
Abstract
Dysregulated expression of long-stranded non-coding RNAs is strongly associated with carcinogenesis. However, the precise mechanisms underlying their involvement in ovarian cancer pathogenesis remain poorly defined. Here, we found that lncRNA RUNX1-IT1 plays a crucial role in the progression of ovarian cancer. Patients with high RUNX1-IT1 expression had shorter survival and poorer outcomes. Notably, knockdown of RUNX1-IT1 suppressed the proliferation, migration and invasion of ovarian cancer cells in vitro, and reduced the formation of peritoneum metastasis in vivo. Mechanistically, RUNX1-IT1 bound to HDAC1, the core component of the NuRD complex, and STAT1, acting as a molecular scaffold of the STAT1 and NuRD complex to regulate intracellular reactive oxygen homeostasis by altering the histone modification status of downstream targets including GPX1. Consequently, RUNX1-IT1 activated NF-κB signaling and altered the biology of ovarian cancer cells. In conclusion, our findings demonstrate that RUNX1-IT1 promotes ovarian malignancy and suggest that targeting RUNX1-IT1 represents a promising therapeutic strategy for ovarian cancer treatment.
Similar content being viewed by others


Introduction
Ovarian cancer is one of the most common and deadly tumors of the female reproductive system, responsible for over 200,000 deaths each year [1]. Due to its small size and deep location within the pelvic cavity, ovarian cancer often develops without obvious symptoms. As a result, most patients are already in the advanced stages of the disease at the time of diagnosis [2]. Despite advancements in treatment options and the development of new drugs, the prognosis for ovarian cancer patients has not significantly improved. Even in countries with abundant medical resources, the 5-year survival rate for ovarian cancer remains below 50% [3, 4]. Therefore, identifying biomarkers associated with ovarian cancer metastasis and screening for effective therapeutic targets are critical for improving the prognosis of ovarian cancer patients.
Long noncoding RNAs (lncRNAs) are transcripts longer than 200 nucleotides that do not encode proteins but participate in the regulation of various biological processes by regulating gene expression at different levels, including transcription, post-transcriptional, and epigenetic levels [5,6,7]. Recent studies have revealed that lncRNAs play multiple roles in cancer development, including proliferation [8], chemoresistance [9], metabolic reprogramming [10], phase separation [11, 12] and metastasis. The involvement and intrinsic mechanisms of lncRNAs in ovarian cancer have been previously reported [13,14,15,16,17]. However, further research is needed to elucidate the specific mechanisms by which lncRNAs contribute to the initiation and development of ovarian cancer.
In this study, by comparing lncRNA expression profile of primary and metastatic tumors, we identified RUNX1-IT1 was upregulated in metastatic tissues and associated with poor prognosis in patients with ovarian cancer. We further demonstrated that RUNX1-IT1 promoted the proliferative and metastatic phenotype of ovarian cancer in vivo and in vitro. Mechanistically, RUNX1-IT1 acted as a scaffold for the STAT1 and NuRD complex to interact with the GPX1 promoter region and suppress GPX1 expression. The reduction of GPX1 led to an increase in intracellular reactive oxygen species (ROS), which subsequently activated NF-κB signaling. Furthermore, we found that treatment with RUNX1-IT1 antisense oligonucleotides (ASOs) or the HDAC inhibitor Entinostat significantly inhibited ovarian cancer progression in mice, and combination therapy showed even better therapeutic effects. In summary, our study revealed the critical role of the RUNX1-IT1/STAT1/NuRD/GPX1 axis in the progression of ovarian cancer and suggested that targeting RUNX1-IT1 could be a potential therapeutic strategy for treating ovarian cancer.
