-cultured with CD4+ PBMCs, compared with PBMC controls (Fig. 5m). However, 1-MT, an inhibitor of IDO, another potential cytokine candidate, had little effect on the regulation of hAESCs in CD4+ subset polarization (Fig. 5j–l and Supplementary Fig. S5e–g). Therefore, hAESCs regulated CD4+ subset polarization in a paracrine mode, in which TGFβ and PGE2 were selectively secreted to mediate Treg elevation and Th1/Th17 inhibition, respectively.
To investigate the impact of hAESCs on the transcriptome of CD4+ T cells, we conducted high-throughput RNA sequencing analysis on CD4+ T cells following coculture of hAESCs. Transcriptome analysis revealed the upregulation of 721 genes, notably including anti-inflammatory genes (FCGBP, GPNMB, GBP1, CISH), in the hAESCs-cocultured group compared to the control group. Conversely, 1220 genes exhibited downregulation in the hAESCs-cocultured group. Among them, SOCS3, IGHG2, DEFA3, and CXCR4 (Supplementary Fig. S6a), recognized for their potential to promote inflammation, contributes to pro-inflammatory responses. Through KEGG analysis, these downregulated genes were enriched in the pathway of PI3K-Akt signaling pathway, TNF signaling pathway, and MAPK signaling pathway, as well as Hippo signaling pathway, Wnt signaling pathway, and calcium signaling pathway, which were reported to induce Th1 and Th17 polarization (Supplementary Fig. S6b) [47,48,49,50,51,52,53].
hAESCs infusion does not impair the GVL effect
Furthermore, to determine whether hAESCs impair the GVL effect in inhibition of aGVHD, leukemia cell lines were employed in co-culture with hAESCs. Flow cytometry of Annexin V/PI indicated that hAESCs did not facilitate the cell growth of the leukemia cells but propelled their apoptosis slightly (Fig. 6a and Supplementary Fig. S7a). Next, the effect of hAESCs on GVL was examined in a leukemia mouse model, which was established by transplanting EGFP-labeled leukemia cells into the NCG mice (Supplementary Fig. S7b). PBMCs infusion in the leukemia mice mitigated the body weight loss, prolonged the survival of mice, and ameliorated local pathological lesions in the liver and spleen, indicating the GVL effect, while co-transplantation of hAESCs with PBMCs demonstrated similar GVL phenotypes in the leukemia mice (Fig. 6b–d). Moreover, comparable cellular growth inhibition of leukemia cells in vivo was detected in the GVL group and the GVL+hAESCs group (Fig. 6e–g and Supplementary Fig. S7c), corresponding to the in vitro results (Fig. 6a). All the data above suggested that hAESCs infusion did not disturb the GVL effect.

a hAESCs were co-cultured with HL60, U937, Kasumi or Nalm6 cell lines for 48 h. The leukemia cell lines were collected for flow cytometry detection of Annexin V/PI. b–g EGFP-labeled leukemia cells (5 × 106/mouse) alone or with PBMCs (1.5 × 107/mouse) were transplanted into NCG mice, set as Leukemia group and Leukemia+PBMC (GVL) group. In another group of mice, hAESCs (2 × 106/mouse) were injected at the time of leukemia and PBMC co-transplantation, set as Leukemia + PBMC (GVL) + hAESCs group. Mice injected with PBS were used as control group. The body weights of each group were recorded every other day (n = 10) (b). The survival rates of each group were recorded every day (n = 10) (c). The disease phenotype and the representative images of spleens and livers 3 week after cell transplantation are shown. Note that hAESCs infusion ameliorates the pathological lesion in spleens and livers (d). The percentage of EGFP-labeled leukemia cells in the peripheral blood of the mice on day 14 after cell transplantation (e). The EGFP-labeled leukemia cells in livers of the mice on day 7 after cell transplantation are shown in representative images of liver sections (f) with cell number quantification (g). Nuclei were stained with DAPI (blue); scale bar = 50 μm. Data are presented as the mean ± SEM, ns no significance; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Clinical outcomes of hAESCs treatment for aGVHD in HRD-HSCT patients
