
Abstract
Background
Androgen-targeted therapy and chemotherapy are currently the mainstay of treatment in metastatic castration resistant prostate cancer (mCRPC). When progression occurs despite these therapeutic strategies, additional FDA-approved treatment options are lacking. However, there is a vast amount of emerging data surrounding novel investigational therapies in this space.
Methods
We reviewed and summarized the body of literature surrounding the current treatment options for mCRPC. Medline and Pubmed as well as abstracts from international congresses were utilized to gather relevant literature surrounding investigational treatment of mCRPC. We highlight the results of recent trials investigating the use of novel strategies to treat mCRPC.
Results
Androgen-targeted therapy and chemotherapy will remain foundational in the treatment of mCRPC. However, heavily pretreated patients who have developed resistance may benefit from novel therapeutic strategies. The use of poly(adenosine diphosphate [ADP]-ribose) polymerase inhibitors (PARPi) has now gained FDA approval for patients with homologous recombination repair (HRR) gene mutations. Novel androgen receptor (AR) degraders and the use of radioligand therapy (RLT) with Lu-PSMA-617 (Lu-PSMA) are under investigation. Immune-directed therapies, including programmed death (PD-1) inhibition, bi-specific T-cell engager (BiTE) technology, and chimeric antigen receptor (CAR) T-cell therapy, have shown promise in early phase trials. Further understanding of resistance mechanisms has led to additional therapeutic targets, including targeting the PI3K-Akt-mTOR pathway and enhancer of zester homolog 2 (EZH2).
Conclusions
Based on our review of the literature, exciting new therapeutic strategies exist for the treatment of mCRPC. In particular, PARPi, AR degraders, PSMA-targeted therapies, immune-directed therapies, and agents targeting resistance mechanisms as monotherapy or in combination could improve patient outcomes. Additional data from randomized trials are necessary to understand the efficacy and tolerability of these treatment strategies.
Similar content being viewed by others


Background
Prostate cancer is the third most common cause of cancer-related death in men [1]. When men are not cured by local therapy, the mainstay of systemic therapy is androgen deprivation therapy (ADT). When the disease progresses despite surgical or pharmaceutical castration, patients are reclassified as having castration-resistant prostate cancer (CRPC), either with or without metastatic disease [2]. Castration resistance is defined as an increase in PSA > 25% and >2 ng/mL above the nadir confirmed on repeat testing >3 weeks apart or radiographic progression by RECIST criteria despite androgen depletion therapy [3]. Approximately 10–20% of patients with prostate cancer will develop castration resistance within 5 years and 84% of patients have metastatic disease at the time of castration resistance. Patients with de novo metastatic disease progress to CRPC more quickly, based on the control arm of the STAMPEDE trial in which men became castration resistant within 11.2 months [4]. Patients with metastatic CRPC (mCRPC) have poor prognosis with median overall survival (OS) of 21.3 months in those with bone metastasis and 16.3 months in those with any visceral disease [5].
The two major pillars of therapy for mCRPC include cytotoxic chemotherapy and androgen-targeted therapies [6]. Cytotoxic therapy with docetaxel has been used for the treatment of mCRPC since 2004, based on the TAX 327 and SWOG 99-16 trials [7, 8]. Androgen-targeted agents, including abiraterone acetate plus prednisone (COU-AA-301 and 302 trials) [9, 10] and enzalutamide (PREVAIL and AFFIRM trials) [11, 12] are also widely used. Of note, with these therapies moving into in the castrate-sensitive and nonmetastatic setting, the role of these agents as monotherapy in mCRPC will continue to decrease, further highlighting the need for novel therapies. Other approved therapies for mCRPC with OS benefits include sipuleucel-T, radium-223, and cabazitaxel [13,14,15]. How best to sequence these available therapies, especially in patients progressing despite ADT plus AR-targeted therapy, remains a dilemma and is an area of active investigation. In the PLATO trial, no clinically meaningful benefit was shown in switching antiandrogen therapy from enzalutamide to abiraterone [16], which was corroborated in a recent phase II trial showing abiraterone or enzalutamide have weak antitumor activity in patients with prior exposure to the other agent [17]. Based on the 2017 Advanced Prostate Cancer Consensus Conference, 90% of experts agreed that in men with mCRPC who have progressed following AR-targeted therapy [18], switching to a taxane, such as docetaxel would be the best treatment option. In those previously treated with docetaxel, switching to cabazitaxel would be preferable based on the CARD trial, which showed that cabazitaxel improved radiographic PFS and OS compared to alternative AR-targeted therapy in patients previously exposed to docetaxel [19]. Ultimately, additional treatment options are needed for the treatment of mCRPC.
Improved understanding of tumor cell biology, resistance mechanisms, and advances in genomic analysis has allowed for the development of new therapeutic strategies. In this review, we will highlight novel therapies including PARPi, PD-1 antibodies, AR degraders, RLT with Lu-PSMA, BiTE therapy, CAR T-cell therapy, PI3K-Akt-mTOR pathway inhibition, and EZH2 inhibition. The goal of this review is to discuss novel therapies that are currently under investigation and explore future directions of treatment of mCRPC.