Results
Upregulation of RUNX1-IT1 correlates with metastasis and poor prognosis in ovarian cancer
To identify potential lncRNAs contributing to ovarian cancer metastasis, we examined dysregulated lncRNAs in 5 pairs of primary and metastatic samples through lncRNA transcriptome sequencing [18], and found that RUNX1-IT1 was consistently upregulated in all metastatic lesions. To further validate these results, we collected 38 pairs of primary and metastatic ovarian cancer lesions and detected RUNX1-IT1 expression by RT-qPCR. The results showed that RUNX1-IT1 was upregulated in 71% (27/38) of metastatic tumor tissues (Fig. 1A). RUNX1-IT1 is annotated as an intronic transcript of RUNX1, and we validated its non-coding status using Coding Potential Assessment Tool (CPAT) [19] and Coding-Non-Coding Identifying Tool (CNIT) [20] (Supplementary Fig. S1C, D). Subsequently, we investigated the expression of RUNX1-IT1 in a panel of ovarian cancer cell lines and observed a heterogeneous expression pattern across the cell lines. However, in all ovarian cancer cell lines tested, the expression level of RUNX1-IT1 was significantly elevated compared to normal ovarian epithelial cell line IOSE80 (Fig. 1B). To evaluate the clinical significance of RUNX1-IT1, we analyzed its expression in two independent ovarian cancer cohorts and found that it was positively correlated with advanced FIGO stages (III and IV) (Fig. 1C, D). Kaplan–Meier survival analysis revealed that patients with high level of RUNX1-IT1 had significant shorter overall survival time and progression free survival time (Fig. 1E, F and Supplementary Fig. S1A, B). Furthermore, we quantified RUNX1-IT1 expression in 127 ovarian cancer tissues using in situ hybridization (ISH), which showed that high expression levels of RUNX1-IT1 were significantly correlated with poor prognosis in ovarian cancer patients (Fig. 1G, H). Additionally, FISH and nuclear fractionation assays demonstrated that RUNX1-IT1 is primarily localized in the nucleus (Supplementary Fig. S1E–G). Taken together, these results suggest that RUNX1-IT1 is a nucleus-localized lncRNA associated with ovarian cancer metastasis.

A Expression of RUNX1-IT1 in 38 paired OC metastatic and primary tissues. Relative expression was calculated using Log2 fold change (metastasis/primary). B RT–qPCR analysis of RUNX1-IT1 expression in OC cell lines. C, D Statistical analysis of RUNX1-IT1 expression in early stage (I and II) and advanced stage (III and IV) OC tissues in the GEO database. E, F The overall survival of OC patients with low and high RUNX1-IT1 expression levels in the GEO database. G Representative images of RUNX1-IT1 expression in ovarian cancer tissue by ISH staining. H Overall survival of 127 OC patients with low and high RUNX1-IT1 expression levels.
RUNX1-IT1 promotes the proliferation and metastasis of ovarian cancer cells
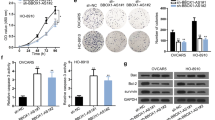
We aimed to investigate the role of RUNX1-IT1 in ovarian cancer progression. To silence its expression, we designed two independent ASOs targeting RUNX1-IT1 and validated their efficiency in ES-2 and OVCA429 cells (Fig. 2A). Additionally, we overexpressed exogenous RUNX1-IT1 in 3AO and SKOV3 cells, which have relatively low RUNX1-IT1 expression levels (Fig. 2B). RUNX1-IT1 is transcribed from an intron of RUNX1, which is a crucial transcription factor involved in the development of many malignancies, especially leukemia [21,22,23]. Despite the important role of RUNX1 in cancer, we observed that silencing RUNX1-IT1 did not affect the expression level of RUNX1 (Supplementary Fig. S2B), which motivated us to investigate the unique role of RUNX1-IT1 in ovarian cancer. CCK-8 assays revealed that silencing RUNX1-IT1 significantly inhibited the proliferation of ES-2 and OVCA429 cells (Fig. 2C, D). In contrast, overexpression of RUNX1-IT1 promoted the proliferation of 3AO and SKOV3 cells (Supplementary Fig. S2C, D). To investigate the impact of RUNX1-IT1 on ovarian cancer metastasis, we performed Transwell assays and found that knockdown of RUNX1-IT1 attenuated the migration and invasion of ES-2 and OVCA429 cells (Fig. 2E, F). Additionally, wound healing assays confirmed that RUNX1-IT1 deficiency reduced cellular motility in vitro (Fig. 2G, H). Conversely, ectopically overexpression of RUNX1-IT1 significantly increased the migration and invasion of 3AO and SKOV3 cells (Supplementary Fig. S2E–H). To further validate the function of RUNX1-IT1 in vivo, we intraperitoneally injected luciferase-labeled OVCA429 cells with stable depletion of RUNX1-IT1 into BALB/c nude mice (Supplementary Fig. S2A). Compared to the control group, depletion of RUNX1-IT1 significantly reduced metastasis in vivo, as evidenced by a marked decrease in the luciferase signal (Fig. 2I, J). Altogether, these results indicate that RUNX1-IT1 promotes both the proliferation and metastasis of ovarian cancer cells.