The detailed features and clinical outcomes of hAESCs treatment for HRD-HSCT patients at high risk of aGVHD are summarized in Table 1. The median follow-up duration for survivors was 6 months. All 10 patients achieved primary engraftment of HSC, white blood cells, and platelets. Eight patients achieved hAESCs engraftment, while no detection was found in the other 2 patients. Among the 10 patients, 3 developed grade I, 2 developed grade II, and 1 developed grade III aGVHD, which was controlled by systemic steroids and/or anti-CD25 antibodies. Interestingly, among the 8 patients who achieved hAESCs engraftment, only 1 developed grade II aGVHD. However, the 2 patients who failed to achieve hAESCs engraftment suffered from grade III and II aGVHD, respectively. No patient relapsed and died from NRM within day +100 post-HSCT. There were no toxicities or AEs attributable to hAESCs infusion. Concerning the viral reactivation, 5 patients (4/8 with hAESCs engraftment, 1/2 without hAESCs engraftment) experienced cytomegalovirus (CMV) reactivation, while only 1 patient (0/8 with hAESCs engraftment, 1/2 without hAESCs engraftment) had asymptomatic EBV reactivation. The outcomes of aGVHD and viral reactivation are summarized in Table 2.
Considering the significant morbidity and mortality associated with CMV reactivation, the observation of CMV reactivation occurring in 4 out of the patients with hAESCs engraftment compared to 1 out of 2 without hAESCs engraftment is noteworthy. In the context of viral immunology, T cell-mediated immunity is essential for controlling CMV reactivation in patients undergoing allo-HSCT. Specially, CMV-specific CD8+ T cells play a crucial role in clearing systemic CMV infection and establishing lifelong immune protection against reactivation in animal CMV infection models [54]. As depicted in Supplementary Fig. S8, we demonstrated that hAESCs did not impact the proportion of CD8+ T cells and did not impair the cytotoxic effect of CD8+ T cells, which may explain the observed equal percentages of CMV reactivation with or without hAESCs engraftment. Therefore, it is likely that hAESCs did not exacerbate viral reactivation, further evidenced by the absence of EBV reactivation.
Reconstitution of immune cell subsets in HRD-HSCT patients with hAESCs engraftment
Immunologic T-cell reconstitution was evaluated at days +14, +30, and +90 in patients with hAESCs engraftment. The numbers of NK cells (Fig. 7a), CD3+ T cells (Fig. 7b), CD8+ T cells (Fig. 7d), NKT cells (Fig. 7e), and monocytes (Fig. 7f) recovered to donors’ levels by day 90 or earlier post transplantation and the numbers of CD4+ T cells (Fig. 7c) also recovered to some extent by day 90. Compared with the donor group, decrease of Th1 proportion of CD4+ T cells was detected at day 30 post transplantation (Fig. 7g) and Treg subset of CD4+ T cells increased at day 14 post transplantation (Fig. 7i), while no significant change was observed in the Th17 subset (Fig. 7h). The higher expression of Ki67 (Fig. 7j) but lower expression of BCL-2 (Fig. 7k) in Treg cells of the patients than donors’ Treg cells indicated that there was rapid proliferation of Treg cells but with reduced anti-apoptosis ability after hAESCs infusion. Additionally, the proportion of HLA-DR+ CD45RA- Treg subset was enhanced (Fig. 7l) and the expression of HELIO in Treg cells was comparable to those of donors’ (Fig. 7m), suggesting the expanded Treg subset was mainly derived from the peripheral blood. Together, the effect of hAESCs on T cell subsets polarization in patient is mostly consistent with the observation in mouse GVHD model.

