
Abstract
Vascular endothelial cells (VECs) are key players in the formation of neovessels and tumor metastasis, the ultimate cause of the majority of cancer-related human death. However, the crosstalk between VECs and metastasis remain greatly elusive. Based on our finding that tumor-associated VECs present significant decrease of Nrdp1 protein which is closely correlated with higher metastatic probability, herein we show that the conditional medium from hypoxia-incubated cancer cells induces extensive Nrdp1 downregulation in human and mouse VECs by vascular endothelial growth factor (VEGF), which activates CHIP, followed by Nrdp1 degradation in ubiquitin-proteasome-dependent way. More importantly, lung metastases of cancer cells significantly increase in conditional VECs Nrdp1 knockout mice. Mechanically, Nrdp1 promotes degradation of Fam20C, a secretory kinase involved in phosphorylating numerous secreted proteins. Reciprocally, deficiency of Nrdp1 in VECs (ecNrdp1) results in increased secretion of Fam20C, which induces degradation of extracellular matrix and disrupts integrity of vascular basement membrane, thus driving tumor metastatic dissemination. In addition, specific overexpression of ecNrdp1 by Nrdp1-carrying adeno-associated virus or chemical Nrdp1 activator ABPN efficiently mitigates tumor metastasis in mice. Collectively, we explore a new mechanism for VEGF to enhance metastasis and role of Nrdp1 in maintaining the integrity of vascular endothelium, suggesting that ecNrdp1-mediated signaling pathways might become potential target for anti-metastatic therapies.
Similar content being viewed by others


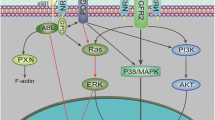
Introduction
Metastasis, a hallmark of cancer and the ultimate cause of the majority of cancer-related death, is orchestrated by a dynamic, multifaceted complex network of biological events [1,2,3]. One such event is the formation of new blood vessels termed as tumor angiogenesis. Tumor cells intravasate from primary tumor into blood circulation and extravasate into distant organ from blood stream. Thus, angiogenesis supports tumor cell metastasis by providing a major route from primary tumors to distant organs besides supplying tumor tissues with nutrients and oxygen for tumor survival and growth. Tumor endothelial cells lining tumor blood vessels, which are key players in the formation of neovessels, differ from normal endothelial cells in many aspects. For instances, they are usually irregular monolayers and impaired endothelial barrier function [4, 5], and the thickness of tumor vascular basement membrane (VBM) is uneven and the association between pericytes and tumor vascular endothelial cells (VECs) is weak, leading to vascular leakiness, which allows cancer cells to escape from the primary site and disseminate to other sites, and contributes to poor drug delivery and enables the immunosurveillance escape of cancers [6].
Crosstalk between cancer cells and VECs in the tumor microenvironment generates a pre-metastatic niche that promotes tumor progression [7, 8]. Disseminated cancer cells exploit factors derived from themselves to incite the formation of neovascularization from existing capillaries [5, 9, 10]. This process is delicately balanced by the production of pro- and anti-angiogenic factors that activate endothelial cells, among which vascular endothelial growth factor-A (VEGF-A, hereafter named VEGF) plays a crucial role in angiogenesis as well as increased vascular permeability for cancer metastasis [11, 12]. Accordingly, neovascularization can be halted by treatment with compounds such as the VEGF inhibitor bevacizumab, the mTOR inhibitor everolimus, or the tyrosine kinase inhibitor pazopanib. However, these drugs did not significantly improve overall patient survival due to adverse events, acquired drug resistance, tumor recurrence, and lack of validated biomarkers [13,14,15,16]. This limitation has prompted a precise investigation into the crosstalk between tumor cells and tumor-associated endothelial cells that accelerates tumor metastasis, particularly in the underlying mechanisms of endothelial-derived factors that contribute to tumor progression [17,18,19,20,21]. However, there is a need to further study the vascular effects of tumor-derived factors to predict and improve the survival of cancer patients [4].
Nrdp1 (Neuregulin receptor degradation protein 1, also known as RNF41 or FLRF), an evolutionarily highly conserved RING finger gene [22], is ubiquitously expressed in human tissues. Originally, Nrdp1 was shown to interact with the cytoplasmic tail of the neuregulin receptors ErbB3 and ErbB4 in an activation-independent manner [23]. The following investigations showed that Nrdp1 is a RING finger-type of ubiquitin E3 ligase, which promotes the ubiquitination and degradation of ErbB3, BRUCE/apollon, and itself, thereby facilitating tumor progression [24,25,26,27]. Nrdp1, whose suppression correlates with poor prognosis of glioblastoma multiforme (GBM), was also reported to reduce GBM cell migration and invasiveness, in which Nrdp1 physically interacts with the Vangl1 and Vangl2 proteins, components of the planar cell polarity arm of non-canonical Wnt signaling, to mediate the K63-linked polyubiquitination of the Wnt pathway protein Dishevelled and suppress the planar cell polarity signaling. Thus, the restoration of Nrdp1 reduces GBM cell migration and invasiveness [28, 29]. Furthermore, the retinoic acid derivative ABPN (A4-amino-2-(butyrylamino)phenyl (2E,4E,6E,8E)-3,7-dimethyl-9- (2,6,6-trimethyl-1-cyclohexenyl)-2,4,6,8-nonatetraenoate) exhibits tremendous anti-cancer activity in pancreatic cancer through inducing Nrdp1 expression [30]. However, roles of the Nrdp1 in carcinogenesis, especially metastasis, remain to be greatly elusive.
Based on the discovery that tumor vascular endothelial cells presented lower Nrdp1 expression, herein we report that cancer cells-derived VEGF triggers Nrdp1 degradation in tumor VECs, while deficiency of Nrdp1 in VECs (ecNrdp1) facilitates cancer cell metastasis in vivo through disrupting the VBM integrity. In accordance, specific overexpression of ecNrdp1 by Nrdp1-carrying adeno-associated virus (AAV) or chemical Nrdp1 activator ABPN efficiently mitigates tumor metastasis in mice.
Results
VECs in tumors present lower expression of Nrdp1 protein
We detected Nrdp1 protein expression by immunohistochemistry (IHC) staining in a tissue microarray of human colorectal cancer tissues (n = 99) with available paired adjacent non-cancerous tissues (n = 74), for which the specificity of anti-Nrdp1 antibody was confirmed (Supplementary Fig. 1). Our results showed that there was no significant difference of Nrdp1 protein levels between cancer and adjacent non-cancerous cells (Fig. 1A, B), which was consistent with analysis of Nrdp1 mRNA levels in colorectal cancer from the The Cancer Genome Atlas (TCGA) COAD cohorts (n = 520) (Fig. 1C). More surprisingly, we found that there was a dramatically reduced Nrdp1 abundance in tumor-associated VECs, as indicated by CD31 staining, in comparison with those from their adjacent normal tissues (Fig. 1A, D). The same phenomenon could also be seen on a multiple-organ tissue chip with lung, renal, and gastric cancers (Fig. 1E). More interestingly, lower ecNrdp1 protein was associated with higher metastatic probability (Fig. 1F), and patients with metastasis exhibited significantly lower ecNrdp1 expression in comparison to patients without metastasis in our cohort of colorectal cancer (Fig. 1G). Furthermore, there was a trend towards shorter progression-free survival in patients with lower ecNrdp1 expression, although statistical difference could not be gotten between cancers with the lower and higher ecNrdp1 expression (Fig. 1H).

A Two representative images of IHC analysis for Nrdp1 protein in CRC samples corresponding primary tumor tissues (Tumor) as well as paired distal colon mucosa (Normal). VECs were estimated by CD31 staining. Scale bar, 10 μm. Vessel (V), normal tissues (N), tumor tissues (T). B Relative quantification of Nrdp1 protein levels in tumor cells and paired epithelial cells from 74 cases of CRC samples. The numbers in parentheses represent those of samples with identical IRS scores. Data were analyzed by paired two-tailed Student’s t-test. C RNA-seq data analysis from TCGA COAD cohorts. Data were analyzed by unpaired two-tailed Student’s t-test. D Relative quantification of ecNrdp1 protein levels between tumor tissues and paired normal tissues from (B). The numbers in parentheses represent those of samples with identical IRS scores. Data were analyzed by paired two-tailed Student’s t-test. E Representative images of IHC staining of Nrdp1 expression on gastric, renal, lung cancer, and corresponding normal tissues. Scale bar, 10 μm. Vessel (V). F, G Analysis of the correlation between ecNrdp1 protein levels and metastasis in CRC patient samples. Data were analyzed by Pearson’s χ2 test. H Kaplan–Meier estimates of progression-free survival of CRC patients with high and low ecNrdp1 expression. Statistical difference was determined by log-rank test.
