
Abstract
The RNA helicase DDX21 is vital for ribosome biogenesis and is upregulated in CRC, but the mechanism by which DDX21 is dysregulated and by which DDX21 promotes tumorigenesis in CRC remains poorly understood. Here, we showed that DDX21 is a direct transcriptional target gene of β-catenin and mediates the protumorigenic function of β-catenin in CRC. DDX21 expression is correlated with the expression and activity of β-catenin, and high DDX21 expression is associated with a poor prognosis in CRC patients. Loss of DDX21 leads to cytoplasmic translocation and decreased transcriptional activity of YAP and suppresses the proliferation and migration of CRC cells, which can be partially rescued by YAP reactivation. Importantly, by using translation elongation inhibitors and DNA intercalators, we showed that ribosomal stress upregulates DDX21 expression and induces the downregulation of LATS and the activation of YAP, probably through the ZAKα-MKK4/7-JNK axis. Overall, our study revealed the transcriptional activation mechanism of DDX21 in CRC and the activation of YAP in the ribosomal stress response, indicating the potential of combination therapy involving the induction of ribosomal stress and YAP inhibition.
Similar content being viewed by others

Introduction
DEAD-box RNA helicases regulate the formation of RNA structures and play a vital role in various biological processes, including ribosome biogenesis, transcription and translation [1]. DDX21 is a nucleolar DEAD-box RNA helicase family member and was first identified as a transcriptional co-factor for c-JUN-dependent transcription [2]. Recent studies have elucidated the mechanism by which DDX21 coordinates gene transcription and ribosomal RNA processing and revealed the crucial role of DDX21 in multiple steps of ribosome biogenesis [3,4,5]. In addition, DDX21 can resolve genomic R loops and cooperate with SIRT7 to maintain genome stability during gene transcription [6]. Interestingly, DDX21 only facilitates double-strand break (DSB) repair at an appropriate expression level. Aberrantly high expression of DDX21 delays homologous recombination (HR) repair and results in genome instability and subsequent tumorigenesis [7]. Pancancer analysis revealed that the expression of the DEAD-box RNA helicase DDX21, which is prominently upregulated in CRC at the mRNA level, promotes CRC metastasis and tumor growth [8,9,10,11]. However, the transcriptional activation mechanism underlying the upregulation of DDX21 mRNA and the downstream effectors of DDX21 in tumorigenesis and tumor progression in CRC remain poorly understood.
The Hippo pathway is a conserved signaling pathway involved in organ size control and tissue homeostasis in mammals [12]. The transcriptional coactivators YAP/TAZ are the major downstream effectors of the Hippo pathway. Dysregulated activation of YAP/TAZ leads to tumorigenesis and an invasive phenotype in various cancers, including CRC [13]. YAP/TAZ can modulate gene transcription and cell behaviors in response to multiple cellular stimuli and stresses, such as osmotic stress, energy stress, ER stress, oxidative stress and heat stress [14,15,16,17]. Ribosomal stress, also referred to as ribotoxic stress, describes the impairment of ribosomal function and biogenesis that ultimately results in the activation of various stress signals, including P53, the ribotoxic stress response (RSR), ribosome-associated quality control (RQC) and the integrated stress response (ISR) [18]. The RSR is a mitogen-activated protein (MAP) kinase signaling cascade initiated by the MAP3K20 kinase ZAKα and activates the stress-associated MAP kinases p38 and JNK [18]. In intestinal stem cells, nutrient deficiency-induced activation of ZAKα kinase activates YAP by enhancing Tyr416 phosphorylation by SRC, ultimately transforming adult ISCs into fetal-like states [19]. However, the biological relevance between the RSR and the Hippo pathway and the regulatory effect of ZAKα on YAP activity in CRC remains obscure.
Wnt signaling is crucial for intestinal homeostasis and the maintenance of intestinal stem cells [20]. Mutations in Wnt pathway tumor suppressors or activating mutations in the CTNNB1 gene, which encodes the transcriptional coactivator β-catenin and the main downstream effector of Wnt signaling, drive the development and tumor progression of CRC by transcriptionally modulating T-cell factor/lymphoid enhancer (TCF/LEF) downstream target genes [20, 21]. In this study, we report that DDX21 is a direct transcriptional target gene of Wnt/β-catenin signaling and is required for the oncogenic function of β-catenin in CRC. The upregulation of DDX21 is positively correlated with the transcriptional activity of β-catenin and increased expression of β-catenin in CRC and is associated with a poor prognosis in CRC patients. Inhibition of DDX21 suppresses the transcriptional activity and oncogenic function of YAP in CRC. Importantly, chemical-induced ribosomal stress activates YAP, partially through the ZAKα-MKK4/7-JNK axis. Thus, our study revealed that β-catenin transcriptionally activates DDX21 in CRC and that YAP/TEAD is activated in response to ribosomal stress, highlighting the therapeutic potential of combination strategies targeting the nucleolar DDX21 and YAP/TEAD in CRC.
Results
DDX21 is transcriptionally activated by β-catenin in CRC cells
As a driver of CRC tumorigenesis and an attractive therapeutic target for CRC treatment, activation of β-catenin promotes the transcription of a series of oncogenes. By re-analyzing the public RNA-seq and microarray datasets of Apc-deficient mice, we found that Ddx21 is transcriptionally upregulated in the intestinal epithelium of Apc-deficient mice, which suggested that β-catenin might transcriptionally activate DDX21 in CRC cells [22, 23] (Fig. 1A). First, we confirmed that DDX21 expression was upregulated in Apcmin/+ mice compared with that in their WT littermates (Fig. 1B, C). In two CRC cell lines with active β-catenin expression, HCT-116 (with an activated β-catenin mutation) and SW480 (with a truncated APC), we found that both the mRNA and protein levels of DDX21 were significantly decreased in β-catenin knockdown HCT-116 and SW480 cells vs. control cells (Fig. 1D, E). Consistently, transient overexpression of Flag-β-catenin moderately increased the mRNA and protein levels of DDX21 in HEK-293T cells and the normal intestinal epithelial cell line HIEC-6 in a dose-dependent manner (Fig. 1F, G). Next, we sought to determine whether β-catenin directly modulates DDX21 gene transcription. The publicly available ChIP-seq datasets of TCF7L1/TCF7L2 revealed that TCF7L1/TCF7L2, which are β-catenin binding partners, could bind to the promoter region of DDX21 in multiple cell lines (Fig. 1H). The ChIP‒qPCR results further confirmed that β-catenin bound to a similar promoter region of DDX21 and the known target gene CCL2 in HCT-116 cells (Fig. 1I) [24]. We further generated a luciferase reporter of the DDX21 promoter and observed that overexpression of β-catenin induced up to 4-fold activation of DDX21 luciferase activity in a dose-dependent manner (Fig. 1J). In addition, bioinformatic analysis via JASPAR identified a possible binding site for TCF7L1/TCF7L2 in the DDX21 promoter (Fig. 1K). Mutation of the TCF7L1/TCF7L2 binding motif largely abolished the activation of the DDX21 luciferase reporter by β-catenin overexpression (Fig. 1L). Taken together, these data suggest that DDX21 is a direct transcriptional target gene of β-catenin in CRC cells.