On day 14, 30, and 90 after HSCT, the peripheral blood samples were collected from patients with hAESCs engraftment, and the counts of NK cells (a), CD3+ T cells (b), CD4+ T cells (c), CD8+ T cells (d), NKT cells (e) and monocytes (f) or percentages of Th1 (g), Th17 (h) and Treg (i–m) subsets in CD4+ cells were analyzed by flow cytometry. Data are presented as the mean ± SEM, ns no significance; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Immunocytokine production in HRD-HSCT patients with or without hAESCs engraftment
To explore the potential role of hAESCs in immunocytokine production in post-HSCT aGVHD, patient plasma samples were collected at day +7 and the concentrations of a series of cytokines were measured. Compared to the patients without hAESCs engraftment, the level of proinflammatory cytokines including IFNγ, CD40L, IL3, IL 33, and IL17 exhibited a decreased trend in patients with hAESCs engraftment and a lower trend in hAESCs-engrafted patients without aGVHD (Fig. 8a–e). On the other hand, a slightly increased trend of anti-inflammatory cytokines including TGFβ, IL4, IL13, and IL1ra in patients with hAESCs engraftment were observed, compared to those in the patients without hAESCs engraftment (Fig. 8f–i).

On day 7 after HSCT, the blood plasma samples were collected from patients with hAESCs infusion. The concentration of pro-inflammatory cytokine IFNγ (a), CD40L (b), IL3 (c), IL33 (d) and IL17 (e) or anti-inflammatory cytokine TGFβ (f), IL4 (g), IL13 (h) and IL1ra (i) was measured by ELISA and shown in the hAESCs non-engrafted group, the hAESCs engrafted without aGVHD group and hAESCs engrafted with aGVHD group respectively. Data are presented as the mean ± SEM, ns no significance; *P < 0.05; **P < 0.01.
Discussion
The amniotic membrane has been used as surgical dressing to promote injury repairing for more than 100 years. Afterwards, the low immunogenicity of hAESCs was uncovered and recently further confirmed in our non-serum isolation/culture system, indicating potential immune tolerance after transplantation. Coupled with the high engraftment ratio of hAESCs in patients (8 out of 10), it also indicates that hAESCs transplantation could be accessible in most patients without the concerns of HLA matching and immune disorder. On the other hand, a preclinical safety evaluation in our previous study, following the form of drug safety evaluations, exhibited that hAESCs administration led to neither hemolytic, allergy, toxicity issues, nor tumorigenicity (tumor generation and tumor promotion) [35]. More significantly, multiple high-dose of cGMP-grade hAESCs was successfully transplanted in all the patients without any infusion reactions and did not cause direct toxicity and other AEs within long-term observation in the present study. In addition, hAESCs are readily isolated in large quantities from placentae, which are in the category of newborn postpartum waste. Therefore, the safety and ethical concerns of our cGMP-grade hAESCs could be negligible for GVHD therapy in clinic.
Aiming clinical application, the therapeutic effect of the human-derived hAESCs therapy product was evaluated in a humanized GVHD mouse model and post-HSCT patients. GVHD arises from the incompatibility between donor immune cells and recipient antigens. The activated immune response leads to the secretion of pro-inflammatory cytokines, the so-called “cytokine storm”, and donor T cells expansion, followed by organ damage by infiltrated T cells. In the present study, hAESCs infusion did not affect the PBMC engraftment rate in aGVHD mice but ameliorated the disease progression. More importantly, the hAESCs delivery post-HSCT did not disrupt the T cell and other immune cell reconstitution and demonstrated potential improvement of GVHD in our clinical trial. Thus, these results suggest that hAESCs mitigate GVHD but not at the expense of impairing immune reconstitution. Large-scale clinical trials will be valuable to further determine the details of the therapeutic effect of hAESCs on GVHD and GVL in correlated patients.