Cancer cells-secreted VEGF induces Nrdp1 degradation in VECs
Considering that hypoxia is a common feature of solid tumors mediating the formation of aberrant blood vessels [31], we asked whether ecNrdp1 downregulation in tumor was dependent on the hypoxic tumor environment. To this end, human umbilical vein endothelial cells (HUVECs) were exposed to 1% oxygen for 24 h, and found that the treatment had no impact on ecNrdp1 protein levels together with increased hypoxia-inducible factor 1α (HIF-1α) protein (Fig. 2A). It has been reported that cancer cells can secrete various cytokines and chemokines to communicate with the endothelium upon hypoxia microenvironment, thereby facilitating metastasis by forming factors-engaged pro-metastatic niches [32, 33]. Thus, we evaluated the effect of conditioned medium (CM) from cancer cells on ecNrdp1 expression. More intriguingly, CM from hypoxia-treated human breast cancer cells MDA-MB-231 and prostate cancer cells DU145, which carried higher levels of VEGF, caused extensively Nrdp1 downregulation in HUVECs, compared with that from normoxia-treated cells (Fig. 2B, Supplementary Fig. 2A). Similar efficiency was also observed in freshly isolated mouse lung endothelial cells (MLECs) upon treatment with hypoxia-induced CM from mouse B16 melanoma cells (Supplementary Fig. 2B).

A Western blot analysis for indicated proteins in HUVECs under normoxia and hypoxia for 24 h. B HUVECs were treated with CM from normoxia or hypoxia-induced MDA-MB-231 cells, and VEGF in CM of cancer cells was quantified by ELISA, and the indicated proteins in the corresponding cells were blotted. The relative Nrdp1 levels in HUVECs were shown on the right panel (n = 3). C Western blot analysis of the Nrdp1 protein in HUVECs treated with CM from normoxia (N-CM) or hypoxia (H-CM)-induced MDA-MB-231 cells with or without Bevacizumab. The relative Nrdp1 levels in HUVECs were shown at the bottom (n = 3). D Proteins as indicated were blotted in HUVECs treated with indicated doses of VEGF for 24 h, and the relative Nrdp1 and CHIP levels in HUVECs were shown. E HUVECs were treated by VEGF (40 ng/ml) with or without Bevacizumab or Cabozantinib for 24 h and indicated proteins were blotted. The quantification of Nrdp1 protein bands was shown on the right (n = 3). F Fluorescent staining of CD31 and Nrdp1 in mouse retinal vasculature at postnatal day 6. The yellow and white arrowheads pointed to tip cells and stalk cells, respectively. Scale bar, 50 μm. G qRT-PCR analysis of Nrdp1 mRNA abundances in the indicated doses of VEGF-treated HUVECs. H Western blotting of Nrdp1 proteins (left) and relative quantification of Nrdp1 levels (right) in HUVECs treated with or without VEGF (40 ng/ml) in the presence of 50 μg/ml CHX for indicated times. I HUVECs were treated with or without VEGF (40 ng/ml) and 10 μM MG132 for 8 h, and the indicated proteins were blotted. The relative Nrdp1 levels in HUVECs were shown at the bottom (n = 3). Non-specific bands labelled with *. J qRT-PCR analysis of CHIP mRNA abundances in HUVECs with indicated doses of VEGF stimulation. K Nrdp1 mRNA expression in mouse normal and tumor endothelial cells from GSEA database (GSE118904). Western blot analysis of indicated proteins in 293T cells transfected with gradient concentration of GFP-tagged CHIP (L) or shRNA targeting CHIP along with shNC as a negative control (M). N Western blot analysis of indicated proteins in 293T cells transfected with GFP-tagged CHIP followed by treatment with DMSO, 10 μM MG132, and 50 μM CQ for 8 h. O 293T cells co-transfected with GFP-tagged Nrdp1 and Flag-tagged CHIP were subjected to immunoprecipitation with an anti-Flag antibody. Western blot of Flag- and GFP-tagged proteins in the input and immunoprecipitates were shown. P Lysates from 293T cells transfected with GFP-tagged CHIP and Flag-tagged Nrdp1 under 10 μM MG132 for 8 h were precipitated with anti-Flag antibody, and the precipitates were blotted for protein as indicated. All experiments in A–E, G–J, L–P were repeated three times with similar results, and the numbers under the indicated protein blots indicated relative quantification of protein bands. Error bars denote mean ± SD. Statistical difference was determined by one-way ANOVA with multiple comparisons (C, E, G, I, J) or two-tailed unpaired Student’s t-test (B, K) and P values were shown.
To identify the specific tumor-derived factor(s) responsible for the reduction of ecNrdp1 protein, we initially focused on VEGF, a pivotal transcriptional target of HIF-1 that contributes to vascular formation and permeability and supports tumor progression and metastasis [34]. More interestingly, neutralizing VEGF with bevacizumab, a humanized anti-VEGF antibody, significantly abrogated the reduction of ecNrdp1 upon stimulation by hypoxia-induced CM from MDA-MB-231 and DU145 cells (Fig. 2C, Supplementary Fig. 2C), suggesting that hypoxia-induced tumor-derived VEGF may contribute to ecNrdp1 downregulation. To support this, we revealed that increasing concentrations of the in vitro recombinant human VEGF, which caused persistent phosphorylation at Y1175 of VEGF receptor 2 (VEGFR2) due to the high affinity of VEGF-VEGFR2 interaction as previously reported [35, 36], dose-dependently reduced Nrdp1 protein of HUVECs (Fig. 2D) and MLECs (Supplementary Fig. 2D). Like Bevacizumab, VEGFR2 inhibitor Cabozantinib also antagonized VEGF-induced Nrdp1 reduction (Fig. 2E). Consistent with previous report [37], notably, VEGF in the CM from HUVECs under hypoxia for 24 h was almost undetectable (Supplementary Fig. 2E), which could explain why the hypoxia treatment failed to impact ecNrdp1 protein levels in HUVECs (Fig. 2A). All these results indicate that VEGF activates the VEGF signaling pathway and then induces Nrdp1 downregulation in VECs.
It has been known that during developmental angiogenesis, endothelial tip cells at the leading edge of vascular sprouts in the retina are exposed to higher levels of VEGF than stalk cells [38, 39]. To confirm the impact of VEGF on ecNrdp1 expression in vivo, mouse retinal vasculature was isolated for immunofluorescence analysis of Nrdp1 and VECs marker CD31. The results revealed that tip cell-specific inhibition of Nrdp1 in the retinal vasculature compared to stalk cells (Fig. 2F), indicating that VEGF may account for lower Nrdp1 expression under physiological conditions.
Because VEGF treatment failed to affect Nrdp1 mRNA expression (Fig. 2G), we used the cycloheximide (CHX) assay to quantify the stability of the Nrdp1 protein and demonstrated that Nrdp1 was rapidly degraded upon VEGF treatment (Fig. 2H). In line, VEGF-induced Nrdp1 degradation was completely abrogated by the proteasome inhibitor MG132, which also rescued HIF-1α protein under normoxia (Fig. 2I).
VEGF-induced CHIP activation promotes degradation of Nrdp1
Above observations suggested that VEGF mediates Nrdp1 degradation in a proteasome-dependent manner. To elucidate the underlying mechanisms, we utilized STRING interaction network prediction (https://cn.string-db.org) and focused on the role of ubiquitin E3 ligase CHIP (C-terminus of Hsc70-interacting protein) in modulating Nrdp1 expression. Indeed, increased CHIP mRNA and protein in HUVECs were observed upon VEGF stimulation (Fig. 2D, J). Additionally, the analysis of a single-cell RNA sequencing dataset (GSE118904) generated from mouse tumor and normal endothelial cells revealed that the level of CHIP expression was greatly increased in tumor endothelial cells (Fig. 2K).
Next, we examined the specific roles of CHIP in Nrdp1 expression. As depicted in Fig. 2L, Nrdp1 protein was reduced in a dose-dependent manner upon transiently expressing green fluorescent protein (GFP)-tagged CHIP in 293T cells. Conversely, knockdown of endogenous CHIP by short-hairpin RNAs (shRNAs) increased Nrdp1 protein (Fig. 2M). Of note, GFP-CHIP overexpression in 293T cells did not alter Nrdp1 mRNA level (Supplementary Fig. 2F). Moreover, MG132 but not the autophagy-lysosomal inhibitor Chloroquine (CQ) blocked CHIP-induced Nrdp1 degradation (Fig. 2N), indicating that CHIP is responsible for Nrdp1 protein stability. Furthermore, co-immunoprecipitation (co-IP) with an anti-Flag antibody revealed a significant interaction between transiently overexpressed Flag-CHIP and GFP-Nrdp1 (Fig. 2O), and Flag-Nrdp1 was abundantly ubiquitylated upon CHIP expression (Fig. 2P). Collectively, the stability of ecNrdp1 is mediated by CHIP in a ubiquitin-dependent manner.
