
Abstract
Polypharmacy poses a significant risk for adverse reactions. While there are clinical decision support tools to assist clinicians in medication management, pharmacogenetic testing to identify potential drug–gene or drug–drug–gene interactions is not widely implemented in the clinical setting. A PRISMA-compliant scoping review was performed to determine if pharmacogenetic testing for absorption, distribution, metabolism, and excretion (ADME)-related genetic variants is associated with improved clinical outcomes in patients with polypharmacy. Six studies were reviewed. Five reported improved clinical outcomes, reduced side effects, reduction in the number of drugs used, or reduced healthcare utilization. The reviewed studies varied in methodological quality, risk of bias, and outcome measures. Age, diet, disease state, and treatment adherence also influence drug response, and may confound the relationship between genetic polymorphisms and treatment outcomes. Further studies using a randomized control design are needed. We conclude that pharmacogenetic testing represents an opportunity to improve health outcomes in patients exposed to polypharmacy, particularly in patients with psychiatric disorders and the elderly.
Similar content being viewed by others


Introduction
Healthcare professionals have recognized polypharmacy, the concomitant use of multiple medications, as a topic of increasing concern in recent years [1, 2]. The World Health Organization anticipates increased prevalence of polypharmacy secondary to the aging population and chronic diseases requiring pharmaceutical interventions [1]. A recent analysis of polypharmacy use estimated that ~22% of adults in the US aged 40–79 consume five or more drugs concurrently [3]. The use of multiple medications is associated with an increased risk for adverse drug events (ADEs) as well as increased healthcare costs. Specifically, patients are more likely to suffer from drug–drug interactions, falls, cognitive decline, and poor nutrition, and have more emergency room visits and hospital admissions [1, 2, 4]. Issues such as decreased medication adherence, and the use of over-the-counter medications and supplements, also hinder medication management efforts. While the use of multiple medications is often necessary and effective, there is a need to identify strategies that mitigate the negative health outcomes associated with polypharmacy.
Genetic variants of drug metabolizing enzymes (DMEs) and drug transporters can have significant consequences for the pharmacokinetics of pharmaceuticals. DMEs are categorized as Phase I and Phase II enzymes, according to their roles in drug biotransformation or elimination. Phase I enzymes modify drugs into water-soluble products through the addition of reactive or polar groups, in preparation for excretion [5, 6]. Cytochrome p450 (CYP) enzymes, which represent the most significant and well-known group of Phase I enzymes, play an important role in drug metabolism. Fifty-seven CYP genes, categorized into 18 families have been identified [7]. These enzymes represent a superfamily of hemeproteins found in all tissues, with the most abundant expression in the liver and small intestine [5,6,7]. Approximately 70–80% of drugs are metabolized by CYP enzymes in the CYP1, CYP2, and CYP3 families [8]. Polymorphic enzymes CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5 metabolize roughly 60–70% of prescribed drugs, some of which are prodrugs that are converted to pharmacologically active metabolites [8]. Other CYP genes are important for the synthesis of steroid hormones, cholesterol biosynthesis, and vitamin metabolism [7]. The genetic variants in some of the CYP genes, such as CYP2C19 and CYP2D6, can be used to determine patient metabolizer phenotypes: poor metabolizers (PM), intermediate metabolizers, extensive (normal) metabolizers (EM), or ultrarapid metabolizers (UM) [5, 9]. For instance, CYP2D6 PMs have decreased or no enzyme activity compared to EMs. Codeine, a pro-drug that is metabolized by CYP2D6 to form morphine, is likely to be ineffective in CYP2D6 PMs, since there is less active metabolite [10]. Codeine use by patients classified as CYP2D6 UMs can lead to significant ADEs [10]. Due to increased CYP2D6 enzyme activity in UMs, these patients are at high risk for morphine toxicity. Thus, having a PM or UM phenotype can potentially lead to adverse outcomes.
Phase II metabolism modifies a drug or drug metabolites from Phase I further for elimination [5, 6]. Phase II enzymes, which are transferases, are also of clinical significance. Genetic variants of enzymes such as N-acetyltransferase, uridine 5′-diphspho-glucuronosyltransferases, and thiopurine S-methyltransferases have been associated with ADEs due to alterations in enzyme activity [5].