A Ddx21 is transcriptionally upregulated in the intestinal epithelium in mice with Apc deficiency and β-catenin activation. CTNNB1ex3/ex3: CTNNB1 exon 3 mutation produces a constitutively active mutant of β-catenin. The data were obtained from the Expression Atlas database (www.ebi.ac.uk/gxa). B, C Western blot analysis (B) and representative images of IHC staining (C) of DDX21 and β-catenin in colon tissues harvested from ApcMin/+ mice and their WT littermates. D, E qPCR (D) and western blot (E) analysis of DDX21 and β-catenin in the β-catenin-active CRC cell lines HCT-116 and SW480 with CTNNB1 knockdown. F, G qPCR (F) and western blot (G) analysis of DDX21 and β-catenin in HIEC-6 and HEK-293T cells overexpressing FLAG-β-catenin. H Representative sequencing tracks of the TCF7L1/L2 ChIP-seq data at the DDX21 genomic locus. The data were extracted from the Cistrome database (www.cistrome.org/db/). I ChIP analysis of β-catenin binding to the DDX21 promoter in HCT-116 cells. J Overexpression of β-catenin increased the activity of the DDX21 luciferase reporter. The DDX21 luciferase reporter and control pGL3-vector were cotransfected with increasing amounts of FLAG-β-catenin plasmid (0/50/100/150 ng per well). K Schematic depiction of the DDX21 gene locus showing the potential TCF7L1/L2 binding sites and the corresponding DNA sequence of the mutant DDX21 luciferase reporter. BS: binding site; TSS: transcription start site. L The increase in DDX21 luciferase activity induced by β-catenin was attenuated by mutation of the TCF7L1/L2 binding site. One-way ANOVA with Dunnett’s multiple comparison test and two-tailed Welch’s t test were performed to assess statistical significance for the experiments with >2 groups and 2 groups, respectively, in this figure. One-way ANOVA with Tukey’s multiple comparisons test was used to assess statistical significance in (l). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Correlation and clinical implications of the β-catenin/DDX21 axis in CRC
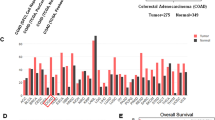
Then, we analyzed the publicly available TCGA CRC dataset and found that the DDX21 mRNA level was positively correlated with the mRNA levels of classical Wnt/β-catenin target genes (CD44, AXIN2, MYC and CCND1), as well as with the mRNA level of CTNNB1 (a gene encoding β-catenin) (Fig. 2A, B). Consistently, according to analysis of the CCLE database, DDX21 mRNA expression was upregulated in CTNNB1-mutant CRC cell lines compared with CTNNB1-WT CRC cell lines (Fig. 2C). Next, we confirmed the high mRNA levels of both DDX21 and CTNNB1 in 40 paired CRC patient tissues (Fig. S1A, B). Similar results were observed in the WB analysis of 24 paired CRC patient tissues (Fig. 2F and S1C). Consistent with the observations in the TCGA dataset, DDX21 expression was positively correlated with CTNNB1 expression in these patients’ cancer tissues at both the mRNA and protein levels (Fig. 2D, G). In addition, the relative increase in the mRNA level of DDX21 (mRNA level in tumors—mRNA level in paired normal mucosa) was also positively correlated with that of CTNNB1 in our cohort (Fig. 2E).

A, B Positive correlations between the mRNA level of DDX21 and the expression levels of canonical WNT/β-catenin downstream target genes (CD44/AXIN2/MYC/CCND1) (A) and CTNNB1 (B) in CRC. The data originated from the TCGA CRC datasets and were analyzed via the GEPIA database (gepia.cancer-pku.cn). The Pearson correlation coefficient was used to assess correlations. C Comparison of the relationship between CTNNB1 hotspot mutations and DDX21 mRNA levels in 71 CRC cell lines. The data were extracted from the DepMap database (depmap.org). Two-tailed Welch’s t test was performed to assess statistical significance. D The mRNA expression levels of CTNNB1 and DDX21 in 40 pairs of CRC and adjacent normal tissues were determined by RT‒qPCR analysis. Pearson’s correlation test was used to evaluate correlations. Dotted lines represent 95% CIs (confidence intervals). E The correlation between increased mRNA levels (mRNA level in tumors—mRNA level in paired normal mucosa) of DDX21 and CTNNB1 in tumors. Pearson’s correlation test was used to examine correlations. F Western blot analysis of β-catenin and DDX21 protein levels in 24 pairs of CRC and paired adjacent normal tissues. G Pearson’s correlation test was used to evaluate the correlation between the protein levels of β-catenin and DDX21 in 24 pairs of CRC tissues and paired adjacent normal tissues.
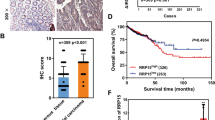
To further assess the clinical relevance of the β-catenin/DDX21 axis in CRC, we performed immunochemical staining of DDX21 and β-catenin in CRC tissue arrays consisting of 169 paired CRC and normal tissues. Representative images of DDX21 and β-catenin at different protein levels are shown in Fig. 3A, C. Consistent with other reports, the protein levels of DDX21 were increased in CRC tissues (Fig. 3B). Furthermore, DDX21 protein levels were significantly correlated with β-catenin protein levels in CRC tissues (Fig. 3D). Next, we evaluated the correlation between DDX21 expression and clinical characteristics in CRC patients (Supplemental Table 1). However, no significant association between high DDX21 expression and T stage, liver metastasis, serum ALB level, or hemoglobin level was found (Supplemental Table 2). However, increased expression of DDX21 was significantly correlated with tumor site (p = 0.003), N stage (p = 0.046), histology (p < 0.001) and increased serum carcinoembryonic antigen (CEA) levels (p = 0.032) (Supplemental Table 2). Both univariate analysis and multivariate Cox regression hazard analysis showed the high expression of DDX21 was associated with poor prognosis (Supplement Table 3). Moreover, Kalan Meier analysis revealed that high protein expression of DDX21 was associated with shorter disease-free survival (DFS) and overall survival (OS) in CRC patients, indicating that it is a marker of a poor prognosis in CRC patients (Fig. 3E, F). In addition, we observed that 28.9% of the CRC tissues exhibited high expression of both DDX21 and β-catenin, and the patients classified as DDX21high/β-cateninhigh had the worst disease outcomes (Fig. 3G, H). These data suggest that activation of the β-catenin/DDX21 axis might promote tumor progression in CRC.

