
Abstract
Androgen deprivation therapies (ADT) are the mainstay treatments for castration-resistant prostate cancer (CRPC). ADT suppresses the androgen receptor (AR) signaling by blocking androgen biosynthesis or inhibiting AR with antiandrogens that target AR’s ligand-binding domain (LBD). However, the ADT’s effect is short-lived, as the AR signaling inevitably arises again, which is frequently coupled with AR-V7 overexpression. AR-V7 is a truncated form of AR that lacks the LBD, thus being constitutively active in the absence of androgens and irresponsive to AR-LBD-targeting inhibitors. Though compelling evidence has tied AR-V7 to drug resistance in CRPC, pharmacological inhibition of AR-V7 is still an unmet need. Here, we discovered a small molecule, SC912, which binds to full-length AR as well as AR-V7 through AR N-terminal domain (AR-NTD). This pan-AR targeting relies on the amino acids 507–531 in the AR-NTD. SC912 also disrupted AR-V7 transcriptional activity, impaired AR-V7 nuclear localization and DNA binding. In the AR-V7 positive CRPC cells, SC912 suppressed proliferation, induced cell-cycle arrest, and apoptosis. In the AR-V7 expressing CRPC xenografts, SC912 attenuated tumor growth and antagonized intratumoral AR signaling. Together, these results suggested the therapeutic potential of SC912 for CRPC.
Similar content being viewed by others


Introduction
Castration-resistant prostate cancer (CRPC) is a relapsed form of prostate cancer from initial androgen deprivation therapies (ADT), its progression manifests reactivated AR signaling activity despite the castration level of androgen. When CRPC develops, second-line ADT is offered to patients for AR signaling blockade [1], including androgen biosynthesis inhibitor abiraterone (Abi) [2]; and the second-generation antiandrogens, such as Enzalutamide (ENZ), which compete for the androgen-binding pocket in the AR ligand-binding domain (LBD) [3]. However, resistance almost always occurs after a brief response period, featuring rising prostate specific antigen (PSA) as a sign of restored AR signaling activity [4, 5]. The underlying mechanisms contributing to Abi and ENZ resistance are AR gene amplification, AR-LBD mutations, and AR splicing variants (AR-Vs) with the LBD truncated. The recently FDA-approved antiandrogen (darolutamide) showed higher LBD binding affinity that was not altered in the LBD mutants [6], which may provide some relief to the drug resistance due to AR amplification and LBD mutation. However, AR-Vs are still undrugged.
Among many documented AR-Vs, AR-V7 is the best-established resistance driver in CRPC [7]. From the molecular perspective, AR-V7 possesses full transcriptional activity derived from the AR N-terminal domain (NTD) and the ability to bind the androgen response elements (ARE) in AR-regulated genes through the DNA binding domain (DBD). Moreover, the loss of LBD unleashes AR-V7 from the constraint of ligand-dependent activation [8], and the acquisition of a unique peptide that reconstitutes NLS permits AR-V7 constant nuclear localization [9]. A recent study revealed that AR-V7 exhibits non-canonical mechanisms of nuclear import [10]. Therefore, unlike full-length AR (AR-FL), whose activation is androgen-dependent and prone to antiandrogen inhibition, AR-V7 is constitutively active regardless of androgen and is irresponsive to any LBD-targeting antiandrogen. From the clinical perspective, AR-V7 mRNA and protein were both found overexpressed in Abi and ENZ-treated CRPC patients and were predictive of worse outcome [11,12,13,14]. The more recent studies uncovered that AR genomic rearrangements, which preferentially give rise to AR-Vs, were detectable in CRPC tissues [15]; these AR intragenic aberrations were particularly enriched in Abi and ENZ-treated patients and were associated with drug resistance [16]. Importantly, AR-V7 functions independently of the AR-FL in prostate cancer cells [17, 18]. Therefore, mounting evidence emphasized the urgency of targeting AR-V7 in CRPC patients.
Tremendous efforts have been dedicated to exploring the pharmacological inhibition of AR-V7. The currently reported approaches can be categorized into: (1) direct targeting AR-V7 via AR-NTD [19] or AR-DBD [20, 21]; (2) reducing the AR-V7 abundance by degrading its protein [22,23,24], disturbing its pre-mRNA splicing [25] or inhibition of AR-V7 mRNA synthesis [26]; (3) inhibiting AR-V7 mediated transcription by targeting the cofactors [27, 28]. Among these approaches, direct targeting is conceptually most desirable as it addresses the origin of the problem. However, it is challenging to directly target the AR-V7, comprising only the NTD and DBD domains. As the AR-DBD is very conserved among hormone receptors, avoiding cross-reactivity with other hormone receptors is difficult [29]. Meanwhile, AR-NTD is composed mostly of “undruggable” intrinsically disordered protein [30, 31].
Since the transcriptional activity of all forms of AR resides mostly in their NTD [32], targeting this domain is still actively pursued despite the technical challenges. To date, EPI compounds are the only class of AR-NTD inhibitor that has entered the clinical trial. Although EPI-506 achieved PSA response in some CRPC patients, the trial was terminated for high pill burden [33]. Concordantly, a high micromolar of EPI-002 was also required to inhibit AR signaling activity in cell-based experiments [34], indicating improved potency is needed for AR-NTD inhibitors. We previously reported a small molecule SC428 that binds to AR-NTD and inhibits AR-FL and AR-V7 biological functions in the 1 – 5 μM range [35]. Importantly, our SC428 work revealed that an AR-NTD inhibitor could indeed sufficiently block AR-V7 mediated AR signaling in CRPC cells [35].
In the present study, we discovered another novel AR-NTD inhibitor SC912 that, has a completely different chemical scaffold from SC428. SC912 is a lead compound of our medicinal chemistry effort to develop novel inhibitors of AR-V7. In this chemical series, we have made 82 derivatives among which SC912 showed the highest potency and selectivity. SC912 inhibited transactivation of AR-FL, AR-LBD mutants, AR-V7, and ARv567es, with the AR-NTD amino acids (aa) 507–531 being indispensable for its target engagement and inhibitory effects. Mechanistically, SC912 impaired AR-V7 mediated transcription, blocked AR-V7 nuclear trafficking, and ARE binding. For the AR-V7 expressing CRPC cells, SC912 antagonized their AR signaling activity and mitigated their castration-resistant growth both in vitro and in vivo, suggesting its therapeutic potential for CRPC.