Drug transport proteins, such as solute carrier (SLC) transporters and ATP-binding cassette transporters act as uptake or efflux transporters to transfer molecules into or out of cells [6]. Polymorphisms in drug transporter genes, such as ATP-binding cassette subfamily B member 1 and solute carrier organic anion transporter family member 1B1, can also affect drug efficacy [5]. Many diagnostic companies use a common panel of pharmacogenetic genes for Food & Drug Administration (FDA) approved drugs with actionable pharmacogenetic drug label annotations (Table 1).
The ability to predict drug response through pharmacogenetic testing has been extremely useful for individualized drug selection and dosing. The drug label for warfarin, an anticoagulant, was the first to be updated by the US FDA to include pharmacogenomics labeling [5, 11]. There are now over 160 drug–gene pairs recognized by the FDA [12]. The FDA, the Clinical Pharmacogenetics Implementation Consortium (CPIC), and the European Medicines Agency, all monitor pharmacogenetics research and offer recommendations for clinicians to improve health outcomes and reduce ADEs [7].
An additional concern is the nongenetic factors known to affect drug metabolism. The concomitant use of DME inhibitors or inducers can impact a patient’s ability to metabolize a particular drug, and will influence drug efficacy. Amiodarone, an antiarrhythmic, is an inhibitor of CYP1A2, CYP2C9, CYP2D6, and CYP3A activities [13]. Co-administration with medications, which are also substrates for these enzymes, may lead to reduced enzyme activity and ADEs. The potential for significant drug interactions with amiodarone have been documented for statins, β-receptor blocking agents, and anticoagulants [13]. Thus, genotyping for absorption, distribution, metabolism, and excretion (ADME)-related genetic variants will not necessarily predict metabolizer phenotype, which has negative implications for the clinical utility of pharmacogenetic testing. The change from the genotype-predicted metabolizer phenotype to a lower or higher metabolizer phenotype is referred to as phenoconversion [14, 15]. The issues of enzyme activity and drug–drug interactions impact the phenoconversion of an enzyme, which can be a key factor affecting the utility of a pharmacogenetic test.
Because patients are increasingly prescribed multiple medications, an evaluation of whether genetic testing could assist in medication management is warranted. Therefore, a scoping review was conducted to assess the impact of pharmacogenetic testing on health outcomes in patients with polypharmacy.
Materials and methods
Protocol
This scoping review was conducted according to the guidelines of the PRISMA Extension for Scoping Reviews [16]. Supplementary Table 1 shows the completed PRISMA-ScR checklist.
Eligibility criteria
Studies that assessed the impact of incorporating pharmacogenetic testing into a clinical decision support tool (CDST) to guide treatment were the targets of the literature search. Documentation of polypharmacy was required. There is no consensus on the number of concurrent medications that qualify as polypharmacy [1, 4]. Due to a limited number of articles that specifically focus on the intersection of polypharmacy and genetic testing, the lower limit of concomitant medications was set at two. Articles that document an average of two or more concomitant medications among study participants also satisfied the criteria for polypharmacy. There was no limitation with regard to patient condition, diagnosis, or variant/phenotype status. Outcomes could be any measure of health, healthcare usage, or ADEs.
Search strategy
The following databases were searched for journal articles published in the English language from 2010 to February 2020: PubMed, Medline, Web of Science, Cochrane CENTRAL. Supplementary Reference 1 includes the strategy and terms used to perform database searches. The references of included articles were also reviewed for relevant studies.
Study selection
The search results were combined and duplicates were removed. Journal article titles and abstracts were reviewed by two authors (ELM and SMS) for exclusion. The full texts for the remaining articles were reviewed for eligibility (ELM). Studies could be prospective, retrospective, or randomized control trials (RCTs). Single case reports and studies that evaluated only one drug therapy were excluded. To address the research question, included studies assessed the impact of pharmacogenetic testing on health outcomes in patients with polypharmacy. Genetic screening should specify the variants tested or the phenotype status of the study participants. Outcomes could be secondary to the study focus.
Data extraction
Data extraction was conducted independently by one author (ELM) using a data extraction spreadsheet and reviewed with the authors (SMS, TKF, CLF). Data collected include: study design, funding, disease/condition, genes and variants included in pharmacogenetic testing panel, number or average number of medications used by the study population, patient population size, patient demographics, and study outcomes. Risk of bias was assessed using guidance from Viswanathan et al. [17].
Synthesis of results
Results were summarized using a narrative format. Tables were prepared for extracted data. The criteria for the assessment of pharmacogenetic studies published by Jorgensen and Williamson and others [18,19,20] were used as a guide to assess study quality.