A Representative images of IHC staining of DDX21 protein in 169 pairs of normal and CRC tissues. B Quantitative analysis of 169 pairs of normal and CRC tissues based on the DDX21 IHC score. The Wilcoxon signed-rank test was used to assess statistical significance. C Representative images of IHC staining of β-catenin in 169 pairs of normal and CRC tissues. D There was a positive correlation between the protein levels of β-catenin and DDX21 in 169 pairs of normal and CRC tissues. Spearman correlation test was used to evaluate the association. E, F Kaplan–Meier plots of the disease-free survival (DFS) (E) and overall survival (OS) (F) of CRC patients stratified by the DDX21 protein level. Statistical significance was assessed by a two-sided log-rank test. G, H Kaplan–Meier plots of the DFS (G) and OS (H) of CRC patients stratified by the protein levels of DDX21 and β-catenin according to two-sided log-rank tests. According to the protein expression levels of the patients, the 169 paired patients were divided into three groups: the group with low expression of both DDX21 and β-catenin (green), the group with high expression of both DDX21 and β-catenin (red) and the group with other samples (yellow).
DDX21 mediates the protumorigenic function of β-catenin in CRC
Consistent with previous studies, by using CCK-8 and colony formation assays, we confirmed the oncogenic function of DDX21 in two CRC cell lines (HCT-116 and RKO) (Fig. 4A, B, S2A). Similar results were observed in the Transwell and wound healing assays in vitro and in the xenograft assay in vivo (Fig. 4C–F, S2B). We also examined the effect of DDX21 knockdown on cell proliferation and colony formation in two normal intestinal epithelial cell lines (HIEC-6 and NCM460) which expressed less DDX21 compared with HCT-116 cells (Figs. S2C, S2D and S2G). Interestingly, contrast to the strong inhibitory effect on cell proliferation and colony formation in HCT-116 cells, knockdown of DDX21 mildly suppressed cell proliferation and colony formation in HIEC-6 cells and NCM460 cells, probably due to the lower expression level of DDX21 in the normal IECs (Fig. S2E, S2F, S2H, S2I).

A CCK-8 analysis of HCT-116 and RKO cells with DDX21 knockdown. B Colony formation assays of HCT-116 and RKO cells with DDX21 knockdown. Scale bars, 35 mm. C Transwell assay showing the migration ability of HCT-116 and RKO cells with DDX21 knockdown. D Wound healing assay of the cell migration ability of HCT-116 cells with DDX21 knockdown. Scale bars, 200 µm. E Representative images of DDX21-knockdown HCT-116 xenograft tumors (n = 6). The weight of each xenograft was used for statistical analysis. F Representative images of IHC staining of the proliferation markers Ki-67 and DDX21 in xenograft tumors derived from HCT-116 cells with DDX21 knockdown. G, H CCK-8 (G) and colony formation (H) assays showing the proliferation and colony formation of CTNNB1-overexpressing HCT-116 cells in the presence or absence of DDX21 knockdown. Scale bars, 35 mm. I Transwell assay of the cell migration ability of CTNNB1-overexpressing HCT-116 cells in the presence or absence of DDX21 knockdown. One-way ANOVA with Dunnett’s multiple comparison test was performed to assess statistical significance in B–F and one-way ANOVA with Tukey’s multiple comparison test was used for statistical analysis in H, I. Data were analyzed by two-way ANOVA with Dunnett’s multiple comparison test in A, G.
To explore whether DDX21 mediates the protumorigenic function of β-catenin in CRC cells, we established HCT-116 cells with stable expression of β-catenin and knockdown of DDX21 (Fig. S3A). We observed that overexpression of β-catenin led to upregulated expression of DDX21 and enhanced cell proliferation, colony formation and cell migration, while knockdown of DDX21 in β-catenin-overexpression cells largely attenuated the enhanced cell proliferation, colony formation and migration induced by overexpression of β-catenin (Fig. 4G–I). Both β-catenin and DDX21 are known to play a vital role in promoting ribosome biogenesis. In consistent to the above observations, we observed that the direct target genes of DDX21, such as RPL7A, RPS24, scaRNA10 and snoRNA62, were upregulated by β-catenin overexpression while downregulated by knockdown of CTNNB1 (Figs. S3B, S3C). More importantly, the upregulated ribosome biogenesis related genes by β-catenin overexpression were abrogated by DDX21 knockdown (S3D). Of note, we found the high mRNA levels of both RPL7A and RPS24 in 40 paired CRC patient tissues, which was positively correlated with the mRNA level of DDX21 (Fig. S3E, S3F). Furthermore, we also established HCT-116 cells with stable expression of shRNA targeting CTNNB1 (Fig. S3G). We observed that downregulation of β-catenin led to reduced expression of DDX21 and attenuated cell proliferation, colony formation and cell migration, while re-expression of DDX21 in CTNNB1 knockdown cells partially attenuated the suppression of cell proliferation, colony formation and migration (Fig. S3H–J). Dependency maps have recently been widely used to identify functionally interacting genes on the basis that a panel of cancer cells whose proliferation is dependent on β-catenin should also be vulnerable to knockdown of DDX21. To this end, we analyzed the Cancer Dependency Map (DepMap) database and found that knockdown of both DDX21 and CTNNB1 suppressed cell proliferation in 46 CRC cell lines (gene effect score<0) (Fig. S3K). In addition, the DDX21 dependency score was positively correlated with the CTNNB1 dependency score (Fig. S3K). Overall, the codependency of DDX21 and CTNNB1 further suggests that DDX21 is a key downstream target gene of β-catenin and plays a vital role in β-catenin-induced tumorigenesis in CRC.