Result
SC912 inhibited the transactivation of AR-FL and AR-V7 through direct binding
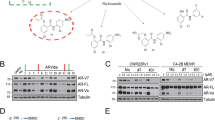
Compound SC912 is a lead compound from our Medicinal Chemistry efforts to identify inhibitors targeting the AR-NTD (Fig. 1A). Using a PSA-luciferase (PSA-Luc) reporter, co-transfected with AR-V7 or ARv567es expressing plasmid into 293 T cells, SC912 potently inhibited both AR-V7 and ARv567es transactivation with an IC50 of 0.36 and 1.01 μM, respectively (Fig. 1B). In contrast, the AR-LBD-targeting agent ENZ showed no inhibitory activity even at 10 μM (Fig. 1B). AR-FL transactivation assays were also performed with wildtype (WT) AR, and a panel of AR-LBD mutants that were previously discovered to confer resistance to antiandrogens: including flutamide-resistant T878A and H875Y, bicalutamide-resistant W742C, and ENZ-resistant F877L [36]. SC912 inhibited dihydrotestosterone (DHT)-induced transactivation of WT AR, T878A, and W742C mutant with a potency comparable to ENZ. Importantly, SC912 was much more potent than ENZ against the F877L mutant (Supplementary Fig. S1).

A Chemical structure of compound SC912. B AR-Vs dependent PSA-Luc reporter assay. HEK293T cells were transiently transfected with PSA-Luc reporter, pRL-TK, and AR-V7 or ARv567es expressing plasmids. Cells were exposed to DMSO vehicle, ENZ, or SC912 at designated doses (0.01 nM–10 μM) for 48 h in androgen-deprived media; Relative PSA-Luc reporter activity was normalized to the DMSO-treated samples. Data represent the average ± SD of two separate experiments. C Hormone receptor-dependent MMTV-Luc reporter assay. PC3 cells were transiently transfected with MMTV-Luc reporter, pRL-TK, and wildtype-AR or PR expressing plasmid, or endogenous GR, as indicated. Cells were pretreated in androgen-deprived media with DMSO vehicle, ENZ, or SC912 at designated doses (0.1 nM–10 μM) for 30 min, followed by the addition of 10 nM DHT, 10 nM Progesterone, or 10 nM Dexamethasone, respectively, for another 24 h. Relative MMTV-Luc reporter activity was normalized to the DMSO-treated samples. Data represent the average ± SD of duplicate samples. Cellular thermal stability assay (CETSA) for assessing the binding of SC912 to AR and AR-V7. LNCaP (D) or 22Rv1 cells (E) were exposed to designated treatments for 1 h followed by 3 min of heat shocks at the indicated temperature. Thermostable AR-FL (D) or AR-V7 (E) was quantified by western blots (relative to β-actin) and normalized to the blot intensity of the 37 °C samples. Data represent the average ± SD of duplicate samples.
To assess the AR specificity of SC912, PC3 cells were transiently transfected with a pan hormone receptor-responsive luciferase reporter MMTV-Luc for the endogenous glucocorticoid receptor (GR) or paired with AR or progesterone receptor (PR)-expressing plasmid, and the transected cells were exposed to the corresponding receptor’ agonist: dexamethasone, DHT, or progesterone, respectively. Within 0.1–10 μM, both SC912 and ENZ potently suppressed AR activation, with IC50 = 0.57 and 0.18 μM, respectively, while they had no inhibitory effect against GR nor PR (Fig. 1C). These results indicated that SC912, like ENZ, is AR-specific. Given that the DBD domain in AR, GR, and PR is highly conserved, we reasoned that SC912 most likely achieves such AR specificity by targeting an AR domain outside the DBD.
Recently, Shaw et al. reported that cellular thermal shift assay (CETSA) is a powerful technique to measure the intracellular binding of ENZ to AR [37]. We, therefore, assessed the direct binding of SC912 to AR in prostate cancer cells by CETSA, with ENZ as a control. In LNCaP cells, 10 nM DHT caused a drastic thermal shift of intracellular AR-FL protein, increasing its Tm from ~45 °C to ~51 °C, whereas the addition of 10 μM ENZ or SC912 decreased that Tm back to ~47 °C (Fig. 1D, Supplementary Fig. S2A). This observation is consistent with Shaw’s report that androgen-induced thermal stabilization of AR-FL is reversed by ENZ binding [37] and demonstrated that SC912, like ENZ, directly binds to AR-FL. Similarly, in 22Rv1 cells, 30 μM SC912 also caused a thermal shift of the endogenous AR-V7, resulting in the Tm dropping from ~51 °C to ~48 °C, whereas, as expected, 30 μM ENZ had no impact (Fig. 1E, Supplementary Fig. S2B). Together, our results indicated that SC912 directly binds to AR-FL and AR-V7 proteins, resulting in their thermal shift.
SC912 targets the AR-NTD and the amino acids 507–531 segment is indispensable for SC912’s antagonistic activity and target engagement
As a first step, we evaluated the binding of SC912 to AR-NTD (aa 2-556). The AR-NTD lacks secondary structure and contains four tryptophan residues (Trp399, Trp435, Trp503, and Trp527). Excitation at 280 nm yield a spectrum that is characterized by emission λmax at ~300 nm due to tryptophans [38], which allowed the structural change of AR-NTD being studied using steady-state fluorescence emission spectroscopy. In the presence of SC912, no fluorescence change was observed in buffer alone nor in the BSA protein tryptophan emission. In contrast, SC912 caused a red shift of λmax from 335 nm to 340 nm with increased quantum yield (Fig. 2A), suggesting AR-NTD undergoes conformational change after binding to SC912 resulting in tryptophan residues becoming more exposed to solvent.