Our results from both preclinical and clinical studies demonstrate that hAESCs infusion directly mediated the polarization of donor CD4+ T cells during aGVHD. Intriguingly, this modulation of hAESCs resulted in a decrease of Th1 and Th17 cells, the major T cell subsets responsible for amplification of the “cytokine storm”; correspondingly, the inhibition of pro-inflammatory cytokines was observed. By contrast, hAESCs enhanced Treg cells, the key modulators of immune tolerance in suppressing aGVHD. This correlated to increased levels of TGFβ and IL10, which, in turn, further facilitated the expansion of Treg cells. Moreover, hAESCs also restrained the aggravated injury in endothelium, the interface between donor T cells and GVHD target organs, as well as consequent pathological damages. Thus, hAESCs infusion has a protective effect on both the first and second stages of GVHD pathogenesis. Of note, we observed that the therapeutic effect of the prevention group was better than the treatment group in mouse GVHD model. Accordingly, we employed the hAESCs administration strategy in advance and at the early stage of HSCT in the clinical trial and observed the potential therapeutic effect of hAESCs on post-HSCT GVHD. Therefore, hAESCs in the prevention strategy may establish an immune rectifying microenvironment that reconstructs immune cell compartments to better minimize the GVHD disorder. To our knowledge, this is the first demonstration of the hAESCs therapy against GVHD.
Regarding the cellular mechanism against GVHD, hAESCs work via non-contact secretion of TGFβ and PGE2, along with other anti-inflammatory cytokines. This mechanism is distinct from the IFNγ-induced IDO up-regulation or TLR induction observed with MSCs, as reported in existing literature [50]. In terms of signaling pathway mechanism, MSCs inhibit the NF-κB and MAPK signaling pathways to suppress Th1 and Th17 activation and Treg induction [50, 55]. On contrast, in our study, transcriptome analysis revealed different regulation under the influence of hAESCs. CD4+ T cells favored shifting polarization from Th1 and Th17 activation to Treg induction likely through the downregulation of the PI3K-Akt, TNF and MAPK signaling pathway as well as Hippo, Wnt, and Calcium signaling pathway. Localization of hAESCs in mice revealed that they could inhabit the major GVHD target organs regardless of the presence of immune cells, while MSCs require pro-inflammatory “licensing” for therapeutic effect [56]. Therefore, in contrast to MSCs, hAESCs maintain a robust survival capability and steady immune modulatory effect in the GVHD environment.
In summary, infusion of cGMP-grade hAESCs can efficiently improve GVHD by repolarizing CD4+ T cells while preserving the GVL effect by inhibiting leukemia cell growth. Importantly, the transplanted hAESCs product does not induce safety issues and affect immune system reconstitution in recipients. hAESCs work to normalize, rather than repress, the immune system in GVHD patients. This notion is supported by the preserved GVL effect and absence of exacerbated viral reactivation in HSCT patients. Therefore, hAESCs infusion could be a promising therapeutic strategy for post-HSCT GVHD and may apply to treatment for other GVHD diseases.
References
Paczesny S. Discovery and validation of graft-versus-host disease biomarkers. Blood. 2013;121:585–94.
Reddy P, Ferrara JLM. Immunobiology of acute graft-versus-host disease. Blood Rev. 2003;17:187–94.
Ferrara JLM, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373:1550–61.
Kanakry CG, Fuchs EJ, Luznik L. Modern approaches to HLA-haploidentical blood or marrow transplantation. Nat Rev Clin Oncol. 2016;13:10–24.
Huang X, Liu D. Related HLA-mismatched/haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion: observations of a single Chinese center. Clin Transpl 2011:237–45.
Wang Y, Liu Q, Xu L, Liu K, Zhang X, Ma X, et al. Haploidentical vs identical-sibling transplant for AML in remission: a multicenter, prospective study. Blood. 2015;125:3956–62.
Ferrara JLM, Reddy P. Pathophysiology of graft-versus-host disease. Semin Hematol. 2006;43:3–10.
Hill GR, Ferrara JL. The primacy of the gastrointestinal tract as a target organ of acute graft-versus-host disease: rationale for the use of cytokine shields in allogeneic bone marrow transplantation. Blood. 2000;95:2754–9.
Ratanatharathorn V, Nash RA, Przepiorka D, Devine SM, Klein JL, Weisdorf D, et al. Phase III study comparing methotrexate and tacrolimus (prograf, FK506) with methotrexate and cyclosporine for graft-versus-host disease prophylaxis after HLA-identical sibling bone marrow transplantation. Blood. 1998;92:2303–14.