Deficiency of ecNrdp1 facilitates cancer cell metastasis in vivo
All above-mentioned observations promoted us to investigate the possible role of ecNrdp1 in tumor metastasis. As for this, we initially utilized gene-targeting strategies to generate Nrdp1 floxed alleles, in which exons 3 and 7 were flanked by loxP sites, to create Nrdp1fl/fl mice [40, 41]. By crossing Nrdp1fl/fl mice with the Tek (Tie2)-cre strain, which drives endothelial deletion of Nrdp1, we successfully obtained Nrdp1Tek-cre mice (ecNrdp1 knockout) (Fig. 3A). The efficient deletion of Nrdp1 was confirmed at mRNA and protein levels in isolated MLECs and aortic endothelium but not liver, heart and brain in Nrdp1Tek-cre mice (Fig. 3B, C, Supplementary Fig. 3A, B). It is worth noting that ecNrdp1-knockout mice were born with normal proportions, and no adverse effects on body weights or survival rates were observed relative to wild-type littermate controls during the experiment. Next, we injected B16 melanoma cells through the tail vein into Nrdp1Tek-cre mice with wild-type mice as controls, and found that three weeks post-injection, ecNrdp1-knockout mice displayed a six-fold increase in lung metastatic nodules compared to wild-type littermate controls, together with a greater abundance of metastatic foci within the lungs, without affecting immune cell infiltrations (Fig. 3D–F, Supplementary Fig. 3C). Similar results could also be seen upon injection with mouse Lewis lung carcinoma cells (LLCs) (Fig. 3G–J) and luciferase-expressing B16F10 melanoma cells (Supplementary Fig. 3D–G). To exclude the possibility that ecNrdp1-knockout impacts tumorigenesis, we also subcutaneously inoculated B16 cells in both WT and Nrdp1Tek-cre mice, and showed that ecNrdp1 deficiency did not affect primary tumor growth (Supplementary Fig. 3H), supporting that ecNrdp1 knockout increases matastasis rather than influences cancer cell growth.

A Schematics of the gene-targeting strategies to generate floxed alleles of RNF41 (Nrdp1) to create Nrdp1fl/fl mice. B Western blot analysis of Nrdp1 protein in freshly isolated MLECs and BM cells from ecNrdp1-knockout and wild-type mice. The symbol * indicated non-specific band. C Representative IHC images of the aorta from wild-type and ecNrdp1-knockout mice. Arrows indicate endothelium. Scale bar, 25 μm. B16 cells (D–F) and LLC cells (G–I) were injected intravenously into ecNrdp1-knockout mice (B16, n = 5; LLC, n = 10) or wild-type littermate controls (B16, n = 5; LLC, n = 10). Macroscopy of lungs with macrometastatic foci (D, G) and quantification of the macrometastatic lung foci per mouse (E, H), and haematoxylin and eosin (H&E) staining of lung metastatic area were shown (F, I). Scale bar, 500 μm. J Statistics of lung metastatic incidence in LLC-bearing Nrdp1fl/fl and Nrdp1Tek-cre mice. K–M B16 cells were injected intravenously into Cdh5-cre-driven ecNrdp1-knockout mice (n = 5) compared with wild-type littermate controls (n = 5). Macroscopy of lungs (K), the number of macrometastatic lung foci (L), and HE staining of metastatic area within the lung (M) were shown. Scale bar, 500 μm. All quantitative data were shown as mean ± SEM. P values were measured by unpaired two-tailed Student’s t-test.
Tek-Cre mouse models also show some degree of Cre recombinase activity in the hematopoietic lineage [41], and a complete knockout of Nrdp1 in the bone marrow (BM) cells of Nrdp1Tek-cre mice could also be seen (Fig. 3B). It has been previously reported that Nrdp1 deficiency does not affect immune cell distribution, but enhances CD8+ T cell activation and interferes with cytokine production in macrophages [42, 43]. In line with these previous reports, Nrdp1Tek-cre mice showed no impact on the distribution of immune cells (Supplementary Fig. 3I), but exhibited heightened levels of antitumor cytokines in CD8+ T cells (IFNγ, IL-2, and GZMB), along with elevated expression of proinflammatory cytokines (IL-6 and TNFα) in macrophages (Supplementary Fig. 3J). Additionally, the function of tumor-infiltrating immune cells was also altered, evidenced by enhanced activation of antitumor CD8+ T cells (as measured by CD69) and increased production of IL-6 and TNFα by macrophages (Supplementary Fig. 3K). Importantly, even though the function of these immune cells was altered, Nrdp1 deficiency did not affect the primary tumor growth.
To exclude the potential interference of hematopoietic cells-derived Nrdp1 on tumor metastasis, we crossed Nrdp1fl/fl mice with the VEC-specific Cdh5 (VE-cadherin)-cre strain to generate Nrdp1Cdh5-cre mice, which showed the specific deficiency of Nrdp1 in VECs (Supplementary Fig. 3L), and parallel metastatic experiments in ecNrdp1-knockout mice with the Cdh5 (VE-Cadheirn)-cre strain were performed. Consistently, the ecNrdp1-knockout mice generated by Cdh5-Cre also exhibited a dramatically increased number of metastatic foci within the lungs compared to the WT group (Fig. 3K–M, Supplementary Fig. 3M–O), without influencing the proportion of tumor-infiltrating immune cells (Supplementary Fig. 3P). By the way, Nrdp1Cdh5-Cre mice did not affect primary tumor growth (Supplementary Fig. 3Q). Collectively, ecNrdp1 deficiency facilitates metastasis of cancer cells.
EcNrdp1 deficiency has no effects on VECs-tumor cell adhesion and VEC gap junctions
To understand the mechanism by which ecNrdp1 deficiency promotes cancer metastasis, we utilized CRISPR-Cas9 with single guide RNA to specifically knock down Nrdp1 in freshly isolated HUVECs, together with scramble sgNC as a control (Fig. 4A). Our results showed that Nrdp1 deficiency in HUVECs did not affect morphology and cell growth, and had also no influence on cell senescence, as estimated by percentage of β-Gal positive cells (Supplementary Fig. 4A–C), which is believed to be responsible for tumor metastasis [17]. Similarly, MLECs from the ecNdrp1 knockout also displayed no differences from those without knockout in terms of the morphology and proliferation capacity of MLECs (Supplementary Fig. 4D, E).

A Western blot analysis of indicated proteins in HUVECs transduced by lentiviruses encoding CAS9 and gNrdp1 along with gNS as a non-specific control. B Red CMTPX-labeled DU145 cells were co-cultured for 30 min with Green CMTPX-labeled HUVEC-gNS and HUVEC-gNrdp1 to assess adhesion ability to HUVEC monolayers. Representative micrographs (left) and quantification (right) are shown (n = 3). Scale bar, 100 μm. C Western blot analysis of proteins as indicated in freshly isolated MLECs from ecNrdp1-knockout and wild-type mice. Immunostaining of VE-cadherin (D), ZO-1 (E), and Phalloidin in HUVECs with Nrdp1 knockout by CRISPR-Cas9 and control. Nuclei were re-stained by DAPI. Scale bar, 20 μm. Immunostaining of VE-cadherin (F), ZO-1 (G), and CD31 in the retinal blood vessels of Nrdp1fl/fl and Nrdp1Tek-cre mice. High magnification of small cappllaies (yellow-boxed) and venules (white-boxed) were shown on the right. Scale bar, 50 μm. The yellow and white arrows pointed to intercellular bridging and intercellular contact, respectively. H Representative images of Nrdp1fl/fl and Nrdp1Tek-cre VECs (CD31) surrounded by pericytes (NG2). Scale bar, 25 μm. Direct contact between endothelial cells and pericytes was quantified by colocalization analysis (right, n = 3). All quantitative data were shown as mean ± SD. Statistical significance was determined by unpaired two-tailed Student’s t-test and P values were shown.
When cancer cells get trapped in blood circulation, the initial step is to facilitate tumor cell adhesion to the endothelial layer [2, 44]. To evaluate whether VECs with Nrdp1 deficiency has a stronger ability to recruit circulating tumor cells, red CMTPX fluorescence-labeled cancer cells were co-cultured with a monolayer state of green CMTPX fluorescence-labeled HUVECs or MLECs with or without Nrdp1 knockout (Fig. 4B, Supplementary Fig. 4F, G), and found that ecNrdp1 deficiency did not foster recruitment and augmentation of interactions of VECs with cancer cells. Consistently, ecNrdp1 deficiency did not also affect expressions of cell adhesive molecules, such as E-selectin, vascular cell adhesion molecule 1 (VCAM-1), and intercellular cell adhesion molecule 1 (ICAM-1) (Fig. 4C). Also, ecNrdp1 deficiency failed to impact protein levels and distributions of VE-cadherin and zonula occludens 1 (ZO-1) in HUVECs (Fig. 4A, D, E) and the retinal blood vessels freshly isolated from ecNrdp1-knockout mice (Fig. 4F, G), which represent the major tight junction and adherens junction molecules that support the endothelial barrier and vessel homeostasis [45, 46]. Considering that the intimate spatial relationship between pericytes and endothelial cells is still crucial for the integrity of capillary structures [47, 48], we further analyzed the coverage of pericytes in NG2-positive retinal blood vessels. As shown in Fig. 4H, retina from ecNrdp1-knockout mice still displayed intact pericyte coverage of the blood vessels, indicating that ecNrdp1 deficiency does not affect the gap junctions between pericytes and VECs.