A Steady-state fluorescence spectroscopy of the AR-NTD. Buffer alone, or 8 μg BSA, or 10 μg AR-NTD (aa 1-556) recombinant protein was preincubated with SC912 (0 μM–10 μM) for 1 h at room temperature. After excitation at 280 nm, the emission spectra was measured between 300 nm to 390 nm. B IRF3-AR transactivation-based ISRE-Luc reporter assays. PC3 cells were transiently transfected with ISRE-Luc reporter, pRL-TK, and a plasmid expressing IRF3, IRF3-AR (1–547), IRF3-AR (1–370), or IRF3-AR (361–547) fusion protein. Cells were exposed to DMSO, ENZ, or SC912 at designated doses (0.01 nM–3.33 μM) for 24 h in androgen-deprived media; Relative ISRE-Luc reporter activity was normalized to the DMSO control. Data represent the average ± SD of two separate experiments. C VP16-AR transactivation-based PSA-Luc reporter assay. PC3 cells were transiently transfected with PSA-Luc, pRL-TK, and a plasmid expressing VP16-AR(507–920), VP16-AR(530–920), or VP16-AR(540–920) fusion protein. Cells were pretreated with DMSO vehicle or SC912 at designated doses (0.01 nM–3.33 μM), in duplicate, in androgen-deprived media for 30 min before adding 10 nM DHT for another 24 h. Relative PSA-Luc reporter activity was normalized to the DMSO vehicle control. Data represent the average ± SD of duplicate samples. D AR-FL transactivation-based PSA-Luc reporter assay. PC3 cells were transfected with PSA-Luc, pRL-TK, and a plasmid expressing WT AR-FL or the corresponding aa 507–530 deleted counterpart: AR-FL-del(507–531). Cells were pretreated with DMSO or SC912 (0.1, 0.3, 1, 3 μM) in androgen-deprived media for 30 min, followed by adding 10 nM DHT for an additional 48 h. Relative PSA-Luc reporter activity was normalized to the DMSO control of AR-FL transfected cells. No DHT-stimulated cells were included as the negative control. Data represent the average ± SD of triplicate samples. E AR-V7 transactivation-based PSA-Luc reporter assay. PC3 cells were transfected with PSA-Luc, pRL-TK, and a plasmid expressing AR-V7 or the corresponding aa 507–530 deleted counterpart: AR-V7-del(507–531). Cells were treated with SC912 (0, 0.1, 0.3, 1, 3 μM) in androgen-deprived media for 48 h. Relative PSA-Luc reporter activity was normalized to AR-V7 in 0 μM SC912 treated cells (DMSO vehicle control), and cells transfected with empty vector were used as the negative control (Mock). Data represent the average ± SD of triplicate samples. F, G Cellular thermal stability assay (CETSA) for assessing the effect of AR-NTD aa 507–531 on the target engagement of SC912. E HEK293T cells transiently transfected with plasmid expressing AR-FL or AR-FL-del(507–531) were exposed to DMSO vehicle or SC912 at designated doses (0.03 μM–33.33 μM) in the presence of 10 nM DHT. F HEK293T cells transiently transfected with plasmid expressing AR-V7 or AR-V7-del(507–531) were treated with DMSO vehicle or SC912 at designated doses (0.03 μM–100 μM). After 1 h of treatment, cells were harvested and subjected to 3 min of heat shock at 46 °C. Thermostable AR-FL or AR-V7 were quantified by Western blots analysis (relative to β-actin) and normalized to the blot intensity of the DMSO vehicle control. Data represent the average ± SD of duplicate samples.
To identify SC912’ interacting motif on AR-NTD, we constructed a series of plasmids expressing chimeric proteins with various fragments of AR-NTD fused to the DBD of the transcriptional factor IRF3. Using an ISRE luciferase (ISRE-Luc) reporter, we observed that, in PC3 cells, SC912 substantially inhibited the transactivation of IRF3-AR1–547 and IRF3-AR361–547, in contrast to no or marginal activity on the WT IRF3 and IRF3-AR1–370 (Fig. 2B). These results suggested that the interacting motif of SC912 on AR is located within the aa 361–547 region at the AR-NTD. To further narrow down this range, we generated another set of plasmids expressing AR proteins which bear different deletions within aa 361–547 and are fused to the transactivation domain of the transcriptional factor VP16. Using PSA-Luc reporter, we found that SC912 attenuated androgen-stimulated transactivation of VP16-AR507–920 in PC3 cells, in contrast to having no impact on VP16-AR530–920 nor VP16-AR540–920 (Fig. 2C). Taken together, these results suggested that SC912 did not target the AR-DBD nor AR-LBD domains within aa 540–920; its targeting motif is likely located in aa 361–547 of AR-NTD, with the possible core segment being aa 507–530.
Next, we investigated if the aa 507–530 segment is essential for the SC912’s antagonistic activity against AR-FL and AR-V7. We constructed plasmids to express the aa 507-531 region-deleted AR-FL and AR-V7 protein, referred to as AR-FL-del(507–531) and AR-V7-del(507–531), respectively. By PSA-Luc reporter assays, with AR-FL as control, we observed that SC912 was inactive against the DHT-induced transactivation of AR-FL-del(507–531) (Fig. 2D). Likewise, aa 507–531 deletion also led to a complete loss of SC912’s inhibitory activity against AR-V7 (Fig. 2E). Interestingly, the transactivation activity of AR-FL-del(507–531) and AR-V7-del(507–531) is comparable to that of their wildtype counterparts (Fig. 2D, E), suggesting that the deletion of aa 507–531 is not deleterious to the overall structure of AR protein. Next, we performed site-directed mutagenesis on AR-V7. The two AR-V7 mutants, P513G and Y531A, had similar transactivation activity to the wildtype but were more resistant to SC912 (Supplementary Fig. S3), implying that P513 and Y531 residues are critically important for the activity of SC912 against AR-V7. These results collectively suggested that the aa 507–531 segment in AR-NTD is indispensable for SC912 to inhibit AR-FL and AR-V7.
We next determined the impact of aa 507–531 on the binding of SC912 to AR. AR-FL or AR-V7 proteins and their aa 507–531 deleted counterparts were transiently expressed in 293 T cells, and CETSA assays were performed to assess SC912’s binding in the concentration range 0.03–100 μM. The result suggested that SC912 bound to AR-FL and AR-V7 with the EC50 of heat-denaturalizing these two proteins being 1.1 and 0.3 μM, respectively (Fig. 2F, G, Supplementary Fig. S2C, S2D). Strikingly, the deletion of aa 507–531 diminished the binding of SC912 to AR-FL and AR-V7, evidenced by no thermal destabilization of AR proteins was detected even at the highest concentration of 100 μM SC912 (Fig. 2F, G). These results indicated that the aa 507–531 segment determines SC912’s target engagement to AR-FL and AR-V7.
SC912 blocked AR-V7-driven AR signaling in CRPC cells
To explore the impact of SC912 treatment on the AR transcriptome in CRPC cells, we performed RNA-sequencing analyses using LNCaP95 cells. LNCaP95 is a subline derived from culturing parental LNCaP in androgen-free media for 95 passages, resulting in overexpression of AR-V7 and dependence on AR-V7 mediated AR signaling activity for proliferation [39]. 2 μM SC912 suppressed AR-regulated genes, especially the canonical AR-regulated genes, that are known to be activated by both AR-FL and AR-V7 (e.g., PSA, TMPRSS2, NKX3.1). More importantly, SC912 strongly inhibited the genes uniquely upregulated by AR-V7 in LNCaP95 (Fig. 3A). Further GSEA analysis revealed that AR pathway, as well as the AR-V7-mediated distinct transcriptional program were significantly blocked by SC912 treatment in LNCaP95 (Fig. 3B). Additionally, qRT-PCR assay verified that the expression of both the canonical AR-regulated gene PSA and the AR-V7 unique gene UBE2C [39] were markedly suppressed by 2 μM SC912, whereas 10 μM ENZ had no effect (Fig. 3C). Together, these results supported that SC912 inhibited AR-V7 transcriptional activity in the ENZ-resistant LNCaP95 cells.