Results
Study selection
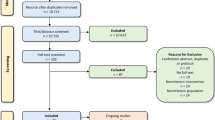
A total of 1097 journal articles were identified during the literature search. Once duplicates were removed, 515 journal titles and abstracts were reviewed to eliminate those that were irrelevant to the research question. Full-text articles were retrieved for the remaining 32 articles. Most were excluded because they did not have the appropriate study design. Additional reasons for article exclusion are listed in the PRISMA [21] flowchart in Fig. 1. Six studies were analyzed [22,23,24,25,26,27].

Flowchart for selection of included studies.
Study characteristics
Study characteristics and results for the articles included in this review are listed in Table 2. Five studies had a prospective study design, and one was a retrospective study. Three studies included patients with diagnoses from multiple therapeutic areas and three focused on psychiatric pharmacotherapy.
Two articles stated research goals that specifically aligned with the purpose of this review [23, 24]. Brixner et al. [23] genotyped CYP variants in study participants who were prescribed three or more medications, one of which was classified as causing ADEs if high risk variants in CYP genes were identified using the YouScript® (Genelex, Washington, USA) CDST. Hospitalizations, emergency department visits, and outpatient visits were documented 4 months after enrollment. The data were compared to an untested control group identified through an administrative claims database (Medical Outcomes Research for Effectiveness and Economics Registry). Elliot et al. [24] were also interested in the influence of pharmacogenetic testing on healthcare resource utilization. They conducted a RCT in which the number of hospitalizations and emergency department visits 30 and 60 days after enrollment was compared between the tested and untested study participants with confirmed polypharmacy. Exploratory outcomes also included number of deaths at 30 and 60 days. This study used the same CDST as Brixner et al. [23].
The remaining four articles did not specifically seek to measure health outcomes related to polypharmacy, but included relevant outcomes as secondary measures [22, 25,26,27]. Blasco-Fontecilla [22] assessed the impact of pharmacogenetic testing in patients diagnosed with psychiatric disorders. Outcomes included measurement of clinical improvement, reduction in number of medications used after pharmacogenetic testing, and reduction in adverse events. Both Winner et al. [26] and Hall-Flavin et al. [27] focused on study participants diagnosed with major depressive disorder. They reported clinical improvement after pharmacogenetic testing using clinical rating instruments that healthcare providers use to assess depression severity (e.g., Hamilton Rating Scale for Depression). van der Wouden et al. [25] assessed the impact of pharmacogenetic testing in a primary care setting. The primary outcomes of the study were unrelated to the purpose of this review. Relevant secondary outcomes included general practitioner consults, emergency room visits, and hospitalizations related to ADEs within 12 weeks of enrollment.
Regarding pharmacogenetic testing, genotyping for CYP2D6 and CYP2C19 variants was universal. Four out of six studies included CYP2C9. Variants and SNPs analyzed differed considerably overall. Genes unrelated to drug ADME were also tested. For example, three studies included testing for HTR2A (5-hydroxytryptamine (serotonin) receptor 2A), a gene of interest in psychiatry and neurology [22, 26, 27]. The most extensive panel was used by Blasco-Fontecilla [22] and has been described in detail by Perez et al. [28]. Brixner et al. [23] categorized study participants as having wild-type status if variant genotyping results were negative. Table 3 lists the genes, variants, and SNPs included in the pharmacogenetic testing panels as described in each study. Only ADME-related genes are listed. Inferred metabolizer phenotypes based on genotyping results were not reported or were described in referenced articles for four out of six studies. Hall-Flavin et al. [27] reported a significant difference in the frequencies of CYP2D6 metabolizer phenotypes between tested and untested patients, but not for CYP2C19 or CYP1A2. Elliott et al. [24] described the CYP metabolizer phenotypes of the study population in order to compare the distribution to another published study.
Polypharmacy among study participants was not defined in the same manner and varied across studies. The number of medications for each patient was reported or the average number of medications per patient was reported. In studies reporting averages, it is possible that a subset of the study participants could be prescribed only one medication. The mean number of medications per patient ranged from 2.2 to 11.6 for all studies analyzed.