EcNrdp1 deletion disrupts the integrity of VBM
Another critical and rate-limiting step in the metastatic process involves the degradation and invasion of the VBM to create an opening for distant localization. VBM, also known as a secondary barrier, is a specialized dense sheet-like protein network predominantly composed of laminin, collagen IV, and proteoglycans, which function as a physical barrier to support endothelial cells and prevent tumor cells from dissemination [49,50,51]. Therefore, we determined whether ecNrdp1 expression can alter VBM integrity, as evaluated by immunofluorescent staining for two VBM components, collagen IV and laminin α5 (a specific laminin isoform) in the retinal blood vessels of ecNrdp1-knockout mice. Consistent with the previous report [52], laminin α5 and collagen IV showed uniform and patchy localization in VBM of postcapillary venules in normal mice. However, larger gaps and more pores upon these two VBM components staining were observed in ecNrdp1-knockout mice (Fig. 5A–C), suggesting that ecNrdp1 deficiency might induce abnormal deposition or dysregulated degradation of VBM components. Further, we exposed HUVECs with or without Nrdp1 knockout on the pre-coated matrigel surface in a confluent monolayer status to simulate the normal vascular wall structure in vitro, and the results demonstrated that pan-laminin and perlecan in the basement membrane were dramatically degraded beneath Nrdp1-deficient endothelial cells (Fig. 5D–F). All these observations verified that ecNrdp1 deletion could degrade the extracellular matrix (ECM), leading to apparent holes in VBM.

Retinal blood vessels were stained (A, B) and the relative degradation % (C) for the Laminin α5 and Collagen IV were measured in Nrdp1fl/fl and Nrdp1Tek-cre mice (n = 6). Scale bar, 20 μm. Immunostaining (D, E) and the relative degradation % (F) for pan-laminin and perlecan in the matrigel beneath the seeded monolayer of HUVECs-gNS and HUVECs-gNrdp1 (n = 6). Nuclei were stained by DAPI. Scale bar, 50 μm. Representative images of HE staining (Scale bar, 50 μm) with immunofluorescence staining of the indicated proteins (Scale bar, 50 μm) (G) and TEM (H) of the aorta from 8-week-old Nrdp1Tek-cre mice and their littermate control Nrdp1fl/fl mice (Scale bar, left, 10 μm; right, 5 μm). L, EC, and EL indicate lumen, endothelial cells, and elastin, respectively. Yellow arrowheads in G pointed to detached endothelial cell from the vessel wall. I Representative images under TEM observation of pulmonary capillary of Nrdp1Tek-cre and control Nrdp1fl/fl mice (Scale bar, 500 nm). Yellow arrowheads pointed to detached endothelial cell from the VBM. EC, EP, and RBC indicate endothelial cells, epithelial cells, and red blood cells, respectively. J Western blot analysis of indicated proteins in HUVECs with or without Nrdp1 knockout, along with retroviruses expressing Flag-Nrdp1. Representative micrographs (K, Scale bar, 100 μm) and Quantification (L, n = 3) of transmigrated DU145 cells in artificial vasculature system. M The fluorescence intensity of dextran was measured in lower chamber normalized to HUVEC-gNS cells (n = 3). All quantitative data were shown as mean ± SD. Statistical significance was determined by two-tailed unpaired Student’s t-test (C, F) and one-way ANOVA with multiple comparisons (L, M). All experiments in K–M repeat three times with each triplicate.
On the other hand, CD31 and alpha-smooth muscle actin staining showed the shedding of endothelial cells from the aorta in ecNrdp1-knockout mice, although no obvious alterations in the cellular structure existed, including the endothelium and smooth muscle layer (Fig. 5G). Furthermore, transmission electron microscopy (TEM) observation on blood vessels showed that the endothelium lining the inner wall of the aorta of WT mice exhibited a flat and smooth surface, while those in ecNrdp1-knockout mice were uneven and rough (Fig. 5H). Under scanning electron microscopy (SEM), an irregular endothelial lamina cover could also be seen in the aorta from ecNrdp1-knockout mice (Supplementary Fig. 5A). Additionally, the tunica intima of the aorta was strikingly disrupted in the ecNrdp1-knockout mice under TEM (Fig. 5H). Similar results were observed in ecNrdp1-knockout mice under the control of the Cdh5 promoter (Supplementary Fig. 5B), indicating that ecNrdp1 deficiency could impair the integrity of the internal elastic membrane and thus contribute to vascular structure abnormalities.
Pulmonary capillaries are regarded as a prime site for tumor cell extravasation during the lung metastatic process [53], and the endothelial cells are essential for maintaining the integrity of the pulmonary vasculature and facilitating gas exchange in the respiratory system. Thus, we also observed the ultrastructure of the lung blood capillaries under TEM. As expected, the ecNrdp1-knockout mice showed significant abnormal detachment between endothelium and VBM compared to wild-type mice, and the shedding of endothelial cells into the vascular lumen resulted in a substantial extrusion of red blood cells (Fig. 5I), consistent with the reduced adhesive capacity of endothelium to the underlying VBM. This was also observed in ecNrdp1-knockout mice under the control of Cdh5 promoter (Supplementary Fig. 5C). All these data indicate that ecNrdp1 deficiency could contribute to VBM degradation, leading to the disruption of the vasculature and detachment of endothelial cells from the VBM. Based on the notion that there might be a direct connection between detached endothelium and tumor metastasis as previously reported [54], these results suggest that ecNrdp1 may provide a crucial molecular signal in driving tumor metastasis.
Deficiency of ecNrdp1 enhances cancer cells to penetrate blood vessels
To further determine whether VBM degradation is highly correlated with tumor metastasis, we performed the in vitro trans-endothelial migration assay to mimic tumor metastasis. For this purpose, HUVECs with or without Nrdp1 were seeded on the pre-coated matrigel surface, allowing them to form a confluent monolayer. Then, DU145 cells were cultured at the top of the confluent monolayer. After 24 h, transmigrated DU145 cells in the bottom layer of the chamber were counted. As depicted in Fig. 5J–L, more DU145 cells infiltrated the artificial vasculature by ecNrdp1-knockout HUVECs than those by wild-type HUVECs, which could be antagonized by re-expression of Nrdp1. Moreover, the permeability of HUVECs monolayer with Nrdp1 deletion was also increased, as detected by 70-kDa FITC-dextran as an intermediator, which could be partially reversed by re-expression of Flag-Nrdp1 (Fig. 5M). Cumulatively, these observations indicated that Nrdp1-deficient endothelial cells may provide heightened molecular signals to actively disrupt the integrity of the VBM, thus facilitating tumor metastasis.
Deletion of ecNrdp1 facilitates metastasis by Fam20C expression
We postulated that Nrdp1-deficient VECs could secrete some molecules to induce VBM degradation and thus cancer metastasis. Indeed, CM from Nrdp1-deficient MLECs and HUVECs significantly promoted the invasion ability of cancer cells, including mouse-derived B16 or B16F10 and human-derived DU145 or HCT116 cells, as estimated by the transwell assay (Fig. 6A–D). To identify the endothelial cells-derived factor(s) that facilitates tumor metastasis, ultra-centrifugal filters were utilized to separate the supernatant with >10-kDa and <10-kDa. We showed that the supernatant from >10-kDa enhanced DU145 cell invasion (Supplementary Fig. 6A, B), demonstrating that molecules greater than 10-kDa in CM contribute to ECM degradation. On the other hand, we performed label-free LC–MS spectrometry analysis to detect the released proteins from the CM from HUVECs and MLECs with 10-kDa centrifugal filters and identified 33 upregulated and 18 downregulated proteins in both Nrdp1-deficient HUVECs and MLECs (Fig. 6E, Supplementary Fig. 6C). In order to identify the key endothelial cells-derived molecules that are responsible for ECM degradation, we knocked down expression of the top 10 and other randomly selected 8 genes among the 33 upregulated genes by their specific shRNAs in HUVECs with Nrdp1 deficiency (Fig. 6E, Supplementary Fig. 6D). The results revealed that knockdown of Fam20C (a typical member of Fam20 family) and Basigin (BSG) almost completely counteract the pro-invasion abilities of CM from Nrdp1-deficient HUVECs (Supplementary Fig. 6E, F). Moreover, recombinant Fam20C protein could significantly enhance metastatic ability of tumor cells (Supplementary Fig. 6G). Fam20C, which encodes a secretory kinase involved in phosphorylating numerous secreted proteins, including ECM, has been reported to drive invasion in several cancer cells [55,56,57]. Therefore, we focused on Fam20C, and the potential role of BSG remained to be illuminated in the future.