A Heatmaps for RNA-seq analysis of AR target genes [50] and AR-V7 target genes [39]. LNCaP95 cells were treated with DMSO or 2 μM SC912 for 24 h (4 biological replicates), AR and AR-V7 regulated genes that were altered in expression are displayed. The gene expression values for each gene were normalized to the standard normal, red and blue coloring indicates upregulated and downregulated relative expression levels, respectively (GSE253122). B GSEA of gene sets comprising PID-AR pathway [50] and AR-V7 gene signature [39]in LNCaP95 cells treated as (A). C qRT-PCR analysis of AR-V7 regulated genes in LNCaP95 cells. LNCaP95 cells were treated with DMSO, 10 μM ENZ, or 2 μM SC912 for 24 h. Data represent the average ± SD of two separate experiments. D Western blot comparision of AR-FL and AR-V7 expression levels in LNCaP, LNCaP95, VCaP, 22Rv1, and LNCaP-AR-V7 cell lines. E qRT-PCR analysis of canonical AR-regulated genes. LNCaP, VCaP, and 22Rv1 cells were cultured in androgen-deprived media for 48 h and then exposed to SC912 at 0, 0.1, 0.3, 1, 3 μM for another 24 h. Data represent the average ± SD of three separate experiments. F qRT-PCR analysis of AR-V7 specifically regulated genes. LNCaP, VCaP, and LNCaP-AR-V7 cells were treated the same as in 3A. Data represent the average ± SD of three separate experiments. G Western blot validation of ShAR-FL 22Rv1 and ShAR-V7 22Rv1 stable cell lines. Cells in androgen-deprived media were exposed to DMSO in the presence or absence of 50 ng/mL doxycycline for 48 h before harvest. H qRT-PCR analysis of canonical AR-regulated genes in ShAR-FL 22Rv1 and ShAR-V7 22Rv1 cells. Cells were processed the same as in (C). Next, cells in refresh media were exposed to DMSO, 10 μM ENZ, or 3 μM SC912 in the presence or absence of 50 ng/mL doxycycline for 24 h. Data represent the average ± SD of three separate experiments.
To evaluate if SC912’s inhibition of AR-V7-mediated AR signaling in LNCaP95 cells is transferable to other CRPC cellular models, three extra prostate cancer cell lines expressing AR-FL and different levels of AR-V7 were exposed to SC912 (Fig. 3D): (i) LNCaP cells can acquire some level of AR-V7 expression at high passage [40]. Here, we used LNCaP cells with ~80 passages in complete media; (ii) VCaP cells are inherently castration-resistant via modest but rapid AR-V7 overexpression after castration or antiandrogen treatments [41]; and (iii) 22Rv1 cells are irresponsive to all of those treatments due to their high intrinsic level of AR-V7 [42]. SC912 did not induce ER stress in these cells (Supplementary Fig. S4), thus ruling out the possibility of downregulating AR signaling through indirect pathway. Using PSA-Luc reporter assay, we observed that the castration-resistant AR signaling in these cells was quite sensitive to SC912 inhibition, with nanomolar IC50 achieved in all three cell lines (IC50: LNCaP 0.11 µM, VCaP 0.37 µM, and 22Rv1 0.42 µM) (Supplementary Fig. S5). When assessed by qPCR assay for three canonical AR-regulated genes (PSA, FKBP5, and TMPRSS2), SC912 dose-dependently (0.3–3 μM) attenuated their transcription in these prostate cancer cell lines (Fig. 3E). Next, we asked if SC912 could also impair the transcription of AR-V7 specifically regulated genes. We quantified the mRNA of three tumor suppressor genes (B4GALT1, SLC30A7, and HIF1A) whose transcription is reported to be specifically repressed by AR-V7 in LNCaP95 [43]. Indeed, SC912 at 3 μM significantly elevated the expression of B4GALT1 and HIF1A in our ~80 passage LNCaP cells (Fig. 3F), while even greater changes were observed in cell lines expressing more AR-V7. In VCaP and our LNCaP-AR-V7 stable cells [35], the mRNA levels of all three AR-V7 repressed genes spiked upon SC912 treatment (Fig. 3F). Collectively, these data indicated that SC912 suppressed AR-V7 transcriptional activity in multiple CRPC cellular models.
Given that AR-V7 is always co-expressed with AR-FL in prostate cancer cells, to evaluate the inhibitory effect of SC912 on AR-V7 alone, we generated two stable 22Rv1 cell lines with doxycycline (DOX)-inducible knockdown of AR-FL or AR-V7, respectively (Fig. 3G). The mRNA quantification of canonical AR-regulated genes (PSA, FKBP5, and TMPRSS2) revealed that AR-FL knockdown did not mitigate AR signaling activity in 22Rv1 cells. In contrast, a drastic decline of such was noted following AR-V7 knockdown (Fig. 3H). These observations aligned with the previous discoveries that AR signaling in 22Rv1 cells is predominantly driven by AR-V7 rather than the AR-FL [17, 44]. More importantly, SC912 at 3 μM markedly reduced AR signaling in AR-FL knockdown cells, but its potency was significantly diminished in AR-V7 knockdown cells. On the contrary, 10 μM of ENZ could not inhibit AR signaling in neither the control nor any of the knockdown cells (Fig. 3H), demonstrating that SC912 but not ENZ was capable of blocking AR-V7 mediated AR signaling in CRPC cells.
SC912 hampered the nuclear localization and chromatin binding of both AR-V7 and AR-FL
AR nuclear translocation and subsequent DNA binding are two prerequisite steps happening before AR-initiated transcription. Since SC912 inhibited AR transcriptional activity, we wondered if such inhibition is also associated with hampering those upper-stream events. By chromatin immunoprecipitation (ChIP) assay, we first examined if SC912 treatment reduced AR chromatin binding in LNCaP cells engineered to stably overexpress WT AR-FL (LNCaP-AR-FL) [35]. We found that androgen-induced AR-FL recruitment to AREs (PSA enhancer and TMPRSS2 enhancer) was mitigated by 3 μM SC912 and 5 μM ENZ (Fig. 4A). Next, we assessed the capability of SC912 in reducing the androgen-independent AR binding to DNA. In LNCaP-AR-V7 cells cultured in androgen-deprived media, 3 μM SC912 lowered the AR chromatin recruitment by more than 50%, whereas ENZ at 5 μM only had a marginal effect (Fig. 4B). Furthermore, in the shFL-22Rv1 cells (with DOX), SC912 potently diminished AR-V7 binding to AREs, whereas ENZ was inactive (Fig. 4C). Likewise, SC912 but not ENZ reduced AR-V7 binding to the enhancer of the AR-V7 specific gene UBE2C [45], suggesting SC912 hampered the androgen-independent chromatin recruitment of AR-V7 (Fig. 4D).