Nash RA, Antin JH, Karanes C, Fay JW, Avalos BR, Yeager AM, et al. Phase 3 study comparing methotrexate and tacrolimus with methotrexate and cyclosporine for prophylaxis of acute graft-versus-host disease after marrow transplantation from unrelated donors. Blood. 2000;96:2062–8.
Macmillan ML, Weisdorf DJ, Wagner JE, Defor TE, Burns LJ, Ramsay NKC, et al. Response of 443 patients to steroids as primary therapy for acute graft-versus-host disease: comparison of grading systems. Biol Blood Marrow Transpl. 2002;8:387–94.
Martin PJ, Schoch G, Fisher L, Byers V, Anasetti C, Appelbaum FR, et al. A retrospective analysis of therapy for acute graft-versus-host disease: initial treatment. Blood. 1990;76:1464–72.
Deeg HJ. How I treat refractory acute GVHD. Blood. 2007;109:4119–26.
Carpenter PA, Sanders JE. Steroid-refractory graft-vs.-host disease: past, present and future. Pediatr Transpl. 2003;7:19–31.
Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010;116:5738–47.
Sockel K, Bornhaeuser M, Mischak-Weissinger E, Trenschel R, Wermke M, Unzicker C, et al. Lenalidomide maintenance after allogeneic HSCT seems to trigger acute graft-versus-host disease in patients with high-risk myelodysplastic syndromes or acute myeloid leukemia and del(5q): results of the LENAMAINT trial. Haematologica. 2012;97:e34–5.
Reshef R, Luger SM, Hexner EO, Loren AW, Frey NV, Nasta SD, et al. Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. N Engl J Med. 2012;367:135–45.
Abouelnasr A, Roy J, Cohen S, Kiss T, Lachance S. Defining the role of sirolimus in the management of graft-versus-host disease: from prophylaxis to treatment. Biol Blood Marrow Transpl. 2013;19:12–21.
Brunstein CG, Miller JS, Cao Q, Mckenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117:1061–70.
Fantini MC, Monteleone G. Update on the therapeutic efficacy of tregs in IBD: thumbs up or thumbs down? Inflamm Bowel Dis. 2017;23:1682–8.
Velaga S, Ukena SN, Höpting M, Ivanyi P, Borchers S, Mischak-Weissinger E, et al. Reconstitution and phenotype of tregs in CMV reactivating patients following allogeneic hematopoietic stem cell transplantation. Immunol Invest. 2013;42:18–35.
Whangbo JS, Antin JH, Koreth J. The role of regulatory T cells in graft-versus-host disease management. Expert Rev Hematol. 2020;13:141–54.
Le Blanc K, Rasmusson I, Sundberg B, Götherström C, Hassan M, Uzunel M, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–41.
Galipeau J. The mesenchymal stromal cells dilemma-does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy. 2013;15:2–8.
Ning H, Yang F, Jiang M, Hu L, Feng K, Zhang J, et al. The correlation between cotransplantation of mesenchymal stem cells and higher recurrence rate in hematologic malignancy patients: outcome of a pilot clinical study. Leukemia. 2008;22:593–9.
Uhlin M, Sairafi D, Berglund S, Thunberg S, Gertow J, Ringden O, et al. Mesenchymal stem cells inhibit thymic reconstitution after allogeneic cord blood transplantation. Stem Cells Dev. 2012;21:1409–17.
Wassmer C, Berishvili E. Immunomodulatory properties of amniotic membrane derivatives and their potential in regenerative medicine. Curr Diab Rep. 2020;20:31.
Magatti M, Caruso M, De Munari S, Vertua E, De D, Manuelpillai U, et al. Human amniotic membrane-derived mesenchymal and epithelial cells exert different effects on monocyte-derived dendritic cell differentiation and function. Cell Transpl. 2015;24:1733–52.
Cargnoni A, Farigu S, Cotti Piccinelli E, Bonassi Signoroni P, Romele P, Vanosi G, et al. Effect of human amniotic epithelial cells on pro-fibrogenic resident hepatic cells in a rat model of liver fibrosis. J Cell Mol Med. 2018;22:1202–13.