Representative images (A, C, Scale bar, 100 μm) and numbers of invasion for cancer cells (B, D) (n = 3) as indicated upon treatment with CM from freshly isolated MLECs of Nrdp1fl/fl and Nrdp1Tek-cre mice (A, B) and HUVECs with or without Nrdp1 knockout (C, D). E Heatmap showing Nrdp1-mediated 33 upregulated proteins in CM from HUVECs and MLECs. Western blot analysis of the indicated proteins in serum-free CM from HUVECs with Nrdp1 and/or Fam20C knockdown (F) and MLECs from Nrdp1fl/fl and Nrdp1Tek-cre mice (G). Representative images of DU145 cell invasion (H, Scale bar, 100 μm) and numbers of invasion for tumor cells (n = 3) (I) upon treatment with CM from HUVECs with Nrdp1 and/or Fam20C knockdown. J Western blot analysis of phosphorylated proteins in matrigel by anti-PY99 antibody upon treatment with serum-free CM from HUVECs with or without Nrdp1 knockout. Recombinant Fam20C protein was added as a positive control. K 293T cells were transfected with Fam20C-Flag and increasing doses of GFP-tagged Nrdp1, and the indicated proteins in cell lysate or CM were blotted. L 293T cells were transfected with increasing doses of GFP-tagged Nrdp1, and Fam20C mRNA was quantified by qRT-PCR (n = 3). 293T cells were transfected with Fam20C-Flag and GFP-Nrdp1 (M) or GFP-Nrdp1-C34S/H36Q (N), followed by treatment with inhibitors. The indicated proteins in cell lysates or CM were blotted. All quantitative data were shown as the mean ± SD. Statistical significance was determined by two-tailed unpaired Student’s t-test (B, D) and one-way ANOVA with multiple comparisons (I, L). All experiments in A–D, H–I were repeated three times with each triplicate. The experiments in G, J, K–N were repeated three times with similar results.
To address roles of Fam20C in ecNrdp1-mediated metastasis, we initially detected the secreted Fam20C from CM by centrifuge concentration. With cystatin C, a kind of cysteine protease inhibitor [58], as a loading control, knockdown of ecNrdp1 significantly enhanced Fam20C secretion in HUVECs and MLECs, which were abrogated by shRNA-mediated Fam20C knockdown (Fig. 6F, G, Supplementary Fig. 6H). In line, Fam20C knockdown inhibited the DU145 cell invasion ability under CMs from HUVECs with and without Nrdp1 deficiency (Fig. 6H, I), suggesting that Fam20C produced by endothelial cells can act as an active signal to promote ecNrdp1 deficiency-mediated pro-metastasis ability. Fam20C has been reported to be necessary for tumor cells to invade the VBM, which predominantly relies on its kinase activity [55]. In line with this, CM with more Fam20C could enhance the phosphorylation level of VBM components (Fig. 6J). These findings indicate that the accelerated tumor metastasis caused by ecNrdp1 deficiency may be attributed to Fam20C expression and its kinase activity.
Nrdp1 promotes Fam20C degradation via the lysosomal pathway
Given that antibody to detect intracellular Fam20C is unavailable, we ectopically expressed Flag-tagged Fam20C into 293T cells, and the results showed that GFP-tagged Nrdp1 expression dose-dependently decreased intracellular and secreted Flag-tagged Fam20C protein without effect on its mRNA (Fig. 6K, L). As Nrdp1 is a RING finger-type of ubiquitin E3 ligase [24,25,26,27, 42], we assessed whether Nrdp1 facilitates Fam20C degradation upon its E3 ligase activity. As for this, 293T cells were transfected with Fam20C-Flag and GFP-Nrdp1, followed by treatment with proteasome inhibitors MG132 and PS341 as well as lysosome inhibitors CQ and NH4CL. The results revealed that Nrdp1 overexpression-induced reduction of Fam20C accumulation and secretion was dramatically blocked under treatment with CQ and NH4CL but not MG132 and PS341 (Fig. 6M), suggesting that Nrdp1-mediated Fam20C degradation is regulated by the lysosome. Notably, previous reports revealed that two residues in the RING finger of Nrdp1 (C34 and H36) regulate its E3 enzyme activity [24]. Here, we identified that C34S/H36Q mutants could still reduce Fam20C expression to a similar degree as wild-type Nrdp1 (Fig. 6N), excluding the possibility that Nrdp1-mediated Fam20C degradation was dependent on its E3 ligase activity.
Increased ecNrdp1 by gene delivery or chemical induction alleviates lung metastasis in mice
To evaluate whether increased ecNrdp1 expression can alleviate cancer cell dissemination, we intravenously injected AAV into mice (Supplementary Fig. 7A), carrying the GFP or mouse Nrdp1-Flag (mNrdp1-Flag) gene under the control of the endothelial cell-specific Tie 2 promoter. On day 21 after AAV infection, specific expression of mNrdp1-Flag protein was confirmed in isolated MLECs (Supplementary Fig. 7B) but not in other organs such as heart, liver, and brain of mice (Supplementary Fig. 7C). To elucidate the role of ecNrdp1 on tumorigenesis, we subcutaneously transplanted B16 cells into C57BL/6 mice and showed that gain-of-function of ecNrdp1 has rare influence on primary tumor growth (Supplementary Fig. 7D, E) and the proportions of tumor-infiltrating immune cells (Supplementary Fig. 7F). Subsequently, we injected B16 melanoma cells into the tail veins of C57BL/6 mice. Twenty days later, mice were sacrificed, and found that specific ecNrdp1 overexpression significantly reduced lung metastatic nodules in mice with AAV-mNrdp1-Flag infection compared to control mice with AAV-GFP infection (Fig. 7A, B), which did not affect the infiltration of immune cells (Supplementary Fig. 7G). The similar result could also be seen by HE staining for pathological evaluation and in BALB/c mice (Fig. 7C, D, Supplementary Fig. 7H, I), which are known to be more susceptible to metastasis of B16 cells compared to C57BL/6 mice [59]. Moreover, we intravenously injected B16 cells into ecNrdp1-knockout mice with AAV-mNrdp1-Flag or AAV-GFP infections. Following the procedure shown in Supplementary Fig. 7A, we observed that the increased lung metastatic nodules in ecNrdp1-knockout mice was completely abolished upon ecNrdp1 gain-of-function therapy (Fig. 7E, F).

B16 cells were intravenously injected into C57BL/6 mice (A, B, n = 8) or BALB/c mice (C, D, n = 10) after AAV administration. Macroscopy of lungs (A, C) and numbers of macrometastatic lung foci (B, D) were shown. B16 cells were intravenously injected into Nrdp1Cdh5-cre mice after AAV administration. Macroscopy of lungs (E) and numbers of the macrometastatic lung foci (F) were shown (n = 4). G Western blot analysis of indicated proteins in HUVECs after ABPN administration for 24 h. H Scheme for the tumor model. ABPN was administrated intravenously every other day and followed by melanoma tail vein injection on day 7. Black and yellow Arrows represent time point for ABPN and B16 cells injection, respectively. I Western blot analysis of indicated proteins in freshly isolated MLECs from BALB/c mice after ABPN administration. #1–4 represent four mice upon different treatment. J, K B16 cells were injected intravenously into BALB/c background mice after APBN administration (n = 6). Macroscopy of lungs (J), the number of macrometastatic lung foci (K) were shown. All quantitative data were shown as the mean ± SEM. Statistical significance was determined by two-tailed unpaired Student’s t-test.
Next, we investigated the effect of the Nrdp1 activator, ABPN, which has been shown to induce Nrdp1 expression and exhibit antitumor activity against certain cancer cells [30]. Indeed, ABPN obviously induced Nrdp1 expression in HUVECs without triggering apoptosis, as estimated by caspase 3 activation (Fig. 7G). We further evaluated the in vivo activity of ABPN using the metastatic model (Fig. 7H). Western blot analysis revealed a distinct increase in the Nrdp1 protein levels in isolated MLECs before cancer cell injection (Fig. 7I). In parallel, sustained injection of ABPN every other day significantly reduced lung metastatic foci in BALB/c mice compared to controls (Fig. 7J, K, Supplementary Fig. 7J), with no noticeable alteration in body weights (Supplementary Fig. 7K). To address whether the inhibitory effect of ABPN on metastasis may be attributed to its direct impact on melanoma cells, we exposed B16 cells to increasing concentrations of ABPN and found no discernible influence on apoptosis or Nrdp1 protein levels (Supplementary Fig. 7L). Especially, ABPN treatment presented similar effects on lung metastasis of B16 cells with and without Nrdp1 silence (Supplementary Fig. 7M, N). These findings support the notion that ABPN’s inhibition of tumor metastasis depends on ecNrdp1 rather than tumoral Nrdp1. Interestingly, ABPN treatment seems to have a stronger inhibitory effect on tumor metastasis than ectopic ecNrdp1 expression (Fig. 7C, J), probably because ABPN has other potential targets besides Nrdp1 that may synergistically contribute to antitumor metastatic ability, which remains to be further investigated.