A ChIP assay for androgen-stimulated AR-FL enrichment to AREs. LNCaP-AR-FL cells were cultured in androgen-deprived media for 48 h and then pretreated for 30 min with DMSO, 5 μM ENZ, or 3 μM SC912, followed by the addition of 10 nM DHT and incubation for another 4.5 h before harvest. Data represent the average ± SD of duplicate samples. B ChIP assay for androgen-independent AR-FL and AR-V7 enrichment to AREs. LNCaP-AR-V7 cells were cultured in androgen-deprived media for 48 h before being treated with DMSO, 5 μM ENZ, or 3 μM SC912 for 5 h. Data represent the average ± SD of duplicate samples. C ChIP assay of androgen-independent AR-V7 enrichment to AREs in shAR-FL 22Rv1 cell line. Cells were incubated in androgen-deprived media containing 50 ng/mL doxycycline for 48 h before being treated the same as (B). Data represent the average ± SD of duplicate samples. D ChIP assay of androgen-independent AR-V7 enrichment to AREs in LNCaP95 cell line. Cells were treated the same as (B). Data represent the average ± SD of duplicate samples. Representative confocal image of GFP-tagged AR-FL (E) or AR-V7 (F) in PC3 cells (Scale bar represents 10 μm). E PC3 cells transiently expressing GFP-AR-FL were treated the same as in (A). F PC3 cells transiently expressing GFP-AR-V7 were cultured in androgen-deprived media with the treatment of DMSO, 5 μM ENZ, or 3 μM SC912 for 16 h. Nuclear: cytoplasmic GFP fluorescence intensity of individual cells was quantified (n = 10–15) with Image J. Data represent the average ± SD.
To examine if SC912 also affects the nuclear localization of AR-FL and AR-V7, GFP-tagged AR-FL or AR-V7 were transiently expressed in PC3 cells, respectively, and their subcellular localization was visualized using confocal microscopy. 3 μM SC912 and 5 μM ENZ significantly reduced androgen-stimulated nuclear translocation of AR-FL (Fig. 4E, Supplementary Fig. S6). More importantly, SC912 also markedly attenuated the androgen-independent nuclear localization of AR-V7, which ENZ failed to affect (Fig. 4F, Supplementary Fig. S7). These results suggested that, though androgen-induced nuclear translocation and DNA binding of AR-FL were diminished by both SC912 and ENZ, while these events for androgen-independent AR-V7 were only attenuated by SC912.
Lastly, we also noted that the AR-NTD aa 507–531 deleted AR-FL and AR-V7’s nuclear trafficking was no longer hampered by SC912, though they had almost identical subcellular localization as wildtype (Supplementary Fig. S6, S7). These results supported our findings of amino acids 507–531 segment being indispensable for SC912 binding to AR (Fig. 2F, G).
SC912 caused proliferation arrest and apoptosis in AR-V7 positive CRPC cells
The antiproliferative activity of SC912 in prostate cancer cells was assessed in three AR-positive cell lines: LNCaP, VCaP, and 22Rv1, as well as an AR-negative cell line: PC3. When cells were grown in androgen-deprived media for 6 days, SC912 inhibited the proliferation of LNCaP, VCaP, and 22Rv1 cells with an IC50 of 0.31, 0.60, and 0.86 μM, respectively, whereas much higher IC50 (5.77 μM) was noted in PC3 cells (Fig. 5A, Supplementary Table. S1). Likewise, higher IC50 (3.3–8.2 μM) was also observed in non-prostate cancer cell lines, as well as in the AR-independent neuroendocrine prostate cancer cells (Supplementary Fig. S8, Supplementary Table. S1). ENZ suppressed LNCaP proliferation with an IC50 of 1.76 μM, in contrast to being less potent in VCaP (IC50 = 9.88 μM) and was almost inactive in 22Rv1 and PC3 cells (Fig. 5A, Supplementary Table. S1). Next, we performed cell cycle analysis on LNCaP, VCaP, 22Rv1, and PC3 cell lines using flow cytometry. SC912 at 1 μM caused a substantial amount of LNCaP, VCaP, and 22Rv1 cells to stall at G1/S transition, with little effect on the cell cycling of AR-negative PC3 cells (Fig. 5B, Supplementary Fig. S9). In comparison, 5 μM of ENZ led to G1-phase arrest only in LNCaP but failed to affect other cell lines (Fig. 5B, Supplementary Fig. S9), suggesting SC912 but not ENZ is broadly effective in blocking the proliferation of AR-V7 positive CRPC cell. Concordantly, 1 μM of SC912 also induced apoptosis in the three AR-positive cell lines, evidenced by the emergence of PARP cleavage in cell lysate western blot, which conversely, was not detectable in AR-negative PC3 cells (Fig. 5C). In contrast, ENZ only induced apoptosis in LNCaP but not in the VCaP and 22Rv1 which express more AR-V7 (Fig. 5C). Last, we performed Annexin V staining for flow cytometry to visualize the apoptotic cell population. Consistent with the cleaved-PARP results, SC912 caused ~2-fold increase of apoptotic cell % in all three AR-positive CRPC cell lines, whereas ENZ only induced apoptosis in LNCaP (Fig. 5D). In summary, these findings agreed with previous discoveries that prostate cancer cells and PDX with higher levels of AR-V7 are more resistant to ENZ [46], and also indicated that high AR-V7 expressing cells remain susceptible to SC912 treatment.

A CellTiter-Glo assays for evaluating the antiproliferative effect of SC912 in prostate cancer cells. LNCaP, VCaP, 22Rv1, and PC3 Cells were seeded in androgen-deprived media for 24 h and then exposed to DMSO, ENZ, or SC912 at designated doses (0.1 nM–10 μM). Cell culture media and corresponding treatments were refreshed once on day 3, and cell viability was measured on day 6. Data represent the average ± SD of three separate experiments. B Cell cycle compartment flow cytometry for assessing SC912-induced growth arrest in CRPC cells. LNCaP, VCaP, 22Rv1, and PC3 cells were cultured in androgen-deprived media for 48 h and then exposed to DMSO, 3 μM ENZ, or 1 μM SC912 for 24 h. Cells were pulsed with BrdU for 1 h prior to harvest. G1 and G2/M-phase cells were defined as cells that show no BrdU staining and possess 1 N/2 N DNA content, respectively. S-phase cells were defined as cells that were positive for BrdU staining. C Western blot analysis of SC912-induced apoptosis in CRPC cells. LNCaP, VCaP, 22Rv1, and PC3 cells were cultured in androgen-deprived media for 48 h and treated with 3 μM ENZ or 1 μM SC912 for 0, 12, or 24 h before harvest. Cell apoptosis was determined by Western blot for the cleaved PARP. D Annexin V staining flow cytometry for assessing SC912 induced apoptosis in CRPC cells. Cells were treated the same as (B). Apoptotic cells were defined as Annexin V-positive cells.