Tan JL, Lau SN, Leaw B, Nguyen HPT, Salamonsen LA, Saad MI, et al. Amnion epithelial cell-derived exosomes restrict lung injury and enhance endogenous lung repair. Stem Cells Transl Med. 2018;7:180–96.
Vosdoganes P, Wallace EM, Chan ST, Acharya R, Moss TJM, Lim R. Human amnion epithelial cells repair established lung injury. Cell Transpl. 2013;22:1337–49.
Tan B, Yuan W, Li J, Yang P, Ge Z, Liu J, et al. Therapeutic effect of human amniotic epithelial cells in murine models of Hashimoto’s thyroiditis and systemic lupus erythematosus. Cytotherapy. 2018;20:1247–58.
Li J, Qiu C, Zhang Z, Yuan W, Ge Z, Tan B, et al. Subretinal transplantation of human amniotic epithelial cells in the treatment of autoimmune uveitis in rats. Cell Transpl. 2018;27:1504–14.
Bai X, Liu J, Yuan W, Liu Y, Li W, Cao S, et al. Therapeutic effect of human amniotic epithelial cells in rat models of intrauterine adhesions. Cell Transpl. 2020;29:2138901199.
Yang P, Yuan W, Liu J, Li J, Tan B, Qiu C, et al. Biological characterization of human amniotic epithelial cells in a serum-free system and their safety evaluation. Acta Pharmacol Sin. 2018;39:1305–16.
Kubo M, Sonoda Y, Muramatsu R, Usui M. Immunogenicity of human amniotic membrane in experimental xenotransplantation. Invest Ophthalmol Vis Sci. 2001;42:1539–46.
Hori J, Wang M, Kamiya K, Takahashi H, Sakuragawa N. Immunological characteristics of amniotic epithelium. Cornea. 2006;25:S53–8.
Akle CA, Adinolfi M, Welsh KI, Leibowitz S, Mccoll I. Immunogenicity of human amniotic epithelial cells after transplantation into volunteers. Lancet. 1981;2:1003–5.
Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte JJ, Crawford JM, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–9.
Polchert D, Sobinsky J, Douglas G, Kidd M, Moadsiri A, Reina E, et al. IFN-gamma activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur J Immunol. 2008;38:1745–55.
Qin X, Li G, Qin Y, Wang Y, Wang F, Liu D, et al. Quantitative chimerism: an independent acute leukemia prognosis indicator following allogeneic hematopoietic SCT. Bone Marrow Transpl. 2014;49:1269–77.
Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974;18:295–304.
Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, et al. National institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transpl. 2015;21:389–401.
Blazar BR, Macdonald KPA, Hill GR. Immune regulatory cell infusion for graft-versus-host disease prevention and therapy. Blood. 2018;131:2651–60.
Grégoire C, Ritacco C, Hannon M, Seidel L, Delens L, Belle L, et al. Comparison of mesenchymal stromal cells from different origins for the treatment of graft-vs.-host-disease in a humanized mouse model. Front Immunol. 2019;10:619.
Duffy MM, Ritter T, Ceredig R, Griffin MD. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Res Ther. 2011;2:34.
Mao M, Qian Y, Sun J. Morphine suppresses T helper lymphocyte differentiation to Th1 type through PI3K/AKT pathway. Inflammation. 2016;39:813–21.
Li Y, Fan H, Han X, Sun J, Ni M, Zhang L, et al. PR-957 suppresses Th1 and Th17 cell differentiation via inactivating PI3k/AKT pathway in Alzheimer’s disease. Neuroscience. 2023;510:82–94.
Pesce B, Ribeiro CH, Larrondo M, Ramos V, Soto L, Catalan D, et al. TNF-alpha affects signature cytokines of Th1 and Th17 T cell subsets through differential actions on TNFR1 and TNFR2. Int J Mol Sci. 2022;23:9306.
Zhu H, Yang F, Tang B, Li XM, Chu YN, Liu YL, et al. Mesenchymal stem cells attenuated PLGA-induced inflammatory responses by inhibiting host DC maturation and function. Biomaterials. 2015;53:688–98.