DDX21 promotes the oncogenic function of YAP in CRC tumorigenesis
Previous studies have shown that, as an important component of the 7SK snRNP, DDX21 modulates RNA secondary conformation and ribosomal RNA (rRNA) synthesis and can promote transcriptional elongation [25,26,27]. To explore the transcriptional effect of DDX21 in CRC cells, RNA-seq analysis was performed on DDX21-knockdown and control HCT-116 cells. Sixty-nine genes were downregulated, while 118 genes were upregulated in the DDX21-knockdown HCT-116 cells compared with the control cells (log2FC > 1, p < 0.05) (Supplemental Table 4). Intriguingly, gene set enrichment analysis (GSEA) revealed that the YAP target gene signature was enriched among the differentially expressed genes in DDX21-knockdown HCT-116 cells (Fig. 5A,B, S4A). qPCR analysis further confirmed decreased mRNA levels of classical YAP target genes, such as CTGF, CYR61, THBS1 and AXL, in DDX21-knockdown HCT-116 and RKO cells (Fig. 5C–E). In contrast, stable overexpression of DDX21 increased the expression of YAP target genes (Fig. 5F). Consistent with the above results, western blot analysis revealed increased levels of phosphorylated YAP-Ser127 in DDX21-knockdown cells (Fig. S4B). Similarly, we observed diffuse YAP staining in the cytoplasm of DDX21-knockdown HCT-116 cells, which reflected the cytoplasmic translocation of YAP (Fig. 5G). Of note, we didn’t detect the interaction between DDX21 and YAP/TAZ/TEAD complex (data not shown). Thus, though YAP has been reported to control transcriptional elongation, DDX21 might not directly participate in transcriptional regulation of YAP/TEAD target genes. Activation of YAP frequently occurs in various cancers, including colorectal cancer [28,29,30]. Therefore, we explored whether YAP mediates the protumorigenic effect of DDX21 in CRC. To this end, we established DDX21-knockdown HCT-116 cells overexpressing constitutively active YAP5SA (Fig.S4C). As expected, the suppression of cell proliferation, colony formation and cell migration induced by DDX21 knockdown was reversed by the overexpression of constitutively active YAP5SA in HCT-116 cells (Fig. 5H–K, S4D). In addition, we established DDX21-overexpressing HCT-116 and HIEC-6 cells with or without YAP knockdown (Fig. S4E, S4J). As expected, knockdown of YAP abolished the upregulated expression of YAP target genes induced by DDX21 overexpression (Fig. S4F). Consistently, the enhanced cell proliferation, colony formation and cell migration induced by DDX21 overexpression was dramatically diminished by knockdown of YAP in both HCT-116 and HIEC-6 cells (Fig. S4G–I, S4K and S4L). Taken together, these data indicate that DDX21 plays a protumorigenic role in CRC, likely by activating YAP, the downstream effector of the Hippo pathway.

A, B Blue pink O’Gram of core enriched genes generated by GSEA according to CORDENONSI_YAP_CONSERVED_SIGNATURE (A). GSEA enrichment plots showing that the upregulated genes in control siNC HCT-116 cells were enriched in CORDENONSI_YAP_CONSERVED_SIGNATURE (B). C Quantitative qPCR analysis of canonical YAP target genes in HCT-116 cells transfected with control siRNA or siRNA targeting DDX21. D, E qPCR analysis of CTGF, CYR61 and THBS1 mRNA levels in stable DDX21-knockdown HCT-116 (D) and RKO (E) cell lines. F qPCR analysis was performed to determine the CTGF, CYR61, and THBS1 mRNA levels in HCT-116 cells stably overexpressing DDX21. G Immunofluorescence analysis of DDX21 (green) and YAP (red) in HCT-116 cells. Nuclei were stained with DAPI (blue). HCT-116 cells were transfected with NC siRNA or DDX21 siRNA for 48 hr before processing for immunofluorescence staining. N, nucleus; C, cytoplasm. Scale bars, 10 µm. H. I CCK-8 (H) and colony formation (I) analyses of the proliferation and colony formation of DDX21-knockdown HCT-116 cells in the presence or absence of YAP5SA overexpression. J, K Transwell (J) and wound healing (K) assays showing the migration of DDX21-knockdown HCT-116 cells in the presence or absence of YAP5SA overexpression. Scale bars, 200 µm. One-way ANOVA with Dunnett’s multiple comparison test was performed to assess statistical significance in C–E, G. One-way ANOVA with Tukey’s multiple comparison test was performed to assess statistical significance in I–K. Two-tailed Welch’s t test was performed to assess statistical significance in f. Data were analyzed by two-way ANOVA with Dunnett’s multiple comparison test in H.
Chemical-induced ribosomal stress upregulates DDX21 and activates YAP
DDX21 is a nucleolar RNA helicase that controls multiple steps of ribosome biogenesis and is required for pre-rRNA processing [4, 26]. Impaired ribosome biogenesis has been shown to induce ribosomal stress; thus, we sought to determine whether there is a relationship between the hippo signaling pathway and the ribosomal stress response. We used two DNA intercalators (doxorubicin and actinomycin D) and two protein translation inhibitors (cycloheximide and puromycin) to induce ribosomal stress in CRC cells. These agents all lead to impaired ribosome function through different mechanisms and are well recognized as ribosomal stress inducers [18, 31,32,33]. By immunofluorescence, we found that treatment with the four ribosomal stress inducers led to the nuclear translocation of YAP in a time-dependent manner in serum-starved HCT-116 and RKO cells (Fig. 6A, S5A). Consistently, qPCR analysis of the well-known YAP target genes CTGF, CYR61 and THBS1 revealed the upregulation of YAP target genes, which further indicated enhanced YAP transcriptional activity upon treatment with ribosomal stress inducers (Fig. 6B, S5B). To further assess the dynamic modulation of YAP in response to ribosomal stress, we performed western blot analysis of YAP and its upstream kinase LATS1/2. As a positive control of ribosomal stress, we observed that all four ribosomal stress inducers increased the levels of p-JNK and p-p38 in a time-dependent manner (Fig. 6C). In addition, the level of p-YAP (Ser127) decreased, while the levels of the upstream kinases LATS1/2 decreased in a similar time-dependent manner (Fig. 6C). Actinomycin D can cause translocation of DDX21 from the nucleolus to the cytoplasm. Notably, upon treatment with actinomycin D, doxorubicin, cycloheximide and puromycin, the protein level of DDX21 was markedly increased, while the level of the DDX21 mRNA decreased and the mRNA level of YAP was steady (Fig. 6C, S6A and S6B). In contrast, serum stimulation also led to increased p-JNK and decreased p-YAP levels but had minimal effect on DDX21 protein levels (Fig. 6C). Next, we explored whether upregulated DDX21 played a role in YAP activation in response to chemical-induced ribosomal stress. Consistent to the previous reports, treatment of the DNA intercalators (doxorubicin and actinomycin D) but not the protein translation inhibitors (cycloheximide and puromycin) led to nucleoplasmic translocation of DDX21 from nucleoli (Fig. S6C). Indeed, knockdown of DDX21 partially blocked the nuclear translocation of YAP and transcriptional activation of YAP target genes, which suggested that upregulation of DDX21 is involved in YAP activation during the chemical-induced ribosomal stress (Fig. S6C, S6D). Taken together, these data suggest that ribosomal stress increases the protein level of DDX21 and increases YAP activity, which might be a protective cellular response to stress.

A Immunofluorescence analysis of YAP (red) in HCT-116 cells. Nuclei were stained with DAPI (blue). HCT-116 cells were seeded for 24 h at a low cell density before overnight serum starvation. Then, the cells were treated with doxorubicin (HY-15142, MCE, working concentration: 2 uM), actinomycin D (S8964, Selleck, 20 nM), cycloheximide (239764, Sigma‒Aldrich, 75 µg/ml), and puromycin (A610593, Sangon Biotech, 2 µg/ml) for the indicated times. Scale bars, 10 µm. One-way ANOVA was performed to assess the statistical significance. B Chemical-induced ribosomal stress triggers the upregulation of YAP target genes in HCT-116 cells. The mRNA levels of CTGF, CYR61, and THBS1 were determined by qPCR in HCT-116 cells treated with the indicated chemicals. One-way ANOVA with Dunnett’s multiple comparison test was performed to assess the statistical significance of the differences among the groups. C Western blot analysis of the protein levels of DDX21 and core components of the Hippo pathway in chemically treated HCT-116 cells.