SC912 repressed tumor growth and interrupted AR signaling in AR-V7 expressing CRPC xenografts
In considering SC912’s therapeutic activity in CRPC tumors, we first assessed the effect of SC912 on VCaP xenografts. Male NOD-SCID mice implanted with VCaP cells were subsequently castrated when tumor sizes reached 250–500 mm3. Compounds were given intraperitoneally 5 times a week for 3 weeks once VCaP tumors resumed growing in castrated hosts. Strikingly, the initial three dosages of SC912 (60 mg/kg) already halted the castration-resistant growth of VCaP tumors, which lasted to the endpoint. In contrast, the tumor volume in the vehicle group increased by ~150% on average (Fig. 6A). When comparing by weight, the tumors in the SC912 group were about 40% of the vehicle group (Fig. 6B). Furthermore, SC912 treatment did not cause obvious animal body weight loss (Fig. 6C), indicating that SC912 at 60 mg/kg is effective and well-tolerated.

A–H SC912 abolished the growth of VCaP xenografts in castrated mice. 1 × 107 VCaP cells mixed with 50% matrigel were subcutaneously injected into the right flank of male NOD-SCID mice. When tumor size reached 250–500 mm3, mice were surgically castrated and monitored daily (Day 0). After the tumors resumed growing and reached the size of 300–600 mm3(Day 18), mice were randomized into 2 groups (n = 9 for the vehicle group, n = 10 for the SC912 group), which received 5 days per week treatment (i.p.) of the vehicle or SC912 (60 mg/kg) for 3 weeks before sacrifice (Day 39). A VCaP tumor growth curve. The %Tumor Change was calculated as (Day39- Day0)/Day0%. B Tumor image and weight on Day 39 (n = 9–10). C ELISA assay of human PSA concentration in mouse serum (n = 9–10). D Mouse body weight comparison on Day 0 and Day 39 (n = 9–10). E qRT-PCR analysis of AR-regulated gene expression in tumors (n = 7). F Western blot analysis of AR-FL and AR-V7 protein expression in tumor (n = 7). G Quantification of AR-FL and AR-V7 protein levels (n = 7). H qRT-PCR analysis of AR-FL and AR-V7 mRNA expression in tumors (n = 7). G–J SC912 suppressed the growth of AR-V7 high-expressing 22Rv1 tumors in castrated mice. 5 × 106 22Rv1 cells mixed with 50% matrigel were subcutaneously injected into the right flank of male Nu/Nu mice. When tumor size reached 100–300 mm3, mice were surgically castrated and monitored daily (Day 0). Animals were allowed to recover from surgery for one week, and tumors reached the size of 150–450 mm3. Mice were then randomized into two groups (n = 9/group), which received treatment (i.p.) of the vehicle or SC912 (90 mg/kg) 5 days a week for 3 weeks before sacrifice (Day 30). I 22Rv1 tumor growth curve (n = 9). J Tumor image and weights on Day 30 (n = 9). K Mouse body weight comparison on Day 0 and Day 30 (n = 9).
Next, we assessed how did SC912 treatment influence the AR signaling in VCaP tumors. In the serum of SC912 treated mice, human PSA concentration was significantly lower than the vehicle group (12.7 ng/mL vs. 28.1 ng/mL, respectively) (Fig. 6D). In addition, qPCR analysis of the intratumoral mRNA revealed a significant reduction of PSA expression and an upregulation of AR-V7-repressed gene B4GALT1 (Fig. 6E), Moreover, within SC912 treated tumors, a 3.2-fold increase of AR-FL protein and a 2.6-fold increase of AR-V7 protein was shown by western blot (Fig. 6F–G), coupled with a statistically insignificant but observable bump of AR-FL and AR-V7 mRNA (Fig. 6H). These data collectively implied that CRPC tumors responded to SC912 inhibited AR signaling with even overexpression of both AR-FL and AR-V7.
Lastly, we evaluated SC912’s potency against 22Rv1 xenografts which are more castration-resistant and express a higher amount of AR-V7. As expected, 22Rv1 tumor progression was not paused by castrating host mice; however, it was markedly mitigated by 90 mg/kg of SC912. After 10 days of treatment, the tumor inhibitory effect of SC912 became statistically significant, followed by the p-value dropping below 0.01 in the next 3 days and remaining so to the end of the experiment (Fig. 6I). The average tumor weight for the SC912 group is approximately 45% of that for the vehicle group (Fig. 6J), and no decline in animal body weight was observed (Fig. 6K). These findings demonstrated that SC912 was capable of repressing the growth of CRPC tumors that express high levels of AR-V7.
Discussion
Persistent AR signaling activity is a widely recognized cause of Abi and ENZ resistance in CRPC. Mounting evidence emphasized the role of AR-V7 as a key driver of sustained AR signaling in CRPC [10, 18, 26, 46]. As an adaptive response to AR-targeted therapies, both AR-V7 mRNA and protein were found overexpressed in Abi and ENZ-treated cell lines and xenografts [41, 47] as well as CRPC patients [11,12,13,14]. Notably, AR-V7 functions independently of the AR-FL in prostate cancer cells [17, 18], while its expression appears sufficient to drive AR-FL function within CRPC cells [18]. Although numerous studies have established AR-V7 as a therapeutic target for CRPC, pharmaceutical inhibition of AR-V7 remains clinically unavailable. In this study, our team discovered a novel chemical compound, SC912, which effectively inhibited the functions of both AR-V7 and AR-FL by targeting the AR-NTD. We demonstrated that the AR-NTD’s aa 507–531 region is essential for the binding of SC912 to AR-FL and AR-V7. Moreover, SC912 was found to block AR-V7-mediated AR signaling in CRPC cells in vitro and significantly repressed castration-resistant growth of VCaP and 22Rv1 xenografts in castrated mice.
SC912 bound to AR-NTD recombinant protein in ex vivo setting, moreover, its direct binding to AR-FL and AR-V7 within cells was also confirmed using CETSA assays in three cellular models: LNCaP with endogenous AR-FL, 22Rv1 cells with endogenous AR-V7, and HEK293T cells transiently expressing AR-FL or AR-V7. The EC50 values for heat-denaturalized AR-FL and AR-V7 were found to be 1.1 and 0.3 μM, respectively, indicating the potent binding affinity of SC912. It was observed that the AR-NTD aa 507–531 region dictates the inhibitory activity, as well as the binding of SC912 to AR-FL and AR-V7. Interestingly, we also found that point mutations P513G and Y531A at the AR-NTD confer substantial resistance to SC912, highlighting the importance of P513 and Y531 residues for the activity of SC912. Collectively, these findings suggested that the AR-NTD aa 507–531 region could be part of the binding pocket for the small molecule inhibitor SC912. However, due to the conformational plasticity and lack of folded structure inherent in intrinsically disordered proteins like the AR-NTD [48], it remains unclear if SC912 binds to a pre-formed structure or if the binding pocket spontaneously forms upon encountering SC912. Further investigation is required to determine the precise binding mode of SC912 within and around the aa 507–531 region.