A RT–qPCR analysis of RUNX1-IT1 expression in ES-2 and OVCA429 cells transfected with control or RUNX1-IT1 ASOs. B RT–qPCR analysis of RUNX1-IT1 expression in 3AO and SKOV3 cells expressing the empty vector or RUNX1-IT1. C, D In vitro growth curve of ES-2 and OVCA429 cells transfected with control or RUNX1-IT1 ASOs. E, F Transwell assays to detect migration and invasion of ES-2 and OVCA429 cells transfected with control or RUNX1-IT1 ASOs. Representative images and quantitative analysis are shown in E and F. G, H Wound healing assays to detect cell motility of ES-2 and OVCA429 cells transfected with control or RUNX1-IT1 ASOs. Representative images and quantitative analysis are shown in G and H. I, J In vivo metastasis in mice injected with OVCA429 cells expressing non-targeting sgRNA or sgRNA targeting RUNX1-IT1. Representative images and quantification of the luminescence radiance are shown in I and J. The data are presented as the mean ± SD; two-tailed t test, *p < 0.05, **p < 0.01, ***p < 0.001. n = 3 biological replicates for in vitro assay, n = 8 per group for in vivo assay.
RUNX1-IT1 physically interacts with the NuRD complex in ovarian cancer cells
To investigate the molecular mechanisms underlying the pro-metastatic role of RUNX1-IT1 in ovarian cancer, we performed RNA pulldown and mass spectrometry (MS) to identify proteins associated with RUNX1-IT1 in ovarian cancer cells. The retrieved proteins were subjected to Western blot analysis. The results showed that HDAC1 is a potential protein associated with RUNX1-IT1 (Fig. 3A). Immunofluorescence subsequently showed strong co-localization of HDAC1 and RUNX1-IT1 in ovarian cancer cells (Fig. 3B). Furthermore, RNA pulldown and RIP assays confirmed the interaction between HDAC1 and RUNX1-IT1 (Fig. 3C, D). We also investigated the interaction between RUNX1-IT1 and other components of the NuRD complex using RNA pulldown assays and found that RUNX1-IT1 can bind to various NuRD complex components including GATAD2B, HDAC2, MTA1, and RbAp46 (Supplementary Fig. S3A). Additionally, we examined whether RUNX1-IT1 affects the expression of HDAC1. Interestingly, knockdown or overexpression of RUNX1-IT1 did not show any regulatory effect on HDAC1 mRNA or protein levels. Conversely, knockdown of HDAC1 did not affect the expression of RUNX1-IT1 (Fig. 3E, F, and Supplementary Fig. S3B–D). Taken together, these results indicate that RUNX1-IT1 specifically interacts with the NuRD complex but does not affect its expression.

A Silver staining for RNA pull-down assays performed using biotinylated RUNX1-IT1 sense or antisense sequences. B Confocal microscopy assessment of co-localization of RUNX1-IT1 (red) and HDAC1 (green) in the indicated cells. Nuclei were stained with Hoechst 33342 (blue). Scale bar = 30 μm. C Western blot analysis of HDAC1 in the precipitates from RNA pull-down assays performed using RUNX1-IT1 sense or antisense sequences. D Anti-HDAC1 RIP assay was performed using ES-2 and OVCA429 cells followed by RT‒qPCR analysis to detect the association between RUNX1-IT1 and HDAC1. E RT–qPCR analysis of HDAC1 expression in ES-2 and OVCA429 cells after knockdown of RUNX1-IT1. F Western blot analysis of HDAC1 protein level in ES-2 and OVCA429 cells after knockdown of RUNX1-IT1. The data are presented as the mean ± SD of three independent experiments. Two-tailed t test, *p < 0.05, **p < 0.01, ***p < 0.001.