Quality assessment and outcomes
There were significant differences between studies regarding the study population. The number of patients ranged from 20 to 1025. Blasco-Fontecilla [22] focused on a young patient population with a mean age of 14.6. The mean age for the remaining five studies ranged from ~41 to 77. None of the studies had an ethnically diverse patient population. Patients in five out of six studies were predominantly Caucasian. In general, there were minor differences in gender composition. Only Winner et al. [26] included a much higher proportion of female participants compared to the other studies. All of the studies described the inclusion and exclusion criteria for study participants and the methods used for variant and copy number determination. None of the studies discussed the degree to which the DMEs studied contribute to the metabolism of the drugs prescribed, screening for Hardy–Weinberg equilibrium, or confirmation of patient adherence to prescribed drug regimens. Due to the factors described above, these studies have at least a moderate risk for bias.
Overall, the studies reported the impact of pharmacogenetic testing in terms of improved clinical outcomes [22, 27], reduced side effects [22, 26], reduction in the number of drugs used [22], or reduced healthcare utilization (e.g., reduced hospitalizations and emergency room visits) [23,24,25]. Five out of six studies reported favorable results, meaning that use of a pharmacogenomics panel improved patient outcomes [22,23,24, 26, 27].
Discussion
In recent years, mounting evidence has supported the notion that pharmacogenetic testing can have a positive impact on health outcomes and advance the development of precision medicine [29, 30]. Several institutions in the USA, such as the Mayo Clinic and St. Jude Children’s Research Hospital, have already established programs to incorporate pharmacogenetic testing into clinical practice [20, 31, 32]. In this review, we presented a synthesis of studies, which focus on the clinical implementation of pharmacogenetic testing in order to describe the real-world impact on patient care, as well as similarities and differences that could contribute to the growing effort to make preemptive pharmacogenetic testing mainstream. We chose to focus on patients exposed to polypharmacy, as there is a growing awareness of the impact on patient care and the growing number of patients with comorbidities [1]. Pharmacogenetic testing in patients with polypharmacy appears to have received little coverage in the literature, however, there are multiple studies reporting an association between ADEs and genotypes related to pharmacokinetics [33,34,35,36]. Recently, Licito et al. [33] showed that a genetic variant of SLCO1B1 is associated with the neuromuscular pain in type 2 diabetes mellitus patients with cardiovascular comorbidities. Mugoša et al. [34] looked at the prevalence of CYP2D6 variants associated with the PM phenotype in a hospitalized cardiac patient population taking β-blockers. They found that ADEs caused by β-blockers could be predicted by having a PM phenotype, in addition to concomitant use of other CYP2D6 substrate medications, and length of hospital stay. Five out of six studies included in this review support the use of pharmacogenetic testing as a tool to assist clinicians in medication management. van der Wouden et al. [25] did not report a benefit for tested participants whose healthcare providers followed Dutch Pharmacogenetics Working Group (DPWG) guidelines compared to those with potential DGIs but did not receive pharmacogenetic guided therapy.
The analysis of the literature also revealed the patient populations with polypharmacy, which are likely to benefit from pharmacogenetic testing. Psychiatric patients, as well as elderly oncology and cardiology patients, are often prescribed multiple medications. In addition to improved health outcomes, patients who are provided with proper medication management and patient-centered care will also benefit from overall reductions in healthcare costs. Recent studies have reported estimated healthcare cost savings resulting from the incorporation of pharmacogenetic testing [23, 37,38,39,40]. Maciel et al. [37] estimated annual cost savings of $3962 per patient associated with pharmacogenetic testing in patients diagnosed with major depressive disorder..Saldivar et al. [38] found that 50% of patients exposed to polypharmacy in a long-term care facility could reduce or eliminate one to three medications if testing results were considered in medication management. The estimated annual savings were $621 per patient.
Genetic variants chosen for pharmacogenetic testing panels are critical to successful implementation in drug prescribing and the avoidance of ADEs. The articles reviewed show that clinicians may opt to use panels focused on a particular therapeutic area. In addition, pharmacogenetic testing panels can have several genetic markers in common, however, there may be differences in the variants included. In their study of severe mental disorders among adolescents, Blasco-Fontecella [22] used Neuropharmagen® (AB-Biotics, Barcelona, Spain), a commercial test developed to optimize drug prescribing for psychiatric conditions [28]. Testing for several genes, including HTR2A, brain-derived neurotrophic factor, and opioid receptor mu 1, is included on this panel, in addition to CYP enzymes such as CYP2C9, CYP2C19, and CYP2D6. Winner et al. [26] focused on patients with major depressive disorder, however, the investigators used the GeneSight® (Assurex Health, Ohio, USA) pharmacogenetic test. Table 3 shows that there can be significant variability in pharmacogenetic testing panels. The lack of uniformity highlights the potential need for panel standardization within specific therapeutic areas, and agreement on core genetic variants to be included on a pharmacogenetic testing panel which could be broadly applied across multiple morbidities. To address this issue, van der Wouden et al. [41] introduced a panel of pharmacogenes which they believe can be used for preemptive pharmacogenetic testing based on guidelines from the DPWG, The Pharmaogenomics Knowledgbase, CPIC, and other predefined criteria. Referred to as the pharmacogenetic testing passport, this panel includes 58 variants of 14 genes.