This study demonstrated that SC912 effectively blocks AR-V7-mediated AR signaling in CRPC cells (Fig. 3). SC912 also inhibited androgen-stimulated AR-FL activity (Fig. 1C, Fig. 4A). This is expected, given SC912 targets the AR-NTD, a domain that is essential for the transactivation of both AR-V7 and AR-FL. Furthermore, SC912 effectively suppressed the proliferation of AR-V7 expressing VCaP and 22Rv1 cells, with IC50 values of 0.6 and 0.86 µM, respectively. However, the anti-proliferation IC50 of SC428 in AR-negative PC3 cells is 5.77 µM (Table S1), which is only 7-fold higher than in 22Rv1 cells, indicating the existence of off-target effect. Further chemical optimization on SC912 is needed to improve the selectivity towards AR-positive prostate cancer cells.
We observed that treatment of SC912 induced overexpression of AR and AR-V7 in VCaP tumors (Fig. 6F). This aligned with the previously described negative feedback loop between AR signaling and AR expression [49] as a CRPC cell adaptive response to AR-targeted therapies. It is worth noting that the AR overexpression induced by SC912 differed from that of conventional AR inhibitors. Androgen depletion, ENZ, and Abi were all reported to preferentially upregulate AR-V7 relative to AR-FL in VCaP cells [41, 47], resulting in an increased AR-V7 vs. AR-FL ratio and potentially conferring drug resistance to conventional AR inhibitors. A recent study demonstrated that ENZ resistance consistently correlates with enhanced AR-V7 expression [46]. In contrast, SC912 induced overexpression of AR-FL and AR-V7 equally without substantially changing the AR-V7 vs. AR-FL ratio (Fig. 6G). Moreover, the upregulated AR-FL and AR-V7 expression by SC912 is not expected to drive drug resistance, as SC912 is active against both forms. In line with this notion, SC912 exhibited potent antitumor activity against castration-resistant xenografts of VCaP and 22Rv1, both of which express substantial levels of AR-FL and AR-V7.
To conclude, we identified a novel AR-NTD-targeting inhibitor with efficacy against AR-V7-expressing CRPC models. Our in vitro and in vivo investigations suggested that SC912 effectively suppresses sustained AR signaling in CRPC, indicating its potential to be further optimized for CRPC therapy.
Materials and methods
Dual-luciferase reporter assay
Cells were seeded into 24-well plates at least 24 h before transfection. Luciferase reporter (PSA-Luc, MMTV-Luc or ISRE-Luc) and pRL-TK plasmids as well as a plasmid expressing the designated transcription factor, were transiently transfected into cells, using lipofectamine 3000 (Invitrogen). 5 h after transfection, cell culture media was refreshed, and treatments were added. Luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega) on a GLOMAX 20/20 luminometer (Promega).
qRT-PCR analysis
2×106 cells were seeded in 6 cm dish and cultured in androgen-deprived media for 48 h and then exposed to compounds at designated doses for 24 h. Next, cells were harvested and subjected to total RNA extraction using RNAqueous™ Total RNA Isolation Kit (Applied Biosystems). cDNA was synthesized using iScript cDNA Synthesis Kit (BIO-RAD). cDNA was assessed using GoTaq qPCR Master Mix (Promega). qRT-PCR reaction was performed on 7500 Fast Real-Time PCR System (Applied Biosystems). The primer sequences are available in Supplementary Information.
Chromatin immunoprecipitation
ChIP assay was performed following the Cold Spring Harbor Protocol. Briefly, 5 × 106 cells were seeded in 10 cm dish and cultured in androgen-deprived media for 48 h, and then exposed to compounds for additional 5 h. Chromatin interacting AR were cross-linked with formaldehyde and terminated with glycine. Cells were lysed and subjected to DNA fragmentation by sonication. AR immunoprecipitation was performed using AR antibody (ab74272, abcam), or AR-V7 antibody (ab198394, abcam) together with pre-blocked G agarose bead. The precipitated AR-DNA adducts were cross-link reversed, and the purified DNA was subjected to qRT-PCR analysis. Details are available in Supplementary Information.
Data availability
The data used during this study are available from the corresponding author on request.
References
Cornford P, van den Bergh RCN, Briers E, Van den Broeck T, Cumberbatch MG, De Santis M, et al. EAU-EANM-ESTRO-ESUR-SIOG guidelines on prostate cancer. Part II-2020 update: treatment of relapsing and metastatic prostate cancer. Eur Urol. 2021;79:263–82.
Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017. alpha.-Hydroxylase-C17, 20-lyase): potential agents for the treatment of prostatic cancer. J. Med. Chem. 1995;38:2463–71.
Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90.
de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005.
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97.
Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. 2015;5:12007.
Kanayama M, Lu C, Luo J, Antonarakis ES. AR splicing variants and resistance to AR targeting agents. Cancers. 2021;13:2563.
Uo T, Plymate SR, Sprenger CC. The potential of AR-V7 as a therapeutic target. Expert Opin Ther Targets. 2018;22:201–16.
Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287:19736–49.
Kim S, Au CC, Jamalruddin MAB, Abou-Ghali NE, Mukhtar E, Portella L, et al. AR-V7 exhibits non-canonical mechanisms of nuclear import and chromatin engagement in castrate-resistant prostate cancer. Elife. 2022;11:e73396.
Sharp A, Coleman I, Yuan W, Sprenger C, Dolling D, Rodrigues DN, et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest. 2019;129:192–208.
Welti J, Rodrigues DN, Sharp A, Sun S, Lorente D, Riisnaes R, et al. Analytical validation and clinical qualification of a new immunohistochemical assay for androgen receptor splice variant-7 protein expression in metastatic castration-resistant prostate cancer. Eur Urol. 2016;70:599–608.
Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–38.
Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Nakazawa M, et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015;1:582–91.
Li Y, Yang R, Henzler CM, Ho Y, Passow C, Auch B, et al. Diverse AR gene rearrangements mediate resistance to androgen receptor inhibitors in metastatic prostate cancerar gene rearrangements in prostate cancer. Clin Cancer Res. 2020;26:1965–76.
Annala M, Taavitsainen S, Khalaf DJ, Vandekerkhove G, Beja K, Sipola J, et al. Evolution of castration-resistant prostate cancer in ctDNA during sequential androgen receptor pathway inhibitionprostate cancer evolution during sequential ar inhibition. Clin Cancer Res. 2021;27:4610–23.