Discussion
Tumor metastasis can be portrayed as a multi-step succession of events termed the invasion–metastasis cascade, which eventually lead to the outgrowth of metastatic nodules in distant tissues [60]. As the first part of this process, cancer cells intravasate into nearby vessels and enter the circulation, subsequently entering secondary organ sites. The final step of the metastatic cascade involves the outgrowth and propagation of disseminated tumor cells into overt tumors at the metastatic site. To form invade and metastases, the microenvironment needs to be adapted to favor remodeling of the ECM, immune cell inhibition, and angiogenesis [4, 60]. In these events, tumor endothelial cells and their interaction with cancer cells exert an important role [61]. It has been well known that upon the solid tumor growth, a tumor microenvironment with hypoxia, ischemia, acidosis, and high interstitial pressure is gradually developed, which releases abundant growth factors and cytokines such as VEGF. These chemokines not only stimulate angiogenesis and lymphangiogenesis to meet the needs of tumor growth and metabolism but also act by recruiting various peripheral blood cells from the circulation into the tumor microenvironment. VEGF, a crucial secretory factor that maintains human endothelial function and promotes cell mitosis and vascular permeability, plays an irreplaceable role in tumor growth, angiogenesis, invasion, and metastasis [16, 62]. Based on a dramatically reduced Nrdp1 protein in tumor-associated VECs and its correlation with higher metastatic probability, herein we showed that the CM from hypoxia-treated cancer cells induced extensive Nrdp1 downregulation in HUVECs and MLECs. For this event, we identified that cancer-derived VEGF was a major player because neutralizing VEGF with bevacizumab and VEGFR2 inhibitor Cabozantinib significantly abrogated ecNrdp1 decrease induced by CM from hypoxia-treated cancer cells. Especially, recombinant human VEGF also dose-dependently reduced Nrdp1 protein in HUVECs and MLECs, and VEGF may account for lower ecNrdp1 expression under physiological conditions, as estimated in mouse retinal vasculature. Further, we showed that VEGF degraded Nrdp1 protein in a ubiquitin-proteasome-dependent manner. On the other hand, we showed that VEGF stimulated transcriptional expression of CHIP in HUVECs. A single-cell RNA sequencing dataset also revealed that the level of CHIP expression was greatly increased in mouse tumor endothelial cells than normal endothelial cells. CHIP is a member of a family of U-box ubiquitin E3 ligases that regulates the stability and functions of multiple proteins in different cell types, such as NF-κB-inducing kinase, runt-related transcription factor 2, multiple TRAF family members (TRAF2, TRAF5, and TRAF6), and others, which plays critical roles in neurodegeneration, immunity, inflammation, and bone remodeling and aging, as reviewed [63]. Indeed, CHIP interacted with and triggered ubiquitination of Nrdp1 protein. Notably, how VEGF activates CHIP expression and whether CHIP-induced Nrdp1 ubiquitination is chaperone protein-dependent or –independent remains to be further investigated.
VBM has traditionally been perceived as a static protein network that provides structural support for tissues. Recently, increasing studies have focused on the multiple roles of VBM in development, cell migration, polarity, and tumor metastasis [64, 65]. More interestingly, lung metastasis of cancer cells rather than primary tumor growth was significantly increased in conditional knockdown of Nrdp1 in VECs driven by either Tek (Tie2)-cre or Cdh5-cre. Although ecNrdp1 deficiency did not alter VECs-tumor cell adhesion and gap junctions between pericytes and VECs, we showed that ecNrdp1 knockdown could degrade the ECM, thus disrupting the integrity of VBM and enhancing the ability of cancer cells to penetrate blood vessels in vivo and in vitro. To physically extravasate blood vessels and further seed distant organs, proteases that are produced by several cell types in the tumor environment are required for proteolytic degradation. Despite cleaving cell adhesion molecules and degrading the basement matrix, the proteases can also facilitate cancer metastasis by promoting protease-based cell signaling activation [66]. Based on these findings, we showed that knockdown of Fam20C and BSG almost completely counteract the ability to promote cancer cell invasion of CM from Nrdp1-deficient HUVECs. Because it has been well known that Fam20C can drive invasion in several cancer cells by its kinase activity, which phosphorylates numerous secreted proteins, including ECM [55,56,57], we focused on role of Fam20C. Indeed, ecNrdp1 knockdown significantly enhanced Fam20C secretion in HUVECs and MLECs and inhibited the DU145 cell invasion ability under CM from HUVECs. Finally, we showed that Nrdp1 promotes Fam20C degradation in endothelial cells. However, the degradation was independent E3 ligase activity of Nrdp1, and the lysosomal pathway appeared be required for this. How Nrdp1 induced lysosome-dependent Fam20C degradation remained to be future investigated.
The AAV system with genetic modification has emerged as a leading platform for precise gene delivery in clinical therapy for various diseases. Therefore, we employed this system to achieve endothelial cell-specific overexpression of Nrdp1 in mice. The results showed that AAV-mNrdp1-Flag infection remarkably inhibited tumor metastasis in C57BL/6 or BABL/c mice and also antagonized ecNrdp1 knockout-increased lung metastasis. It is worthwhile to point out that we did not show that ABPN increased Nrdp1 expression and induced death in B16 cells like previously seen in BxPC3 and HPAC pancreatic cancer cell lines [30]. The reasons to cause the discrepancy remain to be investigated. However, we did show that ABPN could upregulate ecNrdp1 expression in vitro and in vivo. In line, ABPN effectively prevented Nrdp1-mediated lung colonization of melanoma cells. All these data support the therapeutic potential to interfere ecNrdp1 expression.
In summary, we explored a new mechanism for VEGF to enhance cancer metastasis by degrading Nrdp1 and increasing Fam20C secretion of vascular endothelial cells, and thus inducing ECM degradation and increasing permeability of endothelium.
Materials and methods
Human colorectal cancer samples
Tissue microarrays of human colorectal tumors and paired adjacent normal tissues were purchased from Shanghai TUFEI Biotech Co. (TF colon-01). The microarray comprises a total of 74 paired patient tissues and 25 independent tumor cases after discarding the damaged sections. A multi-organ microarray was purchased from Shanghai OUTDO Biotech Co. (Horg C120PG04).
Mice
Nrdp1fl/fl mice were established at GemPharmatech (Shanghai, China). Tek (Tie2)-Cre (Strain #: 004128) or Cdh5-Cre mice (Strain #: 006137) were generous gifts from Pro. Jun-Ke Zheng in our laboratory. Nrdp1fl/fl mice were further crossed with Tek (Tie2)-Cre or Cdh5-Cre mice to achieve specifically deletion of Nrdp1 in endothelial cells (termed as ecNrdp1-knockout mice). C57BL/6 mice and BALB/c mice were purchased from GemPharmatech. These mice were randomly assigned to experimental groups. All mice were housed in suitable temperature (22–25 °C with a 12 h light–dark cycle) and humidity environment with regular chow food and water supplement. For treatment of ABPN, which was synthesized by Shanghai Apeptide Co., Ltd according to previous research [67], ABPN (1 mg/kg) was administered intravenously every other day. All Animal care and experiments were performed in strict accordance with the “Guide for the Care and Use of Laboratory Animals” and the “Principles for the Utilization and Care of Vertebrate Animals” and were approved by the committee for human treatment of animals at Shanghai Jiao Tong University School of Medicine.
Lung metastasis assays
To establish mouse tumor metastasis models, suspensions of LLC (5 × 105 cells per mouse), B16 (2.5 × 105 cells per mouse), or B16F10-luc (2 × 105 cells per mouse) were intravenously injected into the tail veins of 6-to 8-week-old ecNrdp1-knockout and wild-type littermate control mice. Twenty days after cell injection, the number of lung metastases was calculated, and lungs with metastatic foci were harvested and further fixed with 4% paraformaldehyde for histological analysis. For metastatic colonization visualization, mice were injected intraperitoneally with D-luciferin at a dose of 150 mg/kg. After 10 min, images and photonic signal intensities were quantified with an IVIS Spectrum.
Plasmids, shRNA, CRISPR-Cas9 and virus construction
Information on the plasmids is provided in Supplementary Table 1. ShRNAs targeting genes or a normal control (NC) shNC were constructed and then delivered using a lentivirus vector. To obtain Nrdp1 knockout cells, sgNrdp1 sequences were constructed in the vector of pLentiV2-puro and packaged into lentivirus. And then HUVECs were infected with lentivirus and subjected to selection with puromycin. The target sequence was listed in Supplementary Table 2. Nrdp1 coding sequence (CDS) was inserted into a retroviral plasmid pBabe-puro and GFP-C1-vector. For rescue experiments, Flag-tagged Nrdp1 CDS with multiple point mutations in sgNrdp1 target sequence was inserted into the retroviral plasmid pBabe-puro. Retrovirus was produced using lipofectamine 2000 transfection method with the packaging plasmids gag-pol and VSVG, while lentiviral was packaged with pMG2G and psPAX2. For AAV, the mNrdp1 cDNA sequence with the Flag sequence was cloned into the GPAAV2-Tie-MCS-tie enhancer-WPRE plasmid. The AAV-mNrdp1-Flag and AAV-GFP constructs were generated by Genomeditech (Shanghai, China). These constructs were delivered through intravenous tail injection into mice at a dose of 5 × 1010 vg/mouse/100 μl.