Chemical-induced ribosomal stress activates YAP via the ZAKα-MKK4/7-JNK axis
The RSR is a MAP kinase cascade, and the upstream kinase, the alpha isoform of ZAK (ZAKα), activates downstream stress-activated protein (SAP) kinases, such as p38 and JNK, in response to impaired ribosome biogenesis [34]. Next, we explored whether ZAKα was involved in YAP activation by ribosomal stress and observed that specific knockdown of ZAKα partially reversed the nuclear translocation of YAP and gene transcription of YAP target genes upon treatment with four ribosomal stress inducers (Figs. 7A, B and S7A). The MAP2K kinases MKK3/6 and MKK4/7 are known to be directly activated by ZAKα in response to ribosomal stress. Intriguingly, knockdown of MKK4/7 but not MKK3/6 partially attenuated the nuclear translocation of YAP and increased the mRNA levels of YAP target genes induced by the four ribosomal stress inducers used in this study (Figs. 7C, D and S7B–E). Furthermore, knockdown of either ZAKα or MKK4/7 attenuated the decrease in the p-YAP level, the decrease in the LATS1/2 protein level and the increase in the DDX21 protein level (Fig. 7E). MKK4/7 primarily activate JNK but not p38 kinases. As expected, a similar suppressive effect of the JNK inhibitor SP600125 was observed on the YAP activation induced by actinomycin D, doxorubicin, cycloheximide and puromycin (Fig. S8A–E). The p38 inhibitor SB-203580 had a minimal effect on the YAP activation induced by ribosomal stress (Fig. S8A–E).

A, C Ribosomal stress-induced YAP nuclear translocation was attenuated upon knockdown of ZAKα (A) and MKK4 + 7 (C). Immunofluorescence analysis of chemical-induced YAP (red) nuclear translocation in HCT-116 cells. HCT-116 cells were transfected with the indicated siRNAs and seeded at a low cell density two days later. The cells were serum-starved overnight and then treated with the indicated drugs for 3 hours before subsequent immunofluorescence analysis. Scale bar, 10 μm. B, D Knockdown of ZAKα (C) and MKK4 + 7 (D) suppressed the upregulation of YAP target genes in HCT-116 cells treated with the indicated drugs. Two-tailed Welch’s t tests were performed to assess the statistical significance of the differences among the experiments. E Western blot analysis of the protein levels of DDX21 and core components of the Hippo pathway in chemically treated HCT-116 cells with ZAKα and MKK4 + 7 knockdown. F A schematic representation of the findings in this study. Two-tailed Welch’s t tests were performed to assess statistical significance of the differences in this figure.
Chemotherapies usually inhibit ribosome biogenesis, thus inducing the RSR [18]. Our finding that ribosomal stress activates YAP led us to hypothesize that the combined use of chemotherapeutic agents that induce ribosomal stress and YAP-TEAD inhibitors might lead to synergistic inhibition of cancer cell survival. To test this hypothesis, we treated two CRC cell lines with the clinically used chemotherapeutic agent doxorubicin in combination with verteporfin, a compound that disrupts the YAP-TEAD interaction. Interestingly, doxorubicin and verteporfin had synergistic inhibitory effects on the proliferation of HCT-116 and RKO cells, as indicated by the combination index (CI) values (CI < 1) (Fig. S9A). Treatment with the combination of doxorubicin and verteporfin resulted in more significant inhibition of cell proliferation than treatment with either single agent in both the CCK-8 and colony formation assays (Fig. S9A, S9B). Additionally, we assessed the inhibitory effect of combination treatment in the normal intestinal epithelial cells. Intriguingly, both doxorubicin and verteporfin showed reduced inhibitory effect on cell proliferation and colony formation in HIEC-6 cells (Fig. S9C, S9D). Furthermore, we didn’t observe a synergistic inhibitory effect of combination treatment in normal HIEC-6 cells (Fig. S9C, S9D). Taken together, these results indicate that ribosomal stress can activate YAP through the ZAKα-MKK4/7-JNK axis and that inhibiting YAP activity can increase the antitumor effect of chemotherapeutic agents that can induce a ribosomal stress response in CRC.
Discussion
Wnt signaling is vital for the maintenance and self-renewal of intestinal stem cells [21]. Dysregulation of the Wnt pathway leads to various serious cancers, including CRC. In tumor cells, hyperactivation of β-catenin can activate TCF/LEF-mediated gene transcription to promote cell proliferation and cell growth. Recent findings have indicated that Wnt/β-catenin plays a vital role in enhancing ribosome biogenesis to support tumor cell growth, mainly through its classic downstream target gene MYC, which regulates ribosome biogenesis via ribosome-related RNA Pol I-II-III gene transcription [35,36,37]. In this study, we identified DDX21 as a new direct target gene of β-catenin that might also mediate the role of β-catenin in ribosome biogenesis during colorectal tumorigenesis. In addition, copy number amplification has been shown to be involved in the upregulation of DDX21 in CRC [7]. Our findings further demonstrated that activation of β-catenin could account for the increased mRNA level of DDX21 in CRC. Notably, the decrease in the DDX21 protein level upon CTNNB1 knockdown was more dramatic than the decrease in the DDX21 mRNA level. Moreover, we found that ribosomal stress promoted DDX21 protein expression but not DDX21 mRNA expression. Thus, we hypothesize that a posttranslational mechanism might also play a crucial role in modulating DDX21 expression during CRC progression and the ribosomal stress response, which needs to be explored in future studies.
In addition to playing a vital role in tumor cell growth by controlling ribosome biogenesis, DDX21 can also promote cell proliferation and tumor metastasis in CRC by transcriptionally activating CDK1 and MCM5, respectively [9, 38]. In addition, DDX21 can directly interact with a cell cycle-related gene, cell division cycle 5-like (CDC5L), to regulate cell cycle arrest and CRC cell proliferation [39]. Our finding that DDX21 promotes YAP nuclear translocation in CRC indicates that activation of YAP also contributes to the protumorigenic function of DDX21 in CRC. In the nucleoplasm, DDX21 can facilitate Pol II-dependent gene transcription as a canonical 7SK snRNP factor, and DDX21 is known to be a transcriptional cofactor for c-JUN-activated transcription [2, 4]. Notably, AP1 transcription factors (c-JUN, JUND) cooperate with YAP/TEAD to activate gene transcription [40]. Thus, it is likely that DDX21 also coordinates with c-JUN to modulate YAP/TEAD transcriptional activity in CRC.