RUNX1-IT1 regulates downstream targets by coordinating the genome-wide localization of NuRD complex
Given the distinctive functionality of the NuRD complex and the absence of any influence of RUNX1-IT1 on HDAC1 expression, we hypothesize that RUNX1-IT1 may have a regulatory impact on the epigenetic regulation that involves the NuRD complex. Hence, we performed chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) analysis for HDAC1 in both control and RUNX1-IT1 overexpressing cells. The results suggest that the overexpression of RUNX1-IT1 leads to increased occupancy of HDAC1 on DNA binding regions, indicating that RUNX1-IT1 participates in the epigenetic regulation mediated by the NuRD complex. (Fig. 4A, B and S4A). To further validate these results, we selected some of the genes with increased HDAC1 binding peaks for verification. Most of the target genes showed significant downregulation after overexpression of RUNX1-IT1, which was also confirmed by knocking down of RUNX1-IT1 (Fig. 4D and Supplementary Fig. S4B). Considering that a majority of the pathways exhibited enrichment in processes related to oxidative stress and cell redox homeostasis, our attention was directed towards GPX1, recognized for its crucial involvement in the regulation of oxidative stress (Fig. 4C) [24, 25]. According to the ChIP-seq data, GPX1 is one of the target genes of RUNX1-IT1/NuRD complex. RT-qPCR and Western blot analysis showed that RUNX1-IT1 knockdown enhanced GPX1 expression at both RNA and protein levels (Supplementary Fig. S4C, D). Furthermore, knockdown of RUNX1-IT1 led to a decrease of HDAC1 occupancy and an increase of histone 3 acetylation (H3Ac) occupancy in the GPX1 promoter region (Fig. 4E, F). In addition, ectopic expression of HDAC1 was found to inhibit GPX1 expression, which was partially restored by knocking down RUNX1-IT1 (Fig. 4G). These observations prompted us to investigate whether RUNX1-IT1 binds directly to the promoter region of GPX1. Using the chromatin isolation by RNA purification (ChIRP) assay, we isolated chromatin fragments bound by RUNX1-IT1. However, ChIRP-qPCR revealed that the binding intensity of RUNX1-IT1 to the GPX1 promoter region did not significantly increase compared to the control group, indicating that RUNX1-IT1 could not directly guide the NuRD complex to suppress GPX1 expression (Fig. 4H, I). The regulation of GPX1 by the RUNX1-IT1/NuRD complex may require the involvement of other members.

A Heatmap of HDAC1 global binding signals with ±2 kb around TSS after RUNX1-IT1 overexpression. B Genomic distribution of the binding targets of HDAC1 based on ChIP-seq after RUNX1-IT1 overexpression. C Top enriched biological processes of genes with increased occupancy by HDAC1. D Relative expression of selected genes with increased HDAC1 binding strength after RUNX1-IT1 overexpression. E, F ChIP-qPCR were performed with antibodies against HDAC1 or H3Ac using OVCA429 cells to detected the association between GPX1 promoter and HDAC1 (E) or H3Ac (F). G Relative expression levels of GPX1 in OVCA429 cells were detected by RT‒qPCR after transfection with ASO-Ctrl, ASO-RUNX1-IT1 and HDAC1 as indicated. H RT-qPCR was performed to analyze the retrieval efficiency of CHIRP probe specific for RUNX1-IT1. I DNA binding by ChIRP probes were analyzed using RT-qPCR and primers targeting GPX1 promoter region. The data are presented as the mean ± SD of three independent experiments. Two-tailed t test, *p < 0.05, **p < 0.01, ***p < 0.001.
The regulation of GPX1 by the RUNX1-IT1/NuRD complex is mediated by STAT1
To further explore how the RUNX1-IT1/NuRD complex inhibits GPX1 expression, we reanalyzed mass spectrometry data and identified STAT1 as a potential binding protein of RUNX1-IT1. As a critical transcription factor, STAT1 exhibits complex functions and regulatory mechanisms in tumorigenesis. Recent studies have indicated its crucial role in the metastasis of ovarian and breast cancer [26, 27]. To confirm the specific interaction between RUNX1-IT1 and STAT1, we conducted RNA pulldown assays and detected STAT1 in ovarian cancer cells using Western blotting. The results showed that the sense strand of RUNX1-IT1 but not the antisense strand, interacted with STAT1 (Fig. 5A). This interaction was further verified by RIP assays (Fig. 5B). To assess whether there is an interaction between STAT1 and the NuRD complex in ovarian cancer cells, we extracted total proteins of ES-2 and OVCA429 cells and performed co-immunoprecipitation using antibodies against STAT1. Immunoblotting with antibodies against HDAC1, HDAC2, MTA1, and RbAp46 showed that all of these proteins were efficiently co-immunoprecipitated with STAT1. Conversely, immunoprecipitation using antibodies against representative components of the NuRD complex and immunoblotting using antibodies against STAT1 also showed STAT1 could efficiently interact with the NuRD complex (Fig. 5C).