Cell lines and cell culture
HUVECs were freshly isolated from the human umbilical cord vein, dispersed with 0.01% (w/v) collagenase/dispase solution (Roche) and 1 μg/ml DNase I at 37 °C for 15 min, and then ECs were collected and suspended in complete ECM medium (Sciencell). HUVECs that were used for VEGF signaling pathway experiments were only cultured for up to 4–6 passages in a complete medium. Primary MLECs were isolated from mice as described [68, 69]. MLECs that were used for experiments were only cultured for up to eight passages in a complete medium. Human cell lines including 293T, DU145, and HCT116, mouse cell lines including LLC, B16, and B16F10-luc were purchased from Cell Bank of Chinese Academy of Sciences, Shanghai. MDA-LM2 and MDA-MB-231 were generous gifts from Pro. Qian Zhao (Shanghai Jiao Tong University School of Medicine). These cells were authenticated and mycoplasma-free. 293T, DU145, HCT116, MDA-LM2, MDA-MB-231, and LLC cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS (Sigma), 100 IU/ml penicillin, and 100 μg/ml streptomycin. B16 and B16F10-Luc cells were cultured in DMEM/F12 (Gibco) with 10% FBS. These cells were humidified incubated at 37 °C under 5% CO2.
Immunoprecipitation and denaturing ubiquitination assay
293T cells were treated with 10 µM of the proteasome inhibitor MG132 for 8 h, harvested and lysed with 1×RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM PMSF, and 1×protease inhibitor cocktail, PH 7.6) after transfection, and briefly sonicated. After centrifugation at 12,000 × g for 10 min, the supernatants of whole-cell lysates were incubated with anti-Flag M2 beads (Sigma-Aldrich) at 4 °C overnight followed by five times wash with RIPA and then subjected to western blotting analysis. For denatured IP ubiquitination assay, similarly, the transfected cells were treated with MG132 for 8 h, and lysed in denatured IP buffer (50 mM Tris-HCl, pH 6.8, 2% SDS). To briefly dissociate protein–protein interactions, cells were sonicated and boiled at 100 °C for 10 min. After centrifugation at 12,000 × g for 10 min at 4 °C, 900 µl of supernatants was diluted by 9.1 ml 1×RIPA buffer and subjected to immunoprecipitation with anti-Flag M2 beads, followed by western blotting analysis to visualize polyubiquitylated protein bands.
Cell-free RNA-conditioned medium and LC–MS
HUVECs or MLECs were plated and incubated in serum-free medium for 24 h. The medium of each group was collected, centrifuged at 2000 × g for 10 min to remove debris, and the supernatants were concentrated using 10-kDa filtration system (Millipore). Total proteins were subjected to liquid chromatography-tandem mass spectrometry (LC–MS/MS) analysis.
The eluted peptides were lyophilized using a SpeedVac (ThermoSavant) and resuspended in 10 μl 1% formic acid 5% acetonitrile. All mass spectrometric experiments are performed on a Thermo Fusion Lumos mass spectrometer connected to an Easy-nLC 1200 via an Easy Spray (Thermo Fisher Scientific). The peptides mixture was loaded onto a 15 cm with 0.075 mm inner diameter column packed with C18 2-μm Reversed Phase resins (PepMap RSLC), and separated within a 60 min linear gradient from 95% solvent A (0.1% formic acid/2% acetonitrile/98% water) to 28% solvent B (0.1% formic acid/80% acetonitrile) at a flow rate of 300 nl/min. The spray voltage was set to 2.1 KV and the temperature of ion transfer capillary was 275 °C, and RF lens was 60%. The mass spectrometer was operated in positive ion mode and employed in the data-dependent mode to automatically switch between MS and MS/MS using the Tune and Xcalibur 4.0.27.19 software package. One full MS scan from 350 to 1500 m/z was acquired at high-resolution R = 60,000 (defined at m/z = 400), followed by fragmentation of the twenty most abundant multiply charged ions (singly charged ions and ions with unassigned charge states were excluded), for ions with charge states 2–7 and collision energy of 30%. Dynamic exclusion was used at auto. For database search and data analysis, all MS/MS ion spectra were analyzed using PEAKS 11.0 (Bioinformatics Solutions) for processing, de novo sequencing, and database searching. The digestion enzyme semiTrypsin allowed for two missed tryptic cleavages, Carbamidomethyl of cysteine specified as a fixed modification, and oxidation of methionine and acetyl of the N-terminus as variable modifications. FDR estimation was enabled. PSM were filtered for −10logP ≥ 20, and proteins were filtered for −10logP ≥ 15 and one unique peptide. For all experiments, this gave an FDR of <1% at the peptide-spectrum match level. Proteins sharing significant peptide evidence were grouped into clusters.
Public databases analysis
Gene expression data of COAD samples from TCGA database was analyzed by R package TCGA biolinks (2.25.3). Figure 2K was generated from the NCBI’s Gene Expression Omnibus (GEO) database GSE118904.
Quantification and statistical analysis
Statistical analysis was performed using Prism 8.0. Quantitative data were presented as the mean ± SD or mean ± SEM. The two-tailed unpaired Student’s t-test, two-sided log-rank test, or two-way ANOVA were used for comparisons between two groups. Multiple group comparisons were conducted by one-way ANOVA plus Fisher’s least significant difference (LSD) test. P < 0.05 was considered statistically significant. All experiments were repeated independently at least three times with similar results.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files, or available from the corresponding author on reasonable request.
References
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46.
Castaneda M, den Hollander P, Kuburich NA, Rosen JM, Mani SA. Mechanisms of cancer metastasis. Semin Cancer Biol. 2022;87:17–31.
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–64.
Preuss SF, Grieshober D, Augustin HG. Systemic reprogramming of endothelial cell signaling in metastasis and cachexia. Physiology. 2023;38:0.
Pasquier J, Ghiabi P, Chouchane L, Razzouk K, Rafii S, Rafii A. Angiocrine endothelium: from physiology to cancer. J Transl Med. 2020;18:52.
Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018;9:115.
Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–52.
Han L, Lin X, Yan Q, Gu CC, Li MS, Pan L, et al. PBLD inhibits angiogenesis via impeding VEGF/VEGFR2-mediated microenvironmental cross-talk between HCC cells and endothelial cells. Oncogene. 2022;41:1851–65.
De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17:457–74.
Strilic B, Yang L, Albarran-Juarez J, Wachsmuth L, Han K, Muller UC, et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536:215–8.
Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400.
Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010;21:21–6.
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–9.
Ghalehbandi S, Yuzugulen J, Pranjol MZI, Pourgholami MH. The role of VEGF in cancer-induced angiogenesis and research progress of drugs targeting VEGF. Eur J Pharmacol. 2023;949:175586.
Ye W. The complexity of translating anti-angiogenesis therapy from basic science to the clinic. Dev Cell. 2016;37:114–25.
Liu ZL, Chen HH, Zheng LL, Sun LP, Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct Target Ther. 2023;8:198.
Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, et al. Endothelial Notch1 activity facilitates metastasis. Cancer Cell. 2017;31:355–67.
Cartier A, Leigh T, Liu CH, Hla T. Endothelial sphingosine 1-phosphate receptors promote vascular normalization and antitumor therapy. Proc Natl Acad Sci USA. 2020;117:3157–66.
Hongu T, Pein M, Insua-Rodriguez J, Gutjahr E, Mattavelli G, Meier J, et al. Perivascular tenascin C triggers sequential activation of macrophages and endothelial cells to generate a pro-metastatic vascular niche in the lungs. Nat Cancer. 2022;3:486–504.
Tavora B, Mederer T, Wessel KJ, Ruffing S, Sadjadi M, Missmahl M, et al. Tumoural activation of TLR3-SLIT2 axis in endothelium drives metastasis. Nature. 2020;586:299–304.
Kong DG, Zhou HB, Neelakantan D, Hughes CJ, Hsu JY, Srinivasan RR, et al. VEGF-C mediates tumor growth and metastasis through promoting EMT-epithelial breast cancer cell crosstalk. Oncogene. 2021;40:964–79.
Abdullah JM, Li X, Nachtman RG, Jurecic R. FLRF, a novel evolutionarily conserved RING finger gene, is differentially expressed in mouse fetal and adult hematopoietic stem cells and progenitors. Blood Cells Mol Dis. 2001;27:320–33.