Actinomycin D is one of the best-studied rRNA synthesis inhibitors and preferentially inhibits RNA Pol I-dependent transcription at low concentrations [33]. Although doxorubicin is known as a DNA topoisomerase II inhibitor, it can also inhibit rDNA transcription and induce nucleolar disruption [32, 33]. Notably, recent studies have reported that the activation of YAP is involved in doxorubicin-induced cardiomyopathy during chemotherapy, but the underlying biological relevance and mechanism have not been revealed [41, 42]. In addition to perturbations in ribosome biogenesis, protein translation inhibitors also result in ribosomal stress. Both puromycin and cycloheximide, which are widely used protein synthesis inhibitors, can abrogate the mRNA translation process and lead to ribosome stalling and collision, which trigger a series of stress responses, including the RSR [18, 31]. Although the four compounds used in this study could have broad effects on cells, here, we revealed that these agents promoted the nuclear translocation of YAP and activated YAP, which suggests that YAP is activated in response to ribosomal stress. Importantly, perturbation of the RSR pathway and the ZAKα-MKK4/7-JNK axis suppressed the nuclear translocation and activation of YAP induced by these four ribosomal stress inducers, which further supports the role of ribosomal stress in positively regulating YAP. Previous studies have revealed that JNK can activate YAP by phosphorylating the AJUBA family protein LIMD1 and promoting the binding of LIMD1 to LATS1 [43, 44]. Furthermore, ribosomal stress induced by both DDX21 knockdown or inhibitor treatment decreased the protein levels of LATS1/2, which could also account for the decreased expression of p-YAP and nuclear translocation of YAP upon ribosomal stress. However, we observed increased p-YAP and downregulation of LATS1/2 in DDX21 knockdown CRC cells. This discrepancy could be due to the long-term ribosomal stress induced by DDX21 knockdown or the possibility that DDX21 might modulate YAP phosphorylation independent of LATS1/2. Notably, ribosomal stress is known to promote DDX21 translocation from the nucleolus to the nucleoplasm, and we noticed that ribosomal stress also upregulated DDX21 expression in CRC cells [2, 45]. Thus, the upregulation of DDX21 might also anticipate YAP activation during the ribosomal stress response. However, further studies are needed to fully elucidate the mechanism by which DDX21 regulates YAP. Cancer cells increase ribosome biogenesis to support sustained proliferation, so inhibition of ribosome biogenesis is considered a promising strategy for cancer treatment [46]. Various clinically approved chemotherapeutic agents have been shown to interfere with ribosome biogenesis [18, 47]. Both p53-dependent and p53-independent stress responses are involved in ribosome biogenesis inhibition-induced cell cycle arrest and apoptosis [18, 47]. However, a recent study indicated that the Wnt/β-catenin pathway can be activated by ribosomal stress, which is thought to be a compensatory mechanism to sustain ribosome biogenesis [48]. Moreover, intestinal loss of Apc attenuates stem cell loss in response to ribosome biogenesis defects in Nle-deficient crypts [49]. Thus, ribosome stress not only induces antitumor signaling but also activates prosurvival pathways, which could reduce the efficacy of cancer drugs. In this regard, our current study indicated that some chemotherapeutic agents can activate YAP/TEAD via the ribosomal stress-induced ZAKα-MKK4/7-JNK axis. Our discovery of the ribosomal stress–YAP axis is in agreement with the activation of YAP during the administration of chemotherapy, radiation therapy and targeted therapies for multiple cancers, which leads to the failure of cancer treatments [29]. Thus, it is rational to combine strategies targeting the nucleolus and those targeting the Hippo pathway for therapeutic purposes.
Methods
Patient samples and immunohistochemistry
All CRC samples were collected from August 2008 to August 2018 in the Department of Colorectal Surgery, Xinhua Hospital, School of Medicine, Shanghai Jiaotong University. Postoperative follow-up information was updated through August 2020. The study was approved by the Ethics Committee, and informed consent was obtained from all patients. Tissue microarrays (TMAs) were prepared by Shanghai TOPGEN Company for further immunohistochemical (IHC) analysis. The IHC score was calculated by multiplying the score for the percentage of stained cells by the intensity score. The detailed scoring method was described in our previous study [50]. In this study, samples with a staining score ≤ 4 were assigned to the low DDX21/CTNNB1 expression group, and samples with a score >4 were considered to have high expression.
Cell culture and transfection
NCM460 cell line was a gift from Dr. Lechi Ye, originally purchased from INCELL Corporation (San Antonio, TX, USA). Other cell lines used in this study were purchased from the American Type Culture Collection (ATCC, Manassas, Virginia). HCT-116, RKO, SW480, NCM460 and HEK-293 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM), and HIEC-6 cells were cultured by using Roswell Park Memorial Institute (RPMI) 1640 medium. All media were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Gibco). The cultivation environment was 37 degrees centigrade and 5% carbon dioxide.
Polyethylenimine (Polysciences) or Lipofectamine 2000 (Invitrogen) was used for transient transfection. Lentivirus (pLKO.1) and retrovirus (pQCXIH) were produced by transient transfection of 293 T cells with the appropriate packaging vectors. After 48 hours, the viral supernatant was collected to transduce the indicated cells, which were selected with puromycin and hygromycin, respectively, before subsequent functional analysis.
RNA sequencing, qPCR and ChIP‒qPCR
HCT-116 cells were transfected with control siRNA or siRNA targeting DDX21. After 48 h of transfection, the cells were plated in a 10 cm dish at a low cell density (7.5 × 105 cells) and then harvested after 24 h of culture. Total RNA was extracted, and RNA-seq analysis was performed by OE Biotech Company (Shanghai). The raw data were deposited in the SRA database (PRJNA1095449). GSEA and MSigDB were used for subsequent data analysis (www.gsea-msigdb.org/gsea). The mRNA levels of the canonical Hippo target genes were confirmed by using a q-PCR kit from Takara. A Magna ChIP kit (Merck, 17-610) was used for chromatin immunoprecipitation. Briefly, the cells were cross-linked, washed, lysed, and fragmented by ultrasonication according to the manufacturer’s protocol. Next, the processed samples were incubated with a β-catenin antibody (A19657, ABclonal) and a rabbit mAb IgG (3900, Cell Signaling Technology) overnight at 4 °C. After the magnetic beads were washed and cross-linked at 65 °C for 2 hours, the precipitated DNA was purified for qPCR. The sequences of primers used are listed in Supplementary Table 5.
Cell proliferation and colony formation assays
The cells were inoculated at a density of 1000 cells/100 µl in a 96-well plate on Day 0. For the CCK-8 assay, 10 µl of CCK-8 reagent was added to each well, the plates were incubated in a dark environment at 37 °C for 2 hours, and then the absorbance was measured at 450 nm. For drug treatment, cells were inoculated into 96-well plates at a density of 3000 cells per well. After 36 hours, drugs were added, and the cells were cultured for another 48 hours before CCK-8 analysis. Drug synergy was analyzed according to the Chou-Talalay combination index (CI) method [51].