A Western blot analysis of STAT1 in precipitates from RNA pulldown using sense and antisense sequences of RUNX1-IT1 in ES-2 and OVCA429 cells. B Anti-STAT1 RIP assay was performed using OVCA429 cells followed by RT‒qPCR to detect the association between RUNX1-IT1 and STAT1. C Immunoblotting using antibodies against the indicated NuRD complex proteins in a co-IP assay with anti-STAT1 antibody (top), or immunoblotting using antibodies against STAT1 in a co-IP assay with antibodies against NuRD complex proteins (bottom) in ES-2 and OVCA429 cells. D Co-IP assays were performed with indicated antibodies in the presence or absence of RNase A (left), or after transfection with ASO-Ctrl or ASO-RUNX1-IT1 (right). E ChIP-reChIP and RT-qPCR were performed to analysis the binding of STAT1 and HDAC1 to the GPX1 promoter in ES-2 and OVCA429 cells. F Sequence features of STAT1 binding sites predicted by the JASPAR database. G ChIP-qPCR were performed with antibodies against STAT1 to detected the association between GPX1 promoter and STAT1. H Western blot analysis of STAT1 expression after knockdown STAT1 in OVCA429 cells. I Relative expression levels of GPX1 in OVCA429 cells were detected by RT‒qPCR after RUNX1-IT1 overexpression or STAT1 knockdown as indicated. The data are presented as the mean ± SD of three independent experiments. Two-tailed t test, *p < 0.05, **p < 0.01, ***p < 0.001.
Previous studies have suggested that lncRNAs are involved in protein-protein interactions, which motivated us to investigate whether RUNX1-IT1 also possesses similar functions. To test this hypothesis, we conducted co-immunoprecipitation assays in ES-2 and OVCA429 cells in the presence or absence of RNase A. Western blotting revealed that HDAC1 could interact with STAT1, but this interaction was only detectable in the absence of RNase A. After treatment with RNase A, this interaction was no longer observed. Furthermore, knockdown of RUNX1-IT1 weakened this interaction (Fig. 5D). These findings support the notion that RUNX1-IT1 plays a crucial role in mediating the interaction between the NuRD complex and STAT1. Next, we aimed to investigate whether STAT1 is involved in the regulation of GPX1 by the RUNX1-IT1/NuRD complex. To address this, we conducted ChIP-reChIP experiments using antibodies against HDAC1 and STAT1. The results showed significant co-occupancy of HDAC1 and STAT1 on the promoter region of GPX1 (Fig. 5E). We then searched the JASPAR database and identified STAT1 binding sites in the GPX1 promoter region (Fig. 5F). Subsequently, we performed ChIP-qPCR with antibody against STAT1 after knockdown RUNX1-IT1 based on the predicted sequence with JASPAR. As expected, knockdown of RUNX1-IT1 reduced the binding affinity of STAT1 to the promoter region of GPX1 (Fig. 5G). In addition, overexpression of RUNX1-IT1 inhibited GPX1 expression, which was partially restored by knockdown of STAT1 (Fig. 5H, I). Based on our findings, it appears that RUNX1-IT1 acts as a scaffold to connect STAT1 and NuRD complex, thereby influencing the downstream target GPX1 in ovarian cancer.
RUNX1-IT1-STAT1-NuRD complex regulates ROS levels to activate the NF-κB signaling pathway and promotes metastasis
To further investigate the specific mechanism by which RUNX1-IT1 promotes invasion and metastasis of ovarian cancer cells, we performed RNA sequencing to analyze the gene expression profile after RUNX1-IT1 knockdown. Gene set enrichment analysis (GSEA) showed significant enrichment of epithelial-mesenchymal transition (EMT) and TNFα signaling via NF-κB in the presence of RUNX1-IT1 (Fig. 6A). This finding is consistent with the enrichment results obtained from the ChIP-seq analysis (Fig. 4C). Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis also revealed that RUNX1-IT1 positively regulates the NF-κB signaling pathway (Supplementary Fig. S5A), indicating that RUNX1-IT1 mediated the activation of the NF-κB signaling pathway. As expected, the downstream targets of the NF-κB signaling were remarkably affected by RUNX1-IT1 depletion (Fig. 6B). We then evaluated the expression of NF-κB signaling and EMT related proteins, including N-cadherin, E-cadherin, NFKBIA and p65, in