Liang J, Wang L, Poluben L, Nouri M, Arai S, Xie L, et al. Androgen receptor splice variant 7 functions independently of the full length receptor in prostate cancer cells. Cancer Lett. 2021;519:172–84.
Roggero CM, Jin L, Cao S, Sonavane R, Kopplin NG, Ta HQ, et al. A detailed characterization of stepwise activation of the androgen receptor variant 7 in prostate cancer cells. Oncogene. 2021;40:1106–17.
Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–46.
Dalal K, Che M, Que NS, Sharma A, Yang R, Lallous N, et al. Bypassing drug resistance mechanisms of prostate cancer with small molecules that target androgen receptor–chromatin interactionstargeting androgen receptor interactions with chromatin. Mol Cancer Ther. 2017;16:2281–91.
Dalal K, Ban F, Li H, Morin H, Roshan-Moniri M, Tam KJ, et al. Selectively targeting the dimerization interface of human androgen receptor with small-molecules to treat castration-resistant prostate cancer. Cancer Lett. 2018;437:35–43.
Ponnusamy S, He Y, Hwang D-J, Thiyagarajan T, Houtman R, Bocharova V, et al. Orally bioavailable androgen receptor degrader, potential next-generation therapeutic for enzalutamide-resistant prostate cancera novel ar degrader for the treatment of prostate cancer. Clin Cancer Res. 2019;25:6764–80.
Liu C, Armstrong CM, Ning S, Yang JC, Lou W, Lombard AP, et al. ARVib suppresses growth of advanced prostate cancer via inhibition of androgen receptor signaling. Oncogene. 2021;40:5379–92.
Lee GT, Nagaya N, Desantis J, Madura K, Sabaawy HE, Kim W-J. et al.Effects of MTX-23, a novel PROTAC of androgen receptor splice variant-7 and androgen receptor, on CRPC resistant to second-line antiandrogen therapy.Mol Cancer Ther. 2021;20:490–9.
Melnyk JE, Steri V, Nguyen HG, Hwang YC, Gordan JD, Hann B, et al. Targeting a splicing-mediated drug resistance mechanism in prostate cancer by inhibiting transcriptional regulation by PKCβ1. Oncogene. 2022;41:1536–49.
Luna Velez MV, Verhaegh GW, Smit F, Sedelaar JPM, Schalken JA. Suppression of prostate tumor cell survival by antisense oligonucleotide-mediated inhibition of AR-V7 mRNA synthesis. Oncogene. 2019;38:3696–709.
Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–82.
Welti J, Sharp A, Brooks N, Yuan W, McNair C, Chand SN, et al. Targeting the p300/CBP axis in lethal prostate cancertargeting the p300/CBP axis in lethal prostate cancer. Cancer Discov. 2021;11:1118–37.
Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci USA. 2004;101:4758–63.
Lavery DN, McEwan IJ. Structural characterization of the native NH2-terminal transactivation domain of the human androgen receptor: a collapsed disordered conformation underlies structural plasticity and protein-induced folding. Biochem. 2008;47:3360–9.
Tsafou K, Tiwari PB, Forman-Kay JD, Metallo SJ, Toretsky JA. Targeting intrinsically disordered transcription factors: changing the paradigm. J Mol Biol. 2018;430:2321–41.
Monaghan AE, McEwan IJ. A sting in the tail: the N-terminal domain of the androgen receptor as a drug target. Asian J Androl. 2016;18:687.
Sadar MD. Discovery of drugs that directly target the intrinsically disordered region of the androgen receptor. Expert Opin Drug Discov. 2020;15:551–60.
Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948–60.
Yi QH, Liu WG, Seo JH, Alaoui-Jamali MA, Luo J, Lin RT, et al. Discovery of a small-molecule inhibitor targeting the androgen receptor N-terminal domain for castration-resistant prostate cancer. Mol Cancer Ther. 2023;22:570–82.
Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11.
Shaw J, Leveridge M, Norling C, Karen J, Molina DM, O’Neill D, et al. Determining direct binders of the androgen receptor using a high-throughput cellular thermal shift assay. Sci Rep. 2018;8:163.
Reid J, Kelly SM, Watt K, Price NC, McEwan IJ. Conformational analysis of the androgen receptor amino-terminal domain involved in transactivation: influence of structure-stabilizing solutes and protein-protein interactions. J Biol Chem. 2002;277:20079–86.
Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–62.
Esquenet M, Swinnen JV, Heyns W, Verhoeven G. LNCaP prostatic adenocarcinoma cells derived from low and high passage numbers display divergent responses not only to androgens but also to retinoids. J Steroid Biochem Mol Biol. 1997;62:391–9.
Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, Nelson PS, et al. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res. 2014;20:1590–1600.
Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77.
Cato L, de Tribolet-Hardy J, Lee I, Rottenberg JT, Coleman I, Melchers D, et al. ARv7 represses tumor-suppressor genes in castration-resistant prostate cancer. Cancer Cell. 2019;35:401–13.e406.
Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate. 2011;71:1656–67.
Chen Z, Wu D, Thomas-Ahner JM, Lu C, Zhao P, Zhang Q, et al. Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc Natl Acad Sci. 2018;115:6810–5.
Zhu Y, Dalrymple SL, Coleman I, Zheng SL, Xu J, Hooper JE, et al. Role of androgen receptor splice variant-7 (AR-V7) in prostate cancer resistance to 2nd-generation androgen receptor signaling inhibitors. Oncogene. 2020;39:6935–49.
Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. 2014;33:3140–50.
Mollica L, Bessa LM, Hanoulle X, Jensen MR, Blackledge M, Schneider R. Binding mechanisms of intrinsically disordered proteins: theory, simulation, and experiment. Front Mol Biosci. 2016;3:52.
Schweizer MT, Antonarakis ES, Wang H, Ajiboye AS, Spitz A, Cao H, et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Sci Transl Med. 2015;7:269ra262.
Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion–resistant growth. Cancer Res. 2009;69:2305–13.
Acknowledgements
This work was supported by operating grants from the Canadian Institutes of Health Research (to JHW). We are grateful to Drs Jun Luo (Johns Hopkins University), Stephen Plymate (University of Washington), Liangnian Song (Columbia University), S. Srivastava (Uniformed Services University), and O. Ogawa (Kyoto University) for generously providing plasmids as a gift for this work.
Author information
Authors and Affiliations
Contributions
QY and JHW designed the research; QY, XH, HGY, HYC, DL, and JS performed the experiments; RL contributed plasmids; QY, GB, and JHW analyzed data; QY and JHW wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yi, Q., Han, X., Yu, H.G. et al. SC912 inhibits AR-V7 activity in castration-resistant prostate cancer by targeting the androgen receptor N-terminal domain. Oncogene 43, 1522–1533 (2024). https://doi.org/10.1038/s41388-024-02944-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-024-02944-2
- Springer Nature Limited