For the colony formation assay, 1000 cells were plated in each well of 6-well plates, fixed with 4% paraformaldehyde after 2 weeks of culture, and then stained with crystal violet. After washing with phosphate-buffered saline (PBS), the cell colonies were photographed and counted. For the drug treatment assay, 3000 cells were plated into each well and cultured for one week before drug treatment for another 3 days.
Wound healing and transwell assays
For the wound healing assay, cells were plated on six-well plates at high cell density and scratched after the cells reached confluence. Then, the cells were cultured in DMEM supplemented with 1% serum until analysis at the indicated times.
For the transwell assay, cells were resuspended in DMEM supplemented with 0.1% FBS, and 1.5 × 105 cells were plated in each transwell chamber. After 48 h, 4% paraformaldehyde was added to the chambers for 30 min, and then the cells were stained with crystal violet. The cells on the upper side of the membrane were removed, and the cells that had passed through the membrane were photographed and counted for statistical analysis.
Luciferase reporter assay
293 T cells were plated on a 24-well plate. Cells were transiently transfected with the indicated plasmids (pGL3-DDX21 reporter (100 ng/well), CMV-Renilla (5 ng/well), pRK7-Flag-CTNNB1 (300 ng/well) and pRK7-vector (300 ng/well)) via PEI. The day after transfection, luciferase activity was measured by using a dual luciferase reporter gene assay (Promega) on a microplate reader and normalized to Renilla luciferase activity.
Immunofluorescence assay
Cells were seeded at a low cell density (3.0 × 104/well) on a coverslip. For immunofluorescence staining, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 for 15 min at room temperature, and blocked with 5% BSA for 30 min. Then, the cells were coated with YAP and DDX21 antibodies in 1% BSA and incubated overnight at 4 °C. After incubation with secondary antibodies (Beyotime, A0423, A0521) for one hour, the cells were stained with DAPI (Invitrogen, P36962), and then the cells were photographed with an inverted fluorescence microscope (Olympus IX81). To analyze the cytonuclear translocation of YAP, 100 cells were counted for each coverslip, and the cells were divided into N = C, N > C, and N < C groups based on the difference in intensity of YAP fluorescence between the nucleus and cytoplasm.
Mouse Xenograft Tumor Model
The Animal Care and Welfare Committee of Xinhua Hospital approved all animal experiments in this study. Male BALB/C nude mice were purchased from Shanghai Ji Hui Experimental Animal Company. Nude mice aged 4-6 weeks were subcutaneously injected with 1 × 106 of the indicated stable HCT-116 cells, randomly. Two weeks later, the mice were euthanized to collect xenograft tumors, which were photographed, weighed and sectioned for subsequent immunohistochemical staining of DDX21 and Ki-67.
Statistical analysis
GraphPad Prism 9 Software and SPSS version 25.0 software were used for data analysis in this study. The quantitative data are all presented as the means ± standard deviations (SDs). Paired Student’s t-test, one-way ANOVA with Dunnett’s multiple comparison test, and one-way ANOVA with Tukey’s multiple comparison test were used to evaluate the statistical significance of differences between two groups and among three or more groups, respectively. Two-way ANOVA with Dunnett’s multiple comparison test was used to analyze the statistical significance of the CCK-8 results. The Wilcoxon signed-rank test was used for IHC analysis of the TMA. Pearson correlation analysis was used to evaluate the statistical significance of the difference between the mRNA levels from the TCGA dataset, and Spearman correlation analysis was used for IHC analysis of the TMA.
Data availability
The raw data of the RNA-seq analysis were deposited in the SRA database (PRJNA1095449). The data in this study are available within the article and related supplementary information files, or by inquiring the corresponding authors.
References
Putnam AA, Jankowsky E. DEAD-box helicases as integrators of RNA, nucleotide and protein binding. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2013;1829:884–93.
Westermarck J, Weiss C, Saffrich R, Kast J, Musti AM, Wessely M, et al. The DEXD/H-box RNA helicase RHII/Gu is a co-factor for c-Jun-activated transcription. EMBO J. 2002;21:451–60.
Zhang Y, Baysac KC, Yee LF, Saporita AJ, Weber JD. Elevated DDX21 regulates c-Jun activity and rRNA processing in human breast cancers. Breast Cancer Res. 2014;16:449.
Calo E, Flynn RA, Martin L, Spitale RC, Chang HY, Wysocka J. RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature. 2015;518:249–53.
Xing YH, Yao RW, Zhang Y, Guo CJ, Jiang S, Xu G, et al. SLERT regulates DDX21 rings associated with Pol I transcription. Cell. 2017;169:664–78.e616.
Song C, Hotz-Wagenblatt A, Voit R, Grummt I. SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 2017;31:1370–81.
Xie J, Wen M, Zhang J, Wang Z, Wang M, Qiu Y, et al. The Roles of RNA Helicases in DNA Damage Repair and Tumorigenesis Reveal Precision Therapeutic Strategies. Cancer Res. 2022;82:872–84.
Hu A, Wang Y, Tian J, Chen Z, Chen R, Han X, et al. Pan-cancer analysis reveals DDX21 as a potential biomarker for the prognosis of multiple tumor types. Front Oncol. 2022;12:947054.
Gao H, Wei H, Yang Y, Li H, Liang J, Ye J, et al. Phase separation of DDX21 promotes colorectal cancer metastasis via MCM5-dependent EMT pathway. Oncogene. 2023;42:1704–15.
Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 2016;6:80–95.
Guo X, Zhao Y, Yan H, Yang Y, Shen S, Dai X, et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 2017;31:247–59.
Liu CY, Yu T, Huang Y, Cui L, Hong W. ETS (E26 transformation-specific) up-regulation of the transcriptional co-activator TAZ promotes cell migration and metastasis in prostate cancer. J Biol Chem. 2017;292:9420–30.
Liu Y, Wang G, Yang Y, Mei Z, Liang Z, Cui A, et al. Increased TEAD4 expression and nuclear localization in colorectal cancer promote epithelial-mesenchymal transition and metastasis in a YAP-independent manner. Oncogene. 2016;35:2789–2800.
Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17:500–10.
Wu H, Wei L, Fan F, Ji S, Zhang S, Geng J, et al. Integration of Hippo signalling and the unfolded protein response to restrain liver overgrowth and tumorigenesis. Nat Commun. 2015;6:6239.
Hong AW, Meng Z, Yuan HX, Plouffe SW, Moon S, Kim W, et al. Osmotic stress-induced phosphorylation by NLK at Ser128 activates YAP. EMBO Rep. 2017;18:72–86.
Luo M, Meng Z, Moroishi T, Lin KC, Shen G, Mo F, et al. Heat stress activates YAP/TAZ to induce the heat shock transcriptome. Nat Cell Biol. 2020;22:1447–59.
Vind AC, Genzor AV, Bekker-Jensen S. Ribosomal stress-surveillance: three pathways is a magic number. Nucleic Acids Res. 2020;48:10648–61.