Onuora S. Immunology: Hippo signalling influences T cell fate. Nat Rev Rheumatol. 2017;13:389.
Hu Y, Chen G, Huang J, Li Z, Li Z, Xie Y, et al. The calcium channel inhibitor nimodipine shapes the uveitogenicT cells and protects mice from experimental autoimmune uveitis through the p38-MAPK signaling pathway. J Immunol. 2021;207:2933–43.
Podojil JR, Miller SD. Cross-linking of CD80 on CD4+ T cells activates a calcium-dependent signaling pathway. J Immunol. 2009;182:766–73.
Liu J, Chang YJ, Yan CH, Xu LP, Jiang ZF, Zhang XH, et al. Poor CMV-specific CD8+ T central memory subset recovery at early stage post-HSCT associates with refractory and recurrent CMV reactivation. J Infect. 2016;73:261–70.
Song JY, Kang HJ, Ju HM, Park A, Park H, Hong JS, et al. Umbilical cord-derived mesenchymal stem cell extracts ameliorate atopic dermatitis in mice by reducing the T cell responses. Sci Rep. 2019;9:6623.
Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–50.
Acknowledgements
We thank Dr. She-long Zhang and Dr. Fang-liang Huang (Equipment and Technology Service Platform, College of Life Sciences, Zhejiang University) for their excellent technical support with microscopy and flow cytometry. This work was supported by the Zhejiang Provincial Key R&D Program of China (2022C03097), the National Key R&D Program of China (2017YFA0104500, 2018YFA0800504), the National Natural Science Foundation of China (82300296, 81621001, 91839104, 81770444 and 81600354), Project of Health Collaborative Innovation of Guangzhou City (201704020214), the Zhejiang Provincial Natural Science Foundation of China (LZ20H020002), the Medical and Health Science and Technology Program of Health Commission of Zhejiang Province, China (2021KY633) and Fundamental Research Funds for the Central Universities of China.
Author information
Authors and Affiliations
Contributions
PJY, XYZ, LHG, JYL, XJH, and LYY designed research; PJY, XYZ, YHK, JL, YYZ, ZDW, WXY, BT, QL, XYR, and YNS performed research; PJY, XYZ, YHK, ZG, JYL, Chen Qiu, YQJ, Cong Qiu, XYR, and YNS analyzed and interpreted data; PJY, XYZ, YHK, JYL, XJH, and LYY drafted and revised manuscript; JYL, XJH and LYY approved the manuscript submission.
Corresponding authors
Ethics declarations
Competing interests
LYY, PJY, WXY, JYL, LHG, JL authored a China patent (No. ZL 2019 1 0310688.6) held by Zhejiang University and Shanghai iCELL Biotechnology Co. Ltd for the “Use of hAESCs for the treatment of GVHD”. WXY and QL are employees of Shanghai iCELL Biotechnology Co. Ltd. The remaining authors declare no competing financial interests.
Ethics approval and consent to participate
The hAESCs were derived from placenta amniotic membrane of unrelated, unmatched healthy donors with informed patient consent approved by the Institutional Patients and Ethics Committee of the Second Affiliated Hospital of Zhejiang University School of Medicine (Ethics Code: 2020-799). All animal studies were approved by the Institutional Animal Care and Use Committee of Zhejiang University (ZJU20210127) and adhered to the Guide for the Care and Use of Laboratory Animals. The clinical trial was approved by the institutional review board of Peking University People’s Hospital (2018PHD006-01) and conducted under an investigational new stem cells application from the National Health Commission of China. All patients and donors provided written informed consent according to the Declaration of Helsinki. The trial was registered at www.clinicaltrials.gov as #NCT03764228.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, Pj., Zhao, Xy., Kou, Yh. et al. Human amniotic epithelial stem cell is a cell therapy candidate for preventing acute graft-versus-host disease. Acta Pharmacol Sin (2024). https://doi.org/10.1038/s41401-024-01283-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41401-024-01283-y
- Springer Nature Singapore Pte Ltd.