
Abstract
Background:
Hundreds of ongoing clinical trials combine radiation therapy, mostly delivered as stereotactic body radiotherapy (SBRT), with immune checkpoint blockade. However, our understanding of the effect of radiotherapy on the intratumoral immune balance is inadequate, hindering the optimal design of trials that combine radiation therapy with immunotherapy. Our objective was to characterize the intratumoral immune balance of the malignant prostate after SBRT in patients.
Methods:
Sixteen patients with high-risk, non-metastatic prostate cancer at comparable Gleason Grade disease underwent radical prostatectomy with (n = 9) or without (n = 7) neoadjuvant SBRT delivered in three fractions of 8 Gy over 5 days completed 2 weeks before surgery. Freshly resected prostate specimens were processed to obtain single-cell suspensions, and immune-phenotyped for major lymphoid and myeloid cell subsets by staining with two separate 14-antibody panels and multicolor flow cytometry analysis.
Results:
Malignant prostates 2 weeks after SBRT had an immune infiltrate dominated by myeloid cells, whereas malignant prostates without preoperative treatment were more lymphoid-biased (myeloid CD45+ cells 48.4 ± 19.7% vs. 25.4 ± 7.0%; adjusted p-value = 0.11; and CD45+ lymphocytes 51.6 ± 19.7% vs. 74.5 ± 7.0%; p = 0.11; CD3+ T cells 35.2 ± 23.8% vs. 60.9 ± 9.7%; p = 0.12; mean ± SD).
Conclusion:
SBRT drives a significant lymphoid to myeloid shift in the prostate-tumor immune infiltrate. This may be of interest when combining SBRT with immunotherapies, particularly in prostate cancer.
Similar content being viewed by others

Introduction
Today, approximately half of all cancer patients receive radiotherapy at some point during their treatment [1]. Radiation-induced tumor cell kill is thought to be primarily the result of the formation of reactive oxygen species and DNA damage. However, there is increasing evidence that radiation has also significant immune modulating activity, altering the intratumoral immune landscape and in some cases favoring lymphocytic infiltration, particularly after hypofractionated regimes (fraction sizes of 7–8 Gy) [2,3,4,5]. For instance, studies in animal models showed that tumor-infiltrating lymphocytes, after an initial decline, increase well above pre-treatment levels over the span of 1 to 2 weeks after high-dose per fraction radiotherapy as lymphocytes migrate into the irradiated tumor [6, 7]. In the clinic, high-dose per fraction radiotherapy is delivered using the technique of Stereotactic Body Radiotherapy (SBRT), which relies on image guidance to deliver dose to target volumes with high precision. Hundreds of ongoing trials listed in clinicaltrials.gov are currently testing the combination of various immunotherapies with radiotherapy, most using SBRT in combination with checkpoint blockade [8]. However, studies directly measuring the representation of infiltrating immune cells in patients after SBRT are few and far between, and none exist in the context of prostate cancer. We therefore sought to interrogate the tumor-immune interface after prostate SBRT using fresh tissue from prostate cancer patients.
Materials and methods
Correlative analyses from a phase I trial of prostate SBRT (three fractions of 8 Gy delivered over 5 days and directed to the prostate and seminal vesicles) 2 weeks +/− 3 days neoadjuvant to radical prostatectomy (NCT02830165) offered the unique opportunity to study fresh prostate tissue after SBRT in patients. The malignant prostates from nine patients with high-risk, non-metastatic prostate cancer on the trial were compared to those from seven patients with similar Gleason grade who underwent radical prostatectomy alone without neoadjuvant therapy.
Fresh bulk prostatectomy tissue containing both tumor and surrounding normal gland was minced and enzymatically digested for 1 h at 37 °C in a gentleMACS dissociator (Miltenyi Biotec Inc., San Diego, CA) with 5 mg/g collagenase D (Sigma-Aldrich, St. Louis, MO) and 0.5 mg/g tissue DNAse (Roche, San Francisco, CA) to obtain single-cell suspensions prior to antibody staining in two separate 14-color panels (Supplementary Table S1) for 30 min at room temperature to capture major T cells subsets (panel 1), as well as B cells, monocytes, myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), and NK cells (panel 2).