Diamonti AJ, Guy PM, Ivanof C, Wong K, Sweeney C, Carraway KL 3rd. An RBCC protein implicated in maintenance of steady-state neuregulin receptor levels. Proc Natl Acad Sci USA. 2002;99:2866–71.
Qiu XB, Goldberg AL. Nrdp1/FLRF is a ubiquitin ligase promoting ubiquitination and degradation of the epidermal growth factor receptor family member, ErbB3. Proc Natl Acad Sci USA. 2002;99:14843–8.
Chen L, Siddiqui S, Bose S, Mooso B, Asuncion A, Bedolla RG, et al. Nrdp1-mediated regulation of ErbB3 expression by the androgen receptor in androgen-dependent but not castrate-resistant prostate cancer cells. Cancer Res. 2010;70:5994–6003.
Yen L, Cao Z, Wu X, Ingalla ER, Baron C, Young LJ, et al. Loss of Nrdp1 enhances ErbB2/ErbB3-dependent breast tumor cell growth. Cancer Res. 2006;66:11279–86.
Qiu XB, Markant SL, Yuan J, Goldberg AL. Nrdp1-mediated degradation of the gigantic IAP, BRUCE, is a novel pathway for triggering apoptosis. EMBO J. 2004;23:800–10.
Wald JH, Hatakeyama J, Printsev I, Cuevas A, Fry WHD, Saldana MJ, et al. Suppression of planar cell polarity signaling and migration in glioblastoma by Nrdp1-mediated Dvl polyubiquitination. Oncogene. 2017;36:5158–67.
VanderVorst K, Hatakeyama J, Berg A, Lee H, Carraway KL 3rd. Cellular and molecular mechanisms underlying planar cell polarity pathway contributions to cancer malignancy. Semin Cell Dev Biol. 2018;81:78–87.
Byun S, Shin SH, Lee E, Lee J, Lee SY, Farrand L, et al. The retinoic acid derivative, ABPN, inhibits pancreatic cancer through induction of Nrdp1. Carcinogenesis. 2015;36:1580–9.
Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. 2014;14:430–9.
Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther. 2020;5:28.
Munaut C, Lorquet S, Pequeux C, Blacher S, Berndt S, Frankenne F, et al. Hypoxia is responsible for soluble vascular endothelial growth factor receptor-1 (VEGFR-1) but not for soluble endoglin induction in villous trophoblast. Hum Reprod. 2008;23:1407–15.
Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med. 2001;7:345–50.
E G, Cao Y, Bhattacharya S, Dutta S, Wang E, Mukhopadhyay D. Endogenous vascular endothelial growth factor-A (VEGF-A) maintains endothelial cell homeostasis by regulating VEGF receptor-2 transcription. J Biol Chem. 2012;287:3029–41.
Mamer SB, Chen S, Weddell JC, Palasz A, Wittenkeller A, Kumar M, et al. Discovery of high-affinity PDGF-VEGFR interactions: redefining RTK dynamics. Sci Rep. 2017;7:16439.
Romon R, Adriaenssens E, Lagadec C, Germain E, Hondermarck H, Le Bourhis X. Nerve growth factor promotes breast cancer angiogenesis by activating multiple pathways. Mol Cancer. 2010;9:157.
Gerhardt H. VEGF and endothelial guidance in angiogenic sprouting. Organogenesis. 2008;4:241–6.
Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–77.
Kim H, Kim M, Im SK, Fang S. Mouse Cre-LoxP system: general principles to determine tissue-specific roles of target genes. Lab Anim Res. 2018;34:147–59.
Payne S, De Val S, Neal A. Endothelial-specific Cre mouse models. Arterioscler Thromb Vasc Biol. 2018;38:2550–61.
Wang C, Chen T, Zhang J, Yang M, Li N, Xu X, et al. The E3 ubiquitin ligase Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon. Nat Immunol. 2009;10:744–52.
Yang M, Chen T, Li X, Yu Z, Tang S, Wang C, et al. K33-linked polyubiquitination of Zap70 by Nrdp1 controls CD8(+) T cell activation. Nat Immunol. 2015;16:1253–62.
Shenoy AK, Lu J. Cancer cells remodel themselves and vasculature to overcome the endothelial barrier. Cancer Lett. 2016;380:534–44.
Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–9.
Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, et al. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol. 2015;208:821–38.
Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–23.
Chiaverina G, di Blasio L, Monica V, Accardo M, Palmiero M, Peracino B, et al. Dynamic interplay between pericytes and endothelial cells during sprouting angiogenesis. Cells. 2019;8:1109.
Paulsson M. Basement membrane proteins: structure, assembly, and cellular interactions. Crit Rev Biochem Mol Biol. 1992;27:93–127.
Jayadev R, Sherwood DR. Basement membranes. Curr Biol. 2017;27:R207–R211.
Yap L, Tay HG, Nguyen MTX, Tjin MS, Tryggvason K. Laminins in cellular differentiation. Trends Cell Biol. 2019;29:987–1000.
Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol. 2001;153:933–46.
Reymond N, d’Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858–70.
Holopainen T, Saharinen P, D’Amico G, Lampinen A, Eklund L, Sormunen R, et al. Effects of angiopoietin-2-blocking antibody on endothelial cell-cell junctions and lung metastasis. J Natl Cancer Inst. 2012;104:461–75.
Tagliabracci VS, Wiley SE, Guo X, Kinch LN, Durrant E, Wen J, et al. A single kinase generates the majority of the secreted phosphoproteome. Cell. 2015;161:1619–32.
Chen X, Zhang J, Liu P, Wei Y, Wang X, Xiao J, et al. Proteolytic processing of secretory pathway kinase Fam20C by site-1 protease promotes biomineralization. Proc Natl Acad Sci USA. 2021;118:e2100133118.
Du S, Guan S, Zhu C, Guo Q, Cao J, Guan G, et al. Secretory pathway kinase FAM20C, a marker for glioma invasion and malignancy, predicts poor prognosis of glioma. OncoTargets Ther. 2020;13:11755–68.
Onopiuk A, Tokarzewicz A, Gorodkiewicz E. Cystatin C: a kidney function biomarker. Adv Clin Chem. 2015;68:57–69.
Foerster F, Boegel S, Heck R, Pickert G, Russel N, Rosigkeit S, et al. Enhanced protection of C57 BL/6 vs Balb/c mice to melanoma liver metastasis is mediated by NK cells. Oncoimmunology. 2018;7:e1409929.
McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol. 2014;16:717–27.
Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–84.
Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69:4–10.
Wang TY, Wang WB, Wang QS, Xie R, Landay A, Chen D. The E3 ubiquitin ligase CHIP in normal cell function and in disease conditions. Ann NY Acad Sci. 2020;1460:3–10.
Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11:5120.
Sekiguchi R, Yamada KM. Basement membranes in development and disease. Curr Top Dev Biol. 2018;130:143–91.
Koblinski JE, Ahram M, Sloane BF. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291:113–35.
Um SJ, Han HS, Kwon YJ, Park SH, Rho YS, Sin HS, et al. Novel retinoic acid derivative ABPN has potent inhibitory activity on cell growth and apoptosis in cancer cells. Int J Cancer. 2003;107:1038–46.
Sokol L, Geldhof V, Garcia-Caballero M, Conchinha NV, Dumas SJ, Meta E, et al. Protocols for endothelial cell isolation from mouse tissues: small intestine, colon, heart, and liver. STAR Protoc. 2021;2:100489.
Xu Z, Guo C, Ye Q, Shi Y, Sun Y, Zhang J, et al. Endothelial deletion of SHP2 suppresses tumor angiogenesis and promotes vascular normalization. Nat Commun. 2021;12:6310.
Acknowledgements
This work is supported by National Key R&D Program of China (2020YFA0803403), National Natural Science Foundation (82293661, 82070893, 92253303, 82200185), Shanghai Scientific and Technological Innovation Action Plan (20JC1410100), Innovative research team of high-level local universities in Shanghai (SHSMU-ZDCX20211800). We thank Yao-Yao Τu for kindly providing human umbilical cord (International Peace Maternity and Child Health Hospital, SJTUSM), Prof. Dr. Sorakin for kindly providing laminin α5 antibody (Institute of Physiological Chemistry and Pathobiochemistry University of Muenster) and Prof. Lei Wang for kindly providing Fam20C antibody (Laboratory of Proteomics, Institute of Biophysics, Chinese Academy of Sciences).
Author information
Authors and Affiliations
Contributions
QL, MG, and GC conceived and designed the project. QL and MG performed most of the experiments. GH, KX, and MZ helped with biochemistry experiments and animal experiments. RS performed the immune experiments. GC, MG, and RS wrote the manuscript and all authors contributed to the writing and/or critical review of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, QQ., Guo, M., He, GH. et al. VEGF-induced Nrdp1 deficiency in vascular endothelial cells promotes cancer metastasis by degrading vascular basement membrane. Oncogene 43, 1836–1851 (2024). https://doi.org/10.1038/s41388-024-03038-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-024-03038-9
- Springer Nature Limited