PARP inhibitors
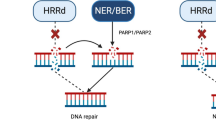
PARP inhibitors function by binding to single-strand DNA breaks and blocking PARP enzyme-mediated DNA damage repair, leading to cancer cell death [20]. Olaparib demonstrated success in the treatment of mCRPC in the phase II TOPARP-A and TOPARP-B trials, which used next-generation sequencing to identify patients with mutations in DNA damage repair genes. All seven patients with BRCA2 and four out of five patients with ATM mutations in TOPARP-A had response [21]. TOPARP-B confirmed responsiveness in patients with BRCA2 mutations and demonstrated need for ongoing evaluation of genomic biomarkers to guide treatment with PARPi [22].
FDA approval of olaparib in mCRPC followed the release of results from the randomized phase III PROfound trial. Patients were split into two cohorts based on alterations in HRR genes and compared with abiraterone or enzalutamide. Cohort A consisted of patients with BRCA1, BRCA2, or ATM alterations, while cohort B included patients with 12 other prespecified gene alterations. Although there was a PFS benefit favoring PARPi in the overall population, this was mostly driven by cohort A. There was improved radiographic progression-free survival (rPFS) in cohort A (7.4 vs. 3.6 months) as well as objective response rates (ORR) of 33% vs. 2%. There was also improvement in PSA response and circulating tumor cell clearance in the PARPi group compared to the AR therapy arm. The final OS analysis was presented at ESMO 2020, showing a significant improvement in OS of 19.1 months with olaparib vs. 17.3 months in the control arm (HR 0.69, 95% CI 0.50–0.97, p = 0.0175) in Cohort A. Common adverse events (AEs) in the olaparib arm included anemia (46%), nausea (41%), and fatigue (41%). Notably, there was 81% crossover to the PARPi group upon confirmation of imaging-based disease progression [23, 24].
Rucaparib has also been approved for the treatment of BRCA-mutated mCRPC based on the results of the phase II TRITON2 study. In this single-arm trial, patients with known DNA damage response gene aberrations and mCRPC progressing after both AR therapy and taxane chemotherapy were administered rucaparib. ORR was 47.5% in patients with BRCA mutations. Objective responses were not seen in patients with ATM, CDK12, or CHEK2 mutations [25, 26]. Thus, PARPi monotherapy has secured a place in the treatment algorithm for progressive mCRPC for those with HRR alterations, especially BRCA alterations.
Olaparib has also been studied with abiraterone versus abiraterone monotherapy in phase II study, which showed rPFS of 13.8 months in the combination arm vs. 8.2 months in the abiraterone arm. Subgroup analyses performed based on HRR showed longer rPFS in the altered HRR group [27]. Further assessment is ongoing with the phase III PROPEL trial [28]. Other ongoing trials with olaparib in mCRPC include the phase II TRAP and olaparib plus testosterone trials [29, 30]. Further studies of combination therapy with androgen-targeted agents, immune-directed therapy, and other novel agents are ongoing (Table 1).
Anti-PD1 antibodies
PD-1 and programmed death ligand (PD-L1) inhibitors, which bind to PD-1 or PD-L1 and prevent interaction between T cells and tumor cells to prevent immune evasion [31], have vastly enhanced treatment options for various malignancies, though results in CRPC have been disappointing. In the phase II KEYNOTE-199 trial, patients with mCRPC previously treated with docetaxel and AR-directed therapy were divided into a first cohort with PD-L1 expression and a second cohort without, each treated with pembrolizumab. The primary endpoint of ORR by RECIST v1.1 was achieved by 7% of men in cohort 1 and 3% in cohort 2, with the secondary endpoint of rPFS of 2.1 months in both cohorts. A third cohort with bone-predominant disease irrespective of PD-L1 expression had rPFS of 3.7 months [32]. Atezolizumab as a single agent in mCRPC has also shown minimal efficacy, with a 9% ORR and a PFS of 3.4 months [33]. The small but real proportion of patients responding to single-agent immunotherapy highlights the need for biomarkers to guide treatments. For example, pembrolizumab is approved in a tumor-agnostic fashion for patients with mismatch repair deficiency and in patients with high tumor mutational burden and can benefit the few mCRPC patients with these biomarkers [34, 35].
Given the limited response to single-agent therapy, multiple combination therapies incorporating immunotherapy are under investigation. The ongoing phase IB/II trial KEYNOTE-365 is investigating combinations with pembrolizumab. In cohort A, docetaxel-pretreated patients with mCRPC received pembrolizumab and olaparib. Of note, 50% of these patients discontinued treatment, with a majority having disease progression or significant toxicity and only 9% having a PSA response [36]. Cohort B examined pembrolizumab in conjunction with docetaxel, with PSA response observed in 33% of patients and ORR of 14% [37]. Cohort C evaluated pembrolizumab and enzalutamide in combination. PSA response was observed in a third of patients with an ORR 20%, including two with complete response (CR) [38]. In a cohort of patients with visceral disease from the KEYNOTE-199 study, the combination of pembrolizumab with enzalutamide resulted in an ORR of 12% with two CRs [39]. In patients with progression on enzalutamide, adding pembrolizumab resulted in a PSA decline ≥50% in 18% patients and a median OS of 21.9 months [40]. However, IMbassador250, a phase III trial of enzalutamide in combination with atezolizumab versus enzalutamide alone showed no survival benefit and no difference in rPFS or time to PSA progression [41]. IMbassador250 was the first phase III trial performed with PD-L1 inhibitors in mCRPC, and the results were disappointing.
Overall, checkpoint inhibitors should only be used in selected patients with mCRPC. The combination of PD-1 and PARP inhibition appears quite toxic with limited efficacy, while combination with chemotherapy or androgen-targeted agents may warrant further investigation in select groups of mCRPC patients.
AR degraders
Androgen receptors (AR) play a critical role in the development and progression of prostate cancer, and therefore, ADT remains as a mainstay of treatment. Briefly, when stimulated, the AR translocates to the nucleus, where it regulates transcription of critical genes involved in cell growth and proliferation, while also affecting genomic stability and DNA repair [42]. ADT blocks testosterone production to decrease AR stimulation. However, the efficacy is limited, and patients will ultimately progress to CRPC [43]. The AR can be additionally targeted by blocking signaling or by degrading the AR [44]. Selective AR degraders (SARD) and PROteolysis Targeting Chimeras (PROTAC) are two classes of compounds that have demonstrated antitumor properties in prostate cancer via proteasome-mediated degradation of AR.
Initial studies with SARDs showed moderate AR downregulation in prostate cancer cells but with putative adverse cardiovascular effects [45]. Further development of SARDs led to the creation of compounds without cardiotoxicity. In phase I studies, AZD3514 demonstrated PSA declines >50% in 13% and objective soft tissue responses in 17% of individuals, with the main side effects being nausea and vomiting [46]. Another SARD, ASC-J9, degraded both wild-type AR and AR-splice-variants in ex vivo models [47]. Further preclinical studies showed activity against AR mutants in enzalutamide-resistant CRPC and counteracted AR enhancement in CRPC cells exposed to docetaxel [48, 49]. In addition, a class of SARDS containing a hydrophobic residue, which mimics a partially denatured protein state and recruits chaperones to induce proteasome-mediated AR degradation, has been shown to overcome enzalutamide resistance in ex vivo models [50].
PROTAC is another promising therapy for mCRPC. These chimeras consist of: (1) a ligand that targets the protein of interest such as AR, (2) a linker region, and (3) a ligand which recruits a specific E3 ubiquitin ligase. Numerous PROTACs have been shown to effectively target AR in preclinical models [51, 52]. Results from a phase I study of ARV-110 administered to heavily pretreated CRPC patient showed that two of eight patients receiving moderate-high dose therapy had PSA declines of >50% [53]. Additional prospective trials will determine the clinical utility of AR degraders moving forward.
PSMA-targeted therapies
Prostate-specific membrane antigen (PSMA) is a transmembrane metallopeptidase that is restricted mainly to the healthy prostate secretory acinar epithelium but is also expressed within the plasma membrane of prostate cancer cells [54]. Thus, neoplastic transformation of the prostate is often accompanied by substantial increase in PSMA levels. The most prominent increase in expression of PSMA is often observed in high-grade, metastatic, and castration resistant disease. Given a fairly restricted expression pattern and correlation with both disease activity and severity in prostate cancer, PSMA-directed therapy is promising. We will discuss emerging data regarding Lu-PSMA, BiTE technology, and CAR-T cells which target PSMA.
Lutetium-PSMA
Lutetium-177 (Lu-177) PSMA therapy utilizes a form of radiotherapy often referred to as peptide receptor radionucleotide therapy. This technique employs a molecule that attaches itself to the PSMA receptor on cancer cells. Prior to administration, the PSMA molecule is bound with Lu-177, which emits beta radiation once attached to prostate cancer cells. This allows for delivery of radiation to the cancer cells directly, while sparing radiation to the entire body [44].
In 2015, a study in which ten patients with advanced mCRPC were treated with Lu-PMSA showed that 70% of patients experienced a PSA decline [55]. Similarly, in a single-arm phase II trial in men with mCRPC who had progressive disease after standard treatments, patients had high PSA response rates, low toxic effects, and reduction in pain [56]. More recently, initial results from the TheraP trial demonstrated that Lu-177 PMSA therapy significantly improved PSA response rate (66% vs. 37%) and had had fewer grade 3/4 AEs when compared to cabazitaxel [57]. Overall, it appears that Lu-177 PSMA therapy is well tolerated, with the most significant AEs being hematologic toxicities.
There are a number of ongoing clinical trials investigating the role of Lu-PSMA therapy. The phase III VISION trial recently completed enrollment. In this study, men with progressive mCRPC previously treated with androgen axis therapy and taxane chemotherapy are randomized to Lu-177 PSMA therapy versus best supportive care. Outcomes include progression-free survival, PSA response, rPFS, as well as safety and tolerability [58]. Thus, this study is expected to help define the role of Lu-177 PSMA therapy moving forward.
BiTE therapy
BiTE technology is an immune-oncology therapy that utilizes patients’ own T cells to attack cancer cells through the generation of molecules that target tumor-specific antigens. BiTE molecules have one domain that recognizes tumor-specific antigens, which can be modified to target any surface antigen to allow for therapies against various tumor types, and a second domain that recognizes T cells to recruit them for elimination of the malignant cells [59]. The first and only FDA approved BiTE molecule is blinatumomab, which targets CD19 surface antigens on B cells in the treatment of acute lymphoblastic leukemia [60].
BiTE technology has been developed to target PSMA in the treatment of mCRPC, using the BiTE molecule pasotuxizumab. In a phase 1 study of pasotuxizumab in mCRPC refractory to standard therapies, a dose-dependent response was seen with PSA decreasing >50% in 19% of patients. Notably, the PSA response in two patients lasted beyond 1 year. All patients had at least 1 AE with most common being fever (94%), chills (69%), and fatigue (50%). Importantly, this therapy requires continuous infusion given its short half-life, which is a limitation [61]. Another agent, AMG 160 is a BiTE molecule that targets PSMA with an extended half-life of ~1 week. In preclinical evaluation, weekly dosing of AMG 160 has shown antitumor activity in a xenograft model and in vivo can induce T-cell activation and cytokine release in blood as well as infiltration into organs known to express PSMA [62]. Phase I data using AMG 160 for in 32 patients with mCRPC was presented at ESMO 2020. The most common adverse event was cytokine release syndrome (84%) with one treatment-related discontinuation and two reversible dose-limiting toxicities. Among those with measurable disease, one patient had a partial response, five had stable disease, and five had progressive disease. PSA reductions in occurred in 63% of patients with confirmed PSA responses in 27% of patients. Treatment with AMG 160 has preliminary efficacy and appears tolerable, although the frequency of CRS will remain an issue. Further exploration of optimal dosing is necessary [63].
CAR T-cell therapy
Chimeric antigen receptor T cells are fusion proteins that consist of an extracellular antigen that can be engineered to recognize specific tumor antigens, a transmembrane zone to fix the extracellular antigen to T cells, and an intracellular signal transduction zone that can activate T cells. These CARs can be introduced into patients using viral transduction [64]. CAR-T cell therapy is effective in treating hematologic malignancies [65], but given the lack of monoclonality of solid tumors, different tumor microenvironments, and physical barriers separating infused CAR-T cells from tumor cells, CAR-T cell therapy has not been as successful in treating solid tumors. Despite these limitations, prostate cancer has become a potential target for the use of CAR-T cell therapy given the understanding of PSMA and its high prostate tissue specificity [66].
Engineered anti-PSMA CAR-T cells can reduce tumor volume in mice [67], and there are several ongoing clinical trials investigating the use of CAR-T cell therapy in mCRPC. One phase I trial resulted in stable disease for >6 months in two patients and showed increase in cytokine levels in CAR-T treated patients, suggesting T-cell activation in vivo [68]. In a second phase I trial, five patients received CAR-T cell therapy targeting PSMA with two patients achieving PSA decline >50% [69]. Because prostate cancer cells can secrete transforming growth factor β (TGFβ) to inhibit immune clearance and allow tumor progression, blocking TGFβ signaling in T cells may allow for enhanced antitumor response. Simultaneously giving CAR-T cell therapy against PSMA while blocking TGFβ activity enhanced T-cell proliferation, cytokine secretion, T-cell survival, and efficacy [70]. Clinical trials using PSMA-directed/TGFβ insensitive CAR-T cells are ongoing [71]. Combinations using CAR-T cell therapy with anti-PD1 antibodies also have promising preclinical activity [72, 73], and clinical studies with CAR-T cell combination regimens are anticipated. One major limitation of CAR-T cells is toxicity. In hematologic malignancies, CAR-T treatment has resulted in neurologic toxicity, cytokine release syndrome, cardiac toxicity, tumor lysis syndrome, and macrophage activation syndrome [74]. In theory, the specificity of PSMA for prostatic tissue may limit toxicity, but this will need to be further evaluated in clinical trials.
Drugs to overcome resistance mechanisms
PI3K-Akt-mTOR pathway inhibition
The PI3K-Akt-mTOR pathway plays a critical role in cell growth and survival. About 40–60% of CRPC metastases have functional loss of PTEN, which is an important tumor suppressor, leading to hyperactivation of the PI3K-Akt-mTOR pathway [75, 76]. PTEN loss may also suppress AR transcriptional activity in tumors and has been associated with poor prognosis and poor response to abiraterone acetate [77]. Therefore, inhibition of PI3K-Akt-mTOR pathway with or without combined inhibition of the AR is a rational strategy in mCRPC. PI3K/mTOR inhibitors have been investigated in early phase trials, including dactolisib, apitolisib, and buparlisib, but have showed limited efficacy with poor toxicity profiles [78,79,80,81].
Akt inhibitors have been developed to target this same pathway, with ipatasertib (GDC-0068) currently under investigation in clinical trials. Cancer cells with PTEN protein loss, Akt mutations, and PIK3CA kinase domain mutations have been associated with sensitivity to ipatasertib [82]. A phase I study of ipatasertib demonstrated an acceptable toxicity profile [83], and data from the phase III IPATential150 trial, randomizing 1101 patients with mCRPC to ipatasertib or placebo plus abiraterone, were recently presented at ESMO 2020. Radiographic PFS was prolonged in the ipatasertib plus abiraterone arm in tumors with PTEN loss (HR 0.77, 95% CI 0.61–0.98, p = 0.0335) but not in the overall intention to treat population. Common AEs in the ipatasertib arm included diarrhea (76%), nausea (52%), vomiting (32%), asthenia (27%), and decreased appetite (25%) [84, 85]. This data suggest that PTEN loss may predict for a group of patients that could benefit from the combination of ipatasertib and abiraterone. Given the success of Akt inhibition with ipatasertib in patients with PTEN loss, there is now a phase I clinical trial assessing the use of a PI3K inhibitor AZD8186 with docetaxel in patients with PTEN mutations [86]. Strategies that target the PI3K-Akt-mTOR pathway will require further phase II and phase III clinical trials to evaluate efficacy.
EZH2 inhibition
EZH2 allows for tumor progression through silencing of tumor suppressors and transcriptional modulation. The EZH2 inhibitor GSK126 inhibits prostate cancer cell migration and invasion [87]. EZH2 is upregulated in enzalutamide-resistant prostate cancer cells and inhibition of EZH2 restores the sensitivity of these cells to enzalutamide in vitro. GSK126 in combination with enzalutamide on this resistant tissue led to increased apoptosis and decreased proliferation compared to enzalutamide alone, indicating that it may help overcome resistance to enzalutamide [88]. Accordingly, phase I trials were initiated to investigate the use of small molecule EZH2 inhibitors in combination with either enzalutamide or with abiraterone plus prednisone in mCRPC [89, 90]. The ProSTAR trial using the EZH2 inhibitor CPI-1205 showed >80% reduction in PSA in AR-V7-negative patients with more than half achieving disease control by RECIST 1.1 criteria for at least 3 months. Commonly AEs were diarrhea (31%), fatigue (26%), and nausea (26%). CPI-1205 at 800 mg PO TID with enzalutamide or abiraterone plus prednisone has been selected for phase II expansion [91]. EZH2 is also overexpressed in docetaxel-resistant prostate cancer cells, and suppression of EZH2 activity restores sensitivity to docetaxel [92]. As such, EZH2 could become a therapeutic target for prostate cancer resistant to both chemotherapy and antiandrogen therapy.
Conclusions
There is a need for novel therapeutic strategies to improve patient outcomes in mCRPC. As highlighted, there are many promising agents with different mechanisms that have shown signals of efficacy in early phase trials. PARPi have secured a place in the treatment algorithm for mCRPC for those with HRR alterations and further analysis of combination therapy with androgen-targeted agents or immune-directed therapy could lead to the expanded use of these agents. Studies of checkpoint inhibition have shown durable responses in a subset of patients. An understanding of which patients are more likely to respond to these agents and investigation of combination therapy with other agents will likely expand utilization. Promising preclinical and early phase data surrounding the efficacy and tolerability of AR degraders, such as the SARD ASC-J9 and the PROTAC ARV-110 provide an exciting option for overcoming resistance to current androgen-targeted therapy. The clinical utility of Lu-PSMA should become clear with the results of the phase III VISION trial. BiTE therapy could gain clinical relevance if difficulties surrounding pharmacodynamics can be overcome, and CAR-T cells could provide durable responses in patients with resistant disease depending on tolerability in later phase trials. Lastly, targeting resistance mechanisms will continue to be a prominent therapeutic strategy. The use of Akt inhibitors in patients with PTEN loss will hopefully be efficacious in phase II/III trials, and we await results from the ProSTAR trial to determine the clinical utility of EZH2 inhibition.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. Ca-a Cancer J Clin. 2018;68:7–30.
Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pr. 2011;65:1180–92.
Scher HI, Morris MJ, Stadler WM, et al. Trial design and objectives for castration-resistant prostate cancer: updated recommendations from the prostate cancer clinical trials working group 3. J Clin Oncol. 2016;34:1402–18.
James ND, Spears MR, Clarke NW, Dearnaley DP, De Bono JS, Gale J, et al. Survival with newly diagnosed metastatic prostate cancer in the “docetaxel era”: data from 917 patients in the control arm of the STAMPEDE trial. Eur Urol. 2105;67:1028–38.
Halabi S, Kelly WK, Ma H, Zhou H, Solomon NC, Fizazi K, et al. Meta-analysis evaluating the impact of site of metastasis on overall survival in med with castration-resistant prostate cancer. J Clin Oncol. 2016;34:1652–9.
He L, Fang H, Chen C, Wu Y, et al. Metastatic castration-resistant prostate cancer: academic insights and perspectives through bibliometric analysis. Medicine. 2020;99:e19760.
Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl J Med. 2004;351:1513–20.
Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl J Med. 2004;351:1502–1.
Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders FA, Sternborg CN, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naïve men with metastatic castration resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomized, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16:152–60.
De Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl J Med. 2011;364:1995–2005.
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl J Med. 2012;367:1187–97.
Beer TM, Armstrong AJ, Rathkopf D, Lorit Y, Sternberg CN, Higano CS, et al. Enzalutamide in men with chemotherapy-naïve metastatic castration resistant prostate cancer: extended analysis of the phase 3 PREVAIL study. Eur Urol. 2017;71:151–4.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration resistant prostate cancer. N. Engl J Med. 2010;363:411–22.
Parker C, Nilsson S, Heinrich D, Helle SI, O’Sullivan JM, Fossa SD, et al. Alphe emitter radium-223 and survival in metastatic prostate cancer. N. Engl J Med. 2013;369:213–23.
Tsao CK, Cutting E, Martin J, Oh WK. The role of cabazitaxel in the treatment of metastatic castration-resistant prostate cancer. Ther Adv Urol. 2014;6:97–104.
Attard G, Borre M, Gurney H, et al. Abiraterone alone or in combination with enzalutamide in mestatatic castration-resistant prostate cancer with rising prostate-specific antigen during enzalutamide treatment. J Clin Oncol. 2018;36:2639–46.
Khalaf DJ, Annala M, Taavitsainen S, et al. Optimal sequencing of enzalutamide and abiraterone acetate plus prednisone in mestatatic castration-resistant prostate cancer: a multicenter, randomized, open-label, phase 2, crossover trial. Lancet Oncol. 2019;20:1730–9.
Gillessen S, Attard G, Beer T, et al. Management of pateints with advanced prostate cancer: the report of the Advanced Prostate Cancer Consensus Conference APCCC 2017. Eur Urol. 2018;73:178–211.
De Wit R, de Bono J, Sternberg CN, et al. Cabazitaxel versus abiraterone or enzalutamide in metastatic prostate cancer. N. Engl J Med. 2019;381:2506–18.
Adashek JJ, Jain, RK, Zhang J. Clinical development of PARP inhibitors in treating metastatic castration-resistant prostate cancer. Cells. 2019;8:860.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and Olaparib in metastatic prostate cancer. N. Engl J Med. 2015;373:1697–708.
Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21:162–74.
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl J Med. 2020;382:2091–102.
De Bono, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Final overall survival analysis of PROfound: Olaparib vs physician’s choice of enzalutamide or abiraterone in patients with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations. Ann Oncol. 2020;31(suppl_4):S507–49.
Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et al. Preliminary results from TRITON2: a phase 2 study of rucaparib in patients with Metastatic Castration-Resistant Prostate Cancer (mCRPC) associated with homologous recombination repair (HRR) gene alternations: updated analyses. Ann Onc. 2019;30:327–8.
Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the Phase II TRITON2 Study. Clin Cancer Res. 2020;26:2487–96.
Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018;19:975–86.
ClinicalTrials.gov. National Library of Medicine (U.S.). (2000, February 29–). Study on Olaparib Plus Abiraterone as First-line Therapy in Men With Metastatic Castration-Resistant Prostate Cancer. Identifier: NCT03732820. Retrieved August 4, 2020 from https://clinicaltrials.gov/ct2/show/NCT03732820.
ClinicalTrials.gov. National Library of Medicine (U.S.). (2000, February 29–). TRAP: Targeting Resistant Prostate Cancer With ATR and PARP Inhibition. Identifier: NCT03787680. Retrieved August 4, 2020 from https://clinicaltrials.gov/ct2/show/NCT03787680.
ClinicalTrials.gov. National Library of Medicine (U.S.). (2000, February 29–). Testosterone and Olaparib in Treating Patients With Castration-Resistant Prostate Cancer. Identifier: NCT03516812. Retrieved August 4, 2020 from https://clinicaltrials.gov/ct2/show/NCT03516812.
Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharm. 2017;8:561.
Antonarakis ES, Piulats JM, Gross-Goupil M, Goh J, Ojamaa K, Hoimes CJ, et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label phase II KEYNOTE-199 study. J Clin Oncol. 2020;38:395–405.
Shaffer DR JW, Massard C, et al. A phase Ia study of safety and clinical activity of atezolizumab (atezo) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2018;36:187–187.
Le DT, Uram NJ, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch repair deficiency. N. Engl J Med. 2015;372:2509–20.
Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair–deficient cancer: results from the phase II KEYNOTE-158 study. Am J Clin Oncol. 2020;38:1–10.
Yu E, Piulats JM, Gravis G, Laguerre B, Arija JA, Oudard S, et al. KEYNOTE-365 cohort A updated results: Pembrolizumab (pembro) plus olaparib in docetaxel-pretreated patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2020;38:100.
Massard C, Retz M, Hammerer P, Quevedo F, Fong PC, Berry WR, et al. Keynote-365 cohort b: pembrolizumab (pembro) plus docetaxel and prednisone in abiraterone (abi) or enzalutamide (enza)-pretreated patients (pts) with metastatic castrate resistant prostate cancer (mCRPC). J Clin Oncol. 2019;37:170.
Fong PCC, Retz M, Drakaki A, Massard C, Berry WR, Romano E, et al. Keynote-365 cohort C: pembrolizumab (pembro) plus enzalutamide (enza) in abiraterone (abi)-pretreated patients (pts) with metastatic castrate resistant prostate cancer (mCRPC). J Clin Oncol. 2019;37:171.
Graff J. KEYNOTE-199: pembrolizumab plus enzalutamide for enzalutamide-resistant mCRPC: cohorts 4-5. J Clin Oncol. 2020;38(6_suppl):15.
Graff JN, Beer TM, Alumkal JJ, Slottke RE, Redmond WL, Thomas GV, et al. A phase II single-arm study of pembrolizumab with enzalutamide in men with metastatic castration-resistant prostate cancer progressing on enzalutamide alone. J Immunother Cancer. 2020;8:e000642.
Sweeney CJ, Gillessen S, Rathkopf D, et al. IMbassador250: A phase III trial comparing atezolizumab with enzalutamide vs enzalutamide alone in patients with metastatic castration-resistant prostate cancer (mCRPC). Presented at: American Association for Cancer Research (AACR) Virtual Annual Meeting I 2020; April 27–28, 2020. Abstract CT014.
Beretta GL, Zaffaroni N. Androgen receptor-directed molecular conjugates for targeting prostate cancer. Front Chem. 2019;7:369.
Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11.
Narayanan R, Ponnusamy S, Miller DD. Destroying the androgen receptor (AR)-potential strategy to treat advanced prostate cancer. Oncoscience. 2017;4:175–77.
Bradbury RH, Hales NJ, Rabow AA, Walker GE, Acton DG, et al. Small-molecule androgen receptor downregulators as an approach to treatment of advanced prostate cancer. Bioorg Med Chem Lett. 2011;21:5442–5.
Omlin A, Jones RJ, van der Noll R, Satoh T, Niwakawa M, Smith SA, et al. AZD3514, an oral selective androgen receptor down-regular in patient with castration-resistant prostate cancer – results of two parallel first-in-human phase I studies. Investig N. Drugs. 2015;33:679–90.
Wang R, Sun Y, Li L, Niu Y, Lin W, Lin C, et al. Preclinical study using Malat1 small interfering RNA or andreogen receptor splicing variant 7 degradation enhancer ASC-J9 to suppress enzalutamide-resistant prostate cancer progression. Eur Urol. 2017;72:835–44.
Wang R, Lin W, Lin C, Li L, Sun Y, Change C. ASC-J9 suppresses castration resistant prostate cancer progression via degrading the enzalutamide induced androgen receptor mutant AR-F876L. Cancer Lett. 2016;379:154–60.
Luo J, Tian J, Chou F, Lin C, Xing EZ, Zuo L, et al. Targeting the androgen receptor (AR) with AR degradation enhancer ASC-J9 led to increase docetaxel sensitivity via suppressing the p21 expression. Cancer Lett. 2019;444:35–44.
Gustafson JL, Neklesa TK, Cox CS, Roth AG, Buckley DL, Tae HS, et al. Small molecule mediated degradation of the androgen receptor through hydrophobic tagging. Angew Chem Int Ed Engl. 2015;54:9659–62.
Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang CY, et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J Med Chem. 2019;62:941–64.
Neklesa TK, Snyder LB, Bookbinder M, Chen X, Crew AP, Crews CM, et al. An oral androgen receptor PROTAC degrader for prostate cancer. J Clin Oncol. 2017;35:273.
Petrylak DP, Gao X, Vogelzang NJ, Garfield MH, Taylor I, Moore MD, et al. First-in-human phase I study of ARV-110, an androgen receptor PROTAC degrader in patients with metastatic castrate-resistant prostate cancer following enzalutamide and/or abiraterone. J Clin Oncol. 2020;38(15_suppl):3500.
Novakova Z, Foss CA, Copeland BT, Morath V, Baranova P, Havlinova B, et al. Novel monoclonal antibodies recognizing human prostate specific membrane antigen (PSMA) as research and theranostic tools. Prostate. 2017;77:749–64.
Ahmadzadehfar H, Rahbar K, Kurpig S, Bogemann M, Claesener M, Eppard E. et al. Early side effects and first results of radioligand therapy with (9177)Lu-DKFZ-617 PSMA of castrate-resistant metastatic prostate cancer: a two-centre study. EJNMMI Res. 2015;5:114.
Hofman MS, Violet J, Hicks RJ, Ferdinandus J, Thang SP, Akhurst T, et al. [177 Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): a single-centre, single-arm, phase 2 study. Lancet Oncol. 2018;19:825–33.
Hofman MS, Emmett L, Sandhu SK, Iravani A, Joshua AM, Goh JC, et al. TheraP: a randomized phase II trial of 177Lu-PSMA-617 theranostic versus cabazitaxel in metastatic castration resistant prostate cancer progressing after docetaxel: initial results (ANZUP protocol 1603). J Clin Oncol. 2020;38(15_suppl):5500.
ClinicalTrials.gov. National Library of Medicine (U.S.). (2000, February 29–). Study of 177Lu-PSMA-617 in metastatic castrate resistant prostate cancer (VISION). Identifier NCT03511664. Retrieved August 4, 2020. from https://clinicaltrials.gov/ct2/show/NCT03511664.
Einsele H, Borghaei H, Orlowski RZ, Subklewe M, Roboz GJ, Zugmaier G, et al. The BiTE (Bispecific T-cell engager) platform: development and future potential of a targeted immune-oncology therapy across tumor types. Cancer. 2020;126:3192–201.
Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl J Med. 2017;376:836–47.
Hummel HD, Kufer P, Grullich C, et al. Phase 1 study of pasotuxizumab (BAY 2010112), a PSMA-targeting bispecific T cell engager (BiTE) immunotherapy for metastatic castration-resistant prostate cancer (mCRPC) [abstract]. J Clin Oncol. 2019;37:5034.
Bailis J, Deegen P, Thomas O, Bogner P, Wahl J, Liao M, et al. Preclinical evaluation of AMG 160, a next generation bispecfic T cell engager (BiTE) targeting the prostate-specific membrane antigen PSMA for metastatic castration-resistant prostate cancer (mCRPC) [abstract]. J Clin Oncol. 2019;37:301.
Tran B, Horvath L, Dorff T, et al. Results from a phase I study of AMG 160, a half-life extended, PSMA-tareted, bispecific T cell engager immune therapy for metastatic castration-resistant prostate cancer. Ann Oncol. 2020;31(suppl_4):S507–49.
Yu H, Pan J, Guo Z, Yang C, Mao L. CART cell therapy for prostate cancer: status and promise. Onco Targets Ther. 2019;12:391–5.
Holstein SA, Lunning MA. CAR T-cell therapy in hematologic malignancies: a voyage in progress. Clin Pharm Ther. 2020;107:112–22.
Schepisi G, Cursano MC, Casadei C, Menna C, Altavilla A, Lolli C, et al. CAR-T cell therapy: a potential new strategy against prostate cancer. J Immunother Cancer. 2019;7:258.
Ma Q, Gomes EM, Lo AS, Junghans RP. Advanced generation anti-prostate specific membrane antigen designer T cells for prostate cancer immunotherapy. Prostate. 2014;74:286–96.
Slovin SF, Wang X, Hullings M, Arauz G, Bartido S, Lewis JS, et al. Chimeric antigen receptor (CAR+) modified T cells targeting prostate-specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). J Clin Oncol. 2013;31:72.
Junghans RP, Qiangzhong M, Rathore R, Gomes EM, Bais AJ, Lo AS, et al. Phase I trial of Anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate. 2016;76:1257–70.
Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26:1855–66.
Narayan V, Gladney W, Plesa G, Vapiwala N, Carpenter E, Maude SL, et al. A phase I clinical trial of PSMA-directed/TGFβ-insensitive CAR-T cells in metastatic castration-resistant prostate cancer. J Clin Oncol. 2019;37(7_suppl).
John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636–46.
Sanchez C, Chan R, Bajgain P, Rambally S, Palapattu G, Mims M, et al. Combining T-cell immunotherapy and anti-androgen therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2013;16:123–31.
Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122:4129–39.
Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–86.
Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804.
Ferraldeschi R, NavaRodrigues D, Riisnaes R, Miranda S, Figueiredo I, Rescigno P, et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur Urol. 2015;67:795–802.
Wise-Draper TM, Moorthy G, Salkeni MA, Karim NA, Thomas HE, Mercer CA, et al. A Phase Ib Study of the Dual PI3K/mTOR Inhibitor Dactolisib (BEZ235) combined with everolimus in patients with advanced solid malignancies. Target Oncol. 2017;12:323–32.
Dolly SO, Wagner AJ, Bendell JC, Kindler HL, Krug LM, Seiwert TY, et al. Phase I Study of Apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2016;22:2874–84.
ClinicalTrials.gov. National Library of Medicine (U.S.). (2000, February 29–). Study of Ipatasertib or Apitolisib With Abiraterone Acetate Versus Abiraterone Acetate in Participants With Castration-Resistant Prostate Cancer Previously Treated With Docetaxel Chemotherapy. Identifier: NCT01485861. Retrieved August 4, 2020 from https://clinicaltrials.gov/ct2/show/NCT01485861.
Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Winters C, Kephart J, et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur J Cancer. 2017;81:228–36.
Lin J, Sampath D, Nannini MA, Lee BB, Degtyarev M, Oeh J, et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res. 2013;19:1760–72.
Saura C, Roda D, Rosello S, Oliveira M, Macarulla T, Perez-Fidalgo JA, et al. A first-in-human phase I study of the ATP-competitive AKT inhibitor ipatasertib demonstrates robust and safe targeting of AKT in patients with solid tumors. Cancer Discov. 2017;7:102–13.
De Bono JS, Giorgi UD, Rodrigues DN, Massard C, Bracarda S, Font A, et al. Randomized phase II study evaluating Akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with or without PTEN loss. Clin Cancer Res. 2019;25:928–36.
De Bono J, Bracarda S, Sternberg CN, et al. IPATential150: Phase III study of ipatasertib plus abiraterone vs placebo plus abi in metastatic castration resistant prostate cancer. ESMO; 2020.
ClinicalTrials.gov. National Library of Medicine (U.S.). (2000, February 29–). PI3Kbeta Inhibitor AZD8186 and Docetaxel in Treating Patients Advanced Solid Tumors With PTEN or PIK3CB Mutations That Are Metastatic or Cannot Be Removed by Surgery. Identifier: NCT03218826. Retrieved August 4, 2020 from https://clinicaltrials.gov/ct2/show/NCT03218826.
Lin W, Chen Y, Zen L, Ying R, Zhu F. Effect of a novel EZH2 inhibitor GSK126 on prostate cancer cells. J Zhejiang Univ. 2016;45:356–63.
Bai Y, Zhang Z, Cheng L, Wang R, Chen X, Kong Y, et al. Inhibition of enhancer of zeste homolog 2 (EZH2) overcomes enzalutamide resistance in castration-resistant prostate cancer. J Biol Chem. 2019;294:9911–23.
ClinicalTrials.gov. Study of Tazemetostat together with enzalutamide or with abiraterone/prednisone in subjects with castration resistant prostate cancer that has spread who have not yet received chemotherapy. ClinicalTrials.gov identifier (NCT number): NCT04179864.
ClinicalTrials.gov. ProSTAR: a study evaluating CPI-1205 in patients with metastatic castration resistant prostate cancer. ClinicalTrials.gov Identifier: NCT03480646.
Taplin ME, Hussain A, Shah S, Shore N, Edenfield WJ, Sartor OA, et al. Abstract CT094: Phase IB results of ProSTAR: CPI-1205, EZH2 inhibitor, combined with enzalutamide (E) or abiraterone/prednisone (A/P) in patients with metastatic castration resistant prostate cancer (mCRPC). Proceedings of the American Association for Cancer Research Annual Meeting 2019; Atlanta, GA. Philadelphia (PA): AACR; Cancer Res 2019;79(13 Suppl).
Qui X, Wang W, Li B, Cheng B, Lin K, Bai J, et al. Targeting EZH2 could overcome docetaxel resistance in prostate cancer cells. BMC Cancer. 2019;19:27.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Labriola, M.K., Atiq, S., Hirshman, N. et al. Management of men with metastatic castration-resistant prostate cancer following potent androgen receptor inhibition: a review of novel investigational therapies. Prostate Cancer Prostatic Dis 24, 301–309 (2021). https://doi.org/10.1038/s41391-020-00299-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41391-020-00299-9
- Springer Nature Limited
This article is cited by
-
Evaluation of the tolerability and safety of [225Ac]Ac-PSMA-I&T in patients with metastatic prostate cancer: a phase I dose escalation study
BMC Cancer (2024)
-
Long-term survival outcomes of salvage [225Ac]Ac-PSMA-617 targeted alpha therapy in patients with PSMA-expressing end-stage metastatic castration-resistant prostate cancer: a real-world study
European Journal of Nuclear Medicine and Molecular Imaging (2023)
-
Vulpinic acid as a natural compound inhibits the proliferation of metastatic prostate cancer cells by inducing apoptosis
Molecular Biology Reports (2021)