Silva J, Alkan F, Ramalho S, Snieckute G, Prekovic S, Garcia AK, et al. Ribosome impairment regulates intestinal stem cell identity via ZAKa activation. Nat Commun. 2022;13:4492.
Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–205.
Bugter JM, Fenderico N, Maurice MM. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat Rev Cancer. 2021;21:5–21.
Meniel V, Song F, Phesse T, Young M, Poetz O, Parry L, et al. Cited1 deficiency suppresses intestinal tumorigenesis. PLoS Genet. 2013;9:e1003638.
Gay DM, Ridgway RA, Muller M, Hodder MC, Hedley A, Clark W, et al. Loss of BCL9/9l suppresses Wnt driven tumourigenesis in models that recapitulate human cancer. Nat Commun. 2019;10:723.
Chen S, Yang L, Dong H, Guo H. Human telomerase reverse transcriptase recruits the beta-catenin/TCF-4 complex to transactivate chemokine (C-C motif) ligand 2 expression in colorectal cancer. Biomed Pharmacother. 2019;112:108700.
Cargill M, Venkataraman R, Lee S. DEAD-Box RNA Helicases and Genome Stability. Genes (Basel) 2021;12.
Koltowska K, Okuda KS, Gloger M, Rondon-Galeano M, Mason E, Xuan J, et al. The RNA helicase Ddx21 controls Vegfc-driven developmental lymphangiogenesis by balancing endothelial cell ribosome biogenesis and p53 function. Nat Cell Biol. 2021;23:1136–47.
Wu M, Xu G, Han C, Luan PF, Xing YH, Nan F, et al. lncRNA SLERT controls phase separation of FC/DFCs to facilitate Pol I transcription. Science. 2021;373:547–55.
Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK, et al. Comprehensive molecular characterization of the hippo signaling pathway in cancer. Cell Rep. 2018;25:1304–1317.e1305.
Calses PC, Crawford JJ, Lill JR, Dey A. Hippo pathway in cancer: aberrant regulation and therapeutic opportunities. Trends Cancer. 2019;5:297–307.
Kulkarni A, Chang MT, Vissers JHA, Dey A, Harvey KF. The hippo pathway as a driver of select human cancers. Trends Cancer. 2020;6:781–96.
Azzam ME, Algranati ID. Mechanism of puromycin action: fate of ribosomes after release of nascent protein chains from polysomes. Proc Natl Acad Sci USA. 1973;70:3866–9.
Wong J, Smith LB, Magun EA, Engstrom T, Kelley-Howard K, Jandhyala DM, et al. Small molecule kinase inhibitors block the ZAK-dependent inflammatory effects of doxorubicin. Cancer Biol Ther. 2013;14:56–63.
Yang K, Yang J, Yi J. Nucleolar Stress: hallmarks, sensing mechanism and diseases. Cell Stress. 2018;2:125–40.
Vind AC, Snieckute G, Blasius M, Tiedje C, Krogh N, Bekker-Jensen DB, et al. ZAKalpha recognizes stalled ribosomes through partially redundant sensor domains. Mol Cell. 2020;78:700–713.e707.
van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10:301–9.
Chaillou T, Kirby TJ, McCarthy JJ. Ribosome biogenesis: emerging evidence for a central role in the regulation of skeletal muscle mass. J Cell Physiol. 2014;229:1584–94.
Madan B, Harmston N, Nallan G, Montoya A, Faull P, Petretto E, et al. Temporal dynamics of Wnt-dependent transcriptome reveal an oncogenic Wnt/MYC/ribosome axis. J Clin Invest. 2018;128:5620–33.
Lu P, Yu Z, Wang K, Zhai Y, Chen B, Liu M, et al. DDX21 interacts with WDR5 to promote colorectal cancer cell proliferation by activating CDK1 expression. J Cancer. 2022;13:1530–9.
Wang K, Li B, Fan P, Ren X, Jiang H. Downregulation of DEAD-box helicase 21 (DDX21) inhibits proliferation, cell cycle, and tumor growth in colorectal cancer via targeting cell division cycle 5-like (CDC5L). Bioengineered. 2021;12:12647–58.
Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218–27.
Berecz T, Yiu A, Vittay O, Orsolits B, Mioulane M, Dos Remedios CG, et al. Transcriptional co-activators YAP1-TAZ of Hippo signalling in doxorubicin-induced cardiomyopathy. ESC Heart Fail. 2022;9:224–35.
She G, Du JC, Wu W, Pu TT, Zhang Y, Bai RY, et al. Hippo pathway activation mediates chemotherapy-induced anti-cancer effect and cardiomyopathy through causing mitochondrial damage and dysfunction. Theranostics. 2023;13:560–77.
Codelia VA, Sun G, Irvine KD. Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr Biol. 2014;24:2012–7.
Sun G, Irvine KD. Ajuba family proteins link JNK to Hippo signaling. Sci Signal. 2013;6:ra81.
Kim DS, Camacho CV, Nagari A, Malladi VS, Challa S, Kraus WL. Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol Cell. 2019;75:1270–1285.e1214.
Pelletier J, Thomas G, Volarevic S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer. 2018;18:51–63.
Zisi A, Bartek J, Lindstrom MS. Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward. Cancers (Basel) 2022;14.
Dannheisig DP, Bachle J, Tasic J, Keil M, Pfister AS. The Wnt/beta-catenin pathway is activated as a novel nucleolar stress response. J Mol Biol. 2021;433:166719.
Raveux A, Stedman A, Coqueran S, Vandormael-Pournin S, Owens N, Romagnolo B, et al. Compensation between Wnt-driven tumorigenesis and cellular responses to ribosome biogenesis inhibition in the murine intestinal epithelium. Cell Death Differ. 2020;27:2872–87.
Zhu Z, Guo Y, Liu Y, Ding R, Huang Z, Yu W, et al. ELK4 Promotes Colorectal Cancer Progression by Activating the Neoangiogenic Factor LRG1 in a Noncanonical SP1/3-Dependent Manner. Adv Sci (Weinh). 2023;10:e2303378.
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 82073201, 82172916, and 82372648) and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (to C-Y. L.).
Author information
Authors and Affiliations
Contributions
C-YL and PD conceived the project. WT, YY, and ZF performed most of the experiments and/or analyzed data. WT and C-YL prepared the manuscript. WX, WO, and FL assisted in performing some experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was approved by the Ethics Committee, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University (XHEC-STCSM-2022-077). Informed consent was obtained from all patients. The Animal Care and Welfare Committee of Xinhua Hospital approved all animal experiments in this study. All methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, W., Yang, Y., Fu, Z. et al. The RNA helicase DDX21 activates YAP to promote tumorigenesis and is transcriptionally upregulated by β-catenin in colorectal cancer. Oncogene (2024). https://doi.org/10.1038/s41388-024-03160-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41388-024-03160-8
- Springer Nature Limited