Tumor single-cell suspensions were prepared with fixable viability stain 510, prior to assaying for surface marker expression levels. Panel 1 was premixed in brilliant stain buffer (BD Horizon/BD Biosciences) containing FITC anti-human CD4, PE anti-human CD25, PE-CF594 anti-human CXCR3, PerCP-Cy5.5 anti-human PD-L1, PE-Cy7 anti-human CD127, APC anti-human CD45RA, Alexa Flour 700 anti-human CD8, APC-H7 anti-human CD45, BV421 anti-human PD-1, BV605 anti-human CCR7, BV650 anti-human CCR6, and BV711 anti-human CD3 (Supplementary Table S1) before mixing with cells in 50 µl 2% FBS/PBS staining buffer. Washed cells were analyzed within 2 h and 1–2 × 105 events collected on a LSRFortessa with UltraComp eBeads compensation (eBioscience, Inc., SanDiego, CA).
The second panel comprised FITC anti-human HLA-DR, PE anti-human CD14, PE-CF594 anti-human CD56, PerCP-Cy5.5 anti-human CD11b, PE-Cy7 anti-human CD19, APC anti-human CD15, Alexa Flour 700 anti-human CD11c, APC-H7 anti-human CD20, BV421 anti-human PD-L1, BV605 anti-human CD45, BV650 anti-human CD16, BV711 anti-human CD3 and BV786 anti-human CD163 (Supplementary Table S1) premixed in brilliant stain buffer as above. Fixable viability stain 510 pre-stained cells were stained in 50 µl 2% FBS/PBS staining buffer for 30 min at room temperature, washed, and submitted to flow cytometry as above. 1–2 × 105 events were accumulated and dead cells excluded based on fixable viability stain 510 uptake (BD Horizon™). Analysis was done with FlowJo, LLC (Ashland, OR) using a gating strategy based on previous experience and illustrated in Supplementary Figs. S1 and S2 [9, 10]. Quality control required ≥2000 viable CD45+ cells.
Statistical analysis
The group comparison was assessed by a two-sided two-sample t-test (Supplementary Table S2). The false-discovery rate was controlled via the Benjamini-Hochberg procedure [11], with a threshold level for adjusted p-values of 0.2. With the explorative aim of this study, a threshold of 0.2 for FDR is considered to be reasonable based on prior literature [12,13,14]. Our null hypothesis was set to be that presurgical hypofractionated radiotherapy does not alter the immune infiltrate in prostate tumors, while the alternative hypothesis to be that presurgical hypofractionated radiotherapy does alter the immune infiltrate in prostate tumors. The hypothesis testing was carried out via two-sided two-sample t-test. No secondary endpoint of our interests was included.
The mean log2 fold changes (log2[mean value irradiated/mean value unirradiated]) were summarized graphically. A heatmap was constructed using all but two patients who did not have a complete set of immune markers run (SBRT patient 1 and control patient 1) by normalizing each immune subset across all subjects for calculation of z-scores.
Results
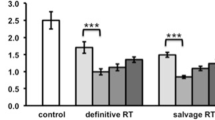
An average of 1.96 ± 1.14 g (range 0.1–4.2) of prostate tissue per patient yielded 3.56 ± 3.13 × 106 viable cells (range 0.04–10.8 × 106) for staining without significant changes in the extent of immune infiltration between treatment groups judged by the fraction of CD45+ cells (mean ± SD SBRT 43.0 ± 24.7% vs. control 52.3 ± 24.5%; p = 0.78). The minimum requirement of 2000 viable CD45+ cells was reached for all specimens. Within the CD45+ population, myeloid cells were enriched after SBRT compared to unirradiated controls, (comprising 48.4 ± 19.7% vs. 25.4 ± 7.0% of the infiltrate; p = 0.11). Similar differences were seen in the percentages of CD14+/hiCD16+DR+ intermediate monocytes/macrophages (8.3 ± 6.3% vs. 1.3 ± 0.7%; p = 0.11). Reciprocally, overall lymphocyte counts were depleted after SBRT (51.6 ± 19.7% vs. 74.5 ± 7.0%; p = 0.11), including fewer CD3 + T cells (35.2 ± 23.8% vs. 60.9 ± 9.7%; p = 0.12), though the CD4/CD8 ratios were similar (0.63 ± 0.36 vs. 0.61 ± 0.56; p = 0.94) (Fig. 1). Levels of PD-1+ on CD8+ T cells or PD-L1+ on myeloid cells were not significantly affected by treatment as both mirrored the change in their respective parent population, i.e., falling CD8+ and rising myeloid cells with a net decrease in PD-1/PD-L1 ratios (1.56 ± 1.59 vs. 3.32 ± 3.93; p = 0.77). In essence, the immune infiltrate in prostate cancer two weeks after SBRT demonstrated a lymphoid to myeloid shift, which is further illustrated by the fact that both groups cluster separately by overall relative abundance, dominated by either lymphoid (control patients) or myeloid subsets (SBRT patients), except for one SBRT patient who clustered within the unirradiated control cohort (Fig. 2). Immune profiles were also notably less uniform overall in irradiated tissue samples when compared to unirradiated prostate tumors (Fig. 2).

Polar graph illustrating the mean log2-fold differences in prostate tumor infiltrates following preoperative, hypofractionated SBRT (n = 9, solid red line) compared to unirradiated prostate tumors (n = 7, black, dotted line). All immune subpopulations were gated on CD45+. Data are log2[mean value irradiated/mean value unirradiated]. Myeloid subsets highlighted in green. Underlined subsets indicate significant differences between irradiated vs. non-irradiated based on adjusted p-value = 0.2 (false-discovery rate threshold).

Heatmap showing the SBRT-treated patient cohort having greater inter-patient variability and a rise in myeloid lineage (green) dominance compared to the unirradiated patient cohort that is overall more uniform and dominated by lymphoid subsets. Heatmap was constructed by normalizing each immune cell subset across the all subjects. Values are z-scores for each immune subset in each patient.
Discussion
This is the first direct clinical evidence that SBRT changes intraprostatic immune milieu by increasing the relative abundance of myeloid cells. Notably, all cell subsets reported here represent a portion of CD45+ cells, i.e., changes are relative, which is a good measure for the overall immune cell balance rather than absolute amounts, that are difficult to measure accurately by these methods. Our finding may be relevant to trials that combine SBRT with immunotherapy in prostate cancer, and potentially other cancer types. Mindful of the caveats in the use of currently available immune biomarkers, prostate cancer tends to be resistant to checkpoint blockade, more so than many other solid tumors [15,16,17] with some notable exceptions such as microsatellite instability-high, mismatch repair-deficient, or CDK12 deficient subtypes [18, 19]. A strong intratumoral T-cell signature is normally a favorable sign of immune recognition and activation [20, 21]. As such, the intratumoral myeloid predominance after SBRT suggests that SBRT may render the tumors even less responsive to checkpoint blockade targeting the PD-1/L1 or CTLA-4 axis, at least at the time point investigated [22, 23]. In addition to correlating to response to checkpoint blockade, in most cancer types, tumor-infiltrating lymphocytes are associated with favorable survival [24]. In prostate cancer, this association may be more complex. A recent report on gene expression profiles in almost 10,000 prostatectomy samples suggests immune signatures within the primary tumor characteristic of T cells and macrophages associated with poor prognosis [25].
Our report has several limitations. First, the number of evaluable patients was limited, leading to restrictive statistical power of our study, by the trial and tissue availability, which prevented functional studies. Functional studies into the polarization and activation status of myeloid cells and T cells would be desirable given that both are malleable, as are their interactions and the net response that ensues [26, 27]. Second, the fresh tissue consisted of a combination of tumor and surrounding normal prostate tissue with spatial relationships amongst cells that cannot be interrogated with our methods. Third, the immune infiltrates two weeks after completion of SBRT may not necessarily be representative of other time points after SBRT. It remains to be seen if these results can be extrapolated to other histologies and radiation dose regimens, e.g. different fractionations and/or target volumes. Of note, one completed trial [28] and many ongoing test combinations of radiotherapy with immunotherapies use a similar SBRT dose regimen. In addition, the intratratumoral immune environment at baseline may impact the response to SBRT, although this was not possible to assess here.
It will be interesting to reassess these data in the context of long-term clinical outcome data when they become available. Ultimately, the optimal design of clinical trials in prostate cancer and meaningful integration of immunotherapies with focal cytotoxic therapies such as SBRT rely on a better understand of these immune signatures in space and time.
References
Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and radiation therapy: current advances and future directions. Int J Med Sci. 2012;9:193–9.
Demaria S, Formenti SC. Radiation as an immunological adjuvant: current evidence on dose and fractionation. Front Oncol. 2012;2:153.
Schaue D, Ratikan JA, Iwamoto KS, McBride WH. Maximizing tumor immunity with fractionated radiation. Int J Radiat Oncol Biol Phys. 2012;83:1306–10.
Frey B, Rückert M, Weber J, Mayr X, Derer A, Lotter M, et al. Hypofractionated irradiation has immune stimulatory potential and induces a timely restricted infiltration of immune cells in colon cancer tumors. Front Immunol. 2017;8:231–231.
Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618.
Lee Y, Auh SL, Wang Y, Burnette B, Meng Y, Beckett M, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114:589–95.
Filatenkov A, Baker J, Mueller AM, Kenkel J, Ahn GO, Dutt S, et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin Cancer Res. 2015;21:3727–39.
Schaue D. A Century of Radiation Therapy and Adaptive Immunity. Front Immunol. 2017;8:431. https://doi.org/10.3389/fimmu.2017.00431.
Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the Human Immunology Project. Nat Rev Immunol. 2012;12:191–200.
Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, et al. Focal irradiation and systemic tgfbeta blockade in metastatic breast cancer. Clin Cancer Res. 2018;24:2493–504.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol). 1995;57:289–300.
Capanu M, Seshan VE. False discovery rates for rare variants from sequenced data. Genet Epidemiol. 2015;39:65–76.
Amar D, Shamir R, Yekutieli D. Extracting replicable associations across multiple studies: Empirical Bayes algorithms for controlling the false discovery rate. PLOS Computational Biol. 2017;13:e1005700.
Efron B. Size, power and false discovery rates. Ann Stat. 2007;35:1351–77.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl J Med. 2012;366:2443–54.
Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–12.
Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol. 2017;35:40–7.
Abida W, Cheng ML, Armenia J, Middha S, Autio KA, Vargas HA, et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 2019;5:471–8.
Wu YM, Cieslik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell. 2018;173:1770–.e1714.
Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A, Avramis E, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res. 2016;4:194–203.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4.
Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120:16–25.
Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol. 2018;9:1310.
Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer. 2011;105:93–103.
Zhao SG, Lehrer J, Chang SL, Das R, Erho N, Liu Y, et al. The immune landscape of prostate cancer and nomination of PD-L2 as a potential therapeutic target. J Natl Cancer Inst. 2019;111:301–10.
Bercovici N, Guérin MV, Trautmann A, Donnadieu E. The Remarkable Plasticity of Macrophages: A Chance to Fight Cancer. Front Immunol. 2019;10:1563. https://doi.org/10.3389/fimmu.2019.01563.
DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro-versus anti-tumor immunity. Cancer Metastasis Rev. 2010;29:309–16.
Theelen W, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts J, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol. 2019;5:1276–82.
Acknowledgements
We acknowledge NIH grants: 5R01CA191234-05 (DS), UCLA Prostate SPORE P50CA0912131 (RR, NN), Prostate Cancer Foundation Young Investigator Award (NN, AK). We would like to express our sincere thanks to all the patients and to the clinical research staff.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Nickols, N.G., Ganapathy, E., Nguyen, C. et al. The intraprostatic immune environment after stereotactic body radiotherapy is dominated by myeloid cells. Prostate Cancer Prostatic Dis 24, 135–139 (2021). https://doi.org/10.1038/s41391-020-0249-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41391-020-0249-8
- Springer Nature Limited
This article is cited by
-
Significant changes in macrophage and CD8 T cell densities in primary prostate tumors 2 weeks after SBRT
Prostate Cancer and Prostatic Diseases (2023)
-
Immunomodulatory effects of carbon ion radiotherapy in patients with localized prostate cancer
Journal of Cancer Research and Clinical Oncology (2023)
-
PSA bounce, prognosis, and clues to the radiation response
Prostate Cancer and Prostatic Diseases (2021)