Studies were accepted for review if they reported outcome measures resulting from the implementation of pharmacogenetic test results. Data analyzed included event counts (e.g., ER visits, hospitalizations) [23, 24] or clinical assessment survey results [22, 26]. The timing of data collection was also important, since investigators must estimate an appropriate time point after pharmacogenetic testing which would be adequate to reveal an impact on health outcomes if an association exists. Elliott et al. [24] recorded rehospitalizations and ED visits. The data approached significance at 30 days after patients were discharged from the hospital, but significant differences were found 60 days after discharge. Winner et al. [26] assessed depression severity after changes in medications at 2, 4, 6, and 10 weeks after patient recruitment. Measured improvement was significant at 6 weeks, but not 2, 4, or 10 weeks. The retrospective study by Blasco-Fontecilla [22] did not state what period of time after pharmacogenetic testing patient records were reviewed. None of the studies measured pharmacokinetic parameters such as elimination half-life (t1/2), clearance (Cl), area under the curve, and maximum concentration (Cmax) [42]. These studies may shed light on another reason why healthcare institutions have been slow to adopt pharmacogenetic testing as a guide for prescribing and dosing [30]. The factors investigators use to document clinical effects or improvement are necessarily different, depending on the study focus, funding for research, and access to data. In a review article on the need for preemptive panel-based pharmacogenetic testing, Weitzel et al. [30] note that lack of awareness regarding the evidence needed to establish analytical validity, clinical validity, and clinical utility of pharmacogenetic testing represents one of many barriers to implementation.
There are several limitations that prevent generalized conclusions related to the benefits of pharmacogenetic testing among patients with polypharmacy, some of which are acknowledged by the investigators included in this review. Risk of bias was an area of concern. Five of the six studies had a small sample size and lacked racial/ethnic diversity. Data collected from medical records may be subject to reporting bias. The articles also did not report testing for Hardy–Weinberg equilibrium, which can be used as a tool to detect genotyping errors [18]. None of the studies specifically described how confirmation of treatment adherence was obtained in the study protocols. And following the advice generated from a CDST is at the discretion of the healthcare providers. Finally, not all study participants were prescribed more than one drug, although the mean number of medications per patient within treatment cohorts was greater than one. Brixner et al. [23] and Elliott et al. [24] stated a specific interest in polypharmacy. Blasco-Fontecilla [22] did not state that polypharmacy was a requirement for patient recruitment, but noted that polypharmacy is common among patients with mental illness, and included reduction in the number of drugs used after pharmacogenetic testing as an outcome measure. The remaining articles did not include polypharmacy as a criterion for patient inclusion. Researchers and clinicians interested in the adverse effects of multiple medications should continue to work toward agreement on what number of concomitant drug therapies constitutes polypharmacy. A systematic review by Masnoon et al. [4] reported 13 different definitions of polypharmacy. Most were numerical, some were numerical and accounted for the duration of therapy, and some were descriptive. Of those studies which used numerical definitions, the range was 2 to over 21. Since polypharmacy is common in the psychiatric patients and the elderly, polypharmacy is an important confounding variable in assessing the utility of pharmacogenetic testing in these populations.
Research focused on identifying gene–drug associations is further complicated by other significant phenomena. As described previously, patients with polypharmacy are susceptible to phenoconversion. There are over ninety drugs known to inhibit CYP enzyme activity [36]. Patients who are normal metabolizers may have a drug response expected for a PM when these drugs are included in a treatment regimen. This is known to contribute to phenoconversion [14, 15, 36]. For example, quinidine, a CYP2D6 inhibitor, can reduce the efficacy of treatment with codeine, tramadol, and oxycodone [36]. The percent contribution of individual CYP genes to drug metabolism can also influence pharmacogenetic associations. Several drugs are known substrates of more than one DME. Phenobarbital, for instance, is metabolized by CYP2C9 and CYP2C19 [36]. CDSTs use algorithms to account for drug–drug–gene interactions, but we are unaware of the extent to which other influences are factored in, such as herbal medicines and diet.
Studies have shown that metabolism and bioavailability of drugs may also be affected by age, gender, ethnicity, disease state, inflammation, and pregnancy [8, 43,44,45,46,47]. In a review on the relationship between age, pharmacokinetics, and pharmacodynamics, Mangoni and Jackson [43] note that reduced renal clearance, liver mass and blood flow, and first-pass metabolism are associated with advanced age. In a study looking at CYP3A4 polymorphisms, Guttman et al. [44] found striking differences in allele frequencies among ethnic groups. Differences for other DMEs between ethnic groups are also described by CPIC [9]. Furthermore, a systematic review on pharmacokinetic changes during pregnancy done by Pariente et al. [42] states that renal clearance is increased during pregnancy due to increased renal blood flow and glomerular filtration rate.. Many of the studies they evaluated showed decreased drug exposure in pregnant women for several classes of drugs. They also cite studies reporting changes in hepatic enzyme activity during pregnancy. The multiplicity of known physiological and genetic influences on drug efficacy have led some to investigate DME phenotyping as another approach to improve drug prescribing and dosing [48,49,50]. Phenotyping involves the administration of probe drugs, which are known substrates of metabolizing enzymes and drug transporters, and the measurement of pharmacokinetic parameters in order to estimate enzyme activity, and by extension, an individual’s likely response to a prescribed drug. For instance, losartan can be used as a probe drug for CYP2C9, and dextromethorphan is used as a probe drug for CYP2D6 [45]. Mariappan et al. [49] note that probe drugs are used in drug development to identify potential DDIs. This is an area of research we hope will gain more attention.
This review assessed the utility of pharmacogenetic testing in patients with polypharmacy. We conclude that the use of pharmacogenomics panels can improve health outcomes, especially among the elderly and patients diagnosed with psychiatric disorders. The reviewed studies illuminate the complexities of pharmacogenomics research, and support the significance of drug–gene associations in personalized medicine. Future studies should include RCTs focusing on the impact of pharmacogenetic testing on health outcomes among patients with polypharmacy, and address, to the greatest extent possible, the multiple sources of potential confounding inherent in research which aims to establish relationships between variants of pharmacokinetic genes and ADEs.
References
World Health Organization. Medication safety in polypharmacy: technical report. Geneva: World Health Organization; 2019. Available from: https://apps.who.int/iris/handle/10665/325454.
Mair A, Wilson M, Dreischulte T. Addressing the challenge of polypharmacy. Annu Rev Pharm Toxicol. 2020;60:661–81.
Hales CM, Servais J, Martin CB, Kohen D. Prescription drug use among adults aged 40-79 in the United States and Canada. NCHS Data Brief. 2019;347:1–8.
Masnoon N, Shakib S, Kalisch-Ellett L, Caughey GE. What is polypharmacy? A systematic review of definitions. BMC Geriatr. 2017;17:230.
Roberts A, Kamdem LK, Weston GS. The pharmacogenetics of drug metabolism. In: Zdanowicz MM, editor. Concepts in pharmacogenomics. 2nd ed. Bethesda, MD: American Society of Health-System Pharmacists; 2017. p. 107–50.
Almazroo OA, Miah MK, Venkataramanan R. Drug metabolism in the liver. Clin Liver Dis. 2017;21:1–20.
Manikandan P, Nagini S. Cytochrome P450 structure, function and clinical significance: a review. Curr Drug Targets. 2018;19:38–54.
Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharm Ther. 2013;138:103–41.
Clinical Pharmacogenetics Implementation Consortium. Retrieved March 2020. https://cpicpgx.org/.
Crews KR, Gaedigk A, Dunnenberger HM, Klein TE, Shen DD, Callaghan JT, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for codeine therapy in the context of cytochrome P450 2D6 (CYP2D6) genotype. Clin Pharm Ther. 2012;91:321–6.
Bottorff MB, Bright DR, Kisor DF. Commentary: should pharmacogenomic evidence be considered in clinical decision making? Focus on select cardiovascular drugs. Pharmacotherapy. 2017;37:1005–13.
US Food & Drug Administration. Table of pharmacogenomic biomarkers in drug labelling. Retrieved March 2020. https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling.
Baxter. Nexterone (amiodarone HCl) Injection for intravenous use [package insert on the Internet]. Deerfield IL: Baxter; 2011. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/022325s002lbl.pdf.
Shah RR, Smith RL. Addressing phenoconversion: the Achilles’ heel of personalized medicine. Br J Clin Pharmacol. 2015;79:222–40.
Klomp SD, Manson ML, Guchelaar HJ, Swen JJ. Phenoconversion of cytochrome P450 metabolism: a systematic review. J Clin Med. 2020;9:2890.
Tricco AC, Lillie E, Zarin W, O’Brien KK, Colquhoun H, Levac D, et al. PRISMA extension for scoping reviews (PRISMA-ScR): checklist and explanation. Ann Intern Med. 2018;169:467–73.
Viswanathan M, Berkman ND, Dryden DM, Hartling L. Assessing risk of bias and confounding in observational studies of interventions or exposures: further development of the RTI item bank [Internet]. Rockville, MD: Agency for Healthcare Research and Quality; 2013. Available from: https://www.ncbi.nlm.nih.gov/books/NBK154461/.
Jorgensen AL, Williamson PR. Methodological quality of pharmacogenetic studies: issues of concern. Stat Med. 2008;27:6547–69.
Ross S, Anand SS, Joseph P, Paré G. Promises and challenges of pharmacogenetics: an overview of study design, methodological and statistical issues. JRSM Cardiovasc Dis. 2012;1:2.
Thorn CF, Whirl-Carrillo M, Hachad H, Johnson JA, McDonagh EM, Ratain MJ, et al. Essential characteristics of pharmacogenomics study publications. Clin Pharm Ther. 2019;105:86–9.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535.
Blasco-Fontecilla H. Clinical utility of pharmacogenetic testing in children and adolescents with severe mental disorders. J Neural Transm. 2019;126:101–7.
Brixner D, Biltaji E, Bress A, Unni S, Ye X, Mamiya T, et al. The effect of pharmacogenetic profiling with a clinical decision support tool on healthcare resource utilization and estimated costs in the elderly exposed to polypharmacy. J Med Econ. 2016;19:213–28.
Elliott LS, Henderson JC, Neradilek MB, Moyer NA, Ashcraft KC, Thirumaran RK. Clinical impact of pharmacogenetic profiling with a clinical decision support tool in polypharmacy home health patients: a prospective pilot randomized controlled trial. PLoS ONE. 2017;12:e0170905.
van der Wouden CH, Bank PCD, Özokcu K, Swen JJ, Guchelaar HJ. Pharmacist-Initiated pre-emptive pharmacogenetic panel testing with clinical decision support in primary care: record of PGx results and real-world impact. Genes. 2019;10:416.
Winner JG, Carhart JM, Altar CA, Allen JD, Dechairo BM. A prospective, randomized, double-blind study assessing the clinical impact of integrated pharmacogenomic testing for major depressive disorder. Disco Med. 2013;16:219–27.
Hall-Flavin DK, Winner JG, Allen JD, Carhart JM, Proctor B, Snyder KA, et al. Utility of integrated pharmacogenomic testing to support the treatment of major depressive disorder in a psychiatric outpatient setting. Pharmacogenet Genomics. 2013;23:535–48.
Pérez V, Salavert A, Espadaler J, Tuson M, Saiz-Ruiz J, Sáez-Navarro C, et al. Efficacy of prospective pharmacogenetic testing in the treatment of major depressive disorder: results of a randomized, double-blind clinical trial. BMC Psychiatry. 2017;17:250.
Relling MV, Evans WE. Pharmacogenomics in the clinic. Nature. 2015;526:343–50.
Weitzel KW, Cavallari LH, Lesko LJ. Preemptive panel-based pharmacogenetic testing: the time is now. Pharm Res. 2017;34:1551–5.
Cavallari LH, Van Driest SL, Prows CA, Bishop JR, Limdi NA, Pratt VM, et al. Multi-site investigation of strategies for the clinical implementation of CYP2D6 genotyping to guide drug prescribing. Genet Med. 2019;21:2255–63.
Luzum JA, Pakyz RE, Elsey AR, Haidar CE, Peterson JF, Whirl-Carrillo M, et al. The pharmacogenomics research network translational pharmacogenetics program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharm Ther. 2017;102:502–10.
Licito A, Marotta G, Battaglia M, Benincasa G, Mentone L, Grillo MR, et al. Assessment of pharmacogenomic SLCO1B1 assay for prediction of neuromuscular pain in type 2 diabetes mellitus and cardiovascular patients: preliminary results. Eur Rev Med Pharm Sci. 2020;24:469–77.
Mugoša S, Djordjević N, Djukanović N, Protić D, Bukumirić Z, Radosavljević I, et al. Factors affecting the development of adverse drug reactions to β-blockers in hospitalized cardiac patient population. Patient Prefer Adherence. 2016;10:1461–9.
Pautas E, Moreau C, Gouin-Thibault I, Golmard JL, Mahé I, Legendre C, et al. Genetic factors (VKORC1, CYP2C9, EPHX1, and CYP4F2) are predictor variables for warfarin response in very elderly, frail inpatients. Clin Pharm Ther. 2010;87:57–64.
Samer CF, Lorenzini KI, Rollason V, Daali Y, Desmeules JA. Applications of CYP450 testing in the clinical setting. Mol Diagn Ther. 2013;17:165–84.
Maciel A, Cullors A, Lukowiak AA, Garces J. Estimating cost savings of pharmacogenetic testing for depression in real-world clinical settings. Neuropsychiatr Dis Treat 2018;14:225–30.
Saldivar JS, Taylor D, Sugarman EA, Cullors A, Garces JA, Oades K, et al. Initial assessment of the benefits of implementing pharmacogenetics into the medical management of patients in a long-term care facility. Pharmgenomics Pers Med. 2016;9:1–6.
Sugarman EA, Cullors A, Centeno J, Taylor D. Contribution of pharmacogenetic testing to modeled medication change recommendations in a long-term care population with polypharmacy. Drugs Aging. 2016;33:929–36.
Verbelen M, Weale ME, Lewis CM. Cost-effectiveness of pharmacogenetic-guided treatment: are we there yet? Pharmacogenomics J. 2017;17:395–402.
van der Wouden CH, van Rhenen MH, Jama WOM, Ingelman-Sundberg M, Lauschke VM, Konta L, et al. Development of the PGx-passport: a panel of actionable germline genetic variants for pre-emptive pharmacogenetic testing. Clin Pharm Ther. 2019;106:866–73.
Pariente G, Leibson T, Carls A, Adams-Webber T, Ito S, Koren G. Pregnancy-associated changes in pharmacokinetics: a systematic review. PLoS Med. 2016;13:e1002160.
Mangoni AA, Jackson SH. Age-related changes in pharmacokinetics and pharmacodynamics: basic principles and practical applications. Br J Clin Pharmacol. 2004;57:6–14.
Guttman Y, Nudel A, Kerem Z. Polymorphism in cytochrome P450 3A4 is ethnicity related. Front Genet. 2019;10:224.
Puris E, Pasanen M, Gynther M, Häkkinen MR, Pihlajamäki J, Keränen T, et al. A liquid chromatography-tandem mass spectrometry analysis of nine cytochrome P450 probe drugs and their corresponding metabolites in human serum and urine. Anal Bioanal Chem. 2017;409:251–68.
Alagiakrishnan K, Mah D, Padwal R. Classic challenges and emerging approaches to medication therapy in older adults. Discov Med. 2018;26:137–46.
2019 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2019 updated AGS beers Criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2019;67:674–94.
Lloret-Linares C, Daali Y, Chevret S, Nieto I, Molière F, Courtet P, et al. Exploring venlafaxine pharmacokinetic variability with a phenotyping approach, a multicentric french-swiss study (MARVEL study). BMC Pharm Toxicol. 2017;18:70.
Mariappan TT, Shen H, Marathe P. Endogenous biomarkers to assess drug-drug interactions by drug transporters and enzymes. Curr Drug Metab. 2017;18:757–68.
Vogl S, Lutz RW, Schönfelder G, Lutz WK. CYP2C9 genotype vs. metabolic phenotype for individual drug dosing–a correlation analysis using flurbiprofen as probe drug. PLoS ONE. 2015;10:e0126329.
Acknowledgements
The authors would like to thank Jenessa McElfresh, Health Sciences Librarian (Clemson University), for her assistance during manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Meaddough, E.L., Sarasua, S.M., Fasolino, T.K. et al. The impact of pharmacogenetic testing in patients exposed to polypharmacy: a scoping review. Pharmacogenomics J 21, 409–422 (2021). https://doi.org/10.1038/s41397-021-00224-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41397-021-00224-w
- Springer Nature Limited
This article is cited by
-
Challenges of pediatric pharmacotherapy: A narrative review of pharmacokinetics, pharmacodynamics, and pharmacogenetics
European Journal of Clinical Pharmacology (2024)
-
Pharmacogenetic profiling via genome sequencing in children with medical complexity
Pediatric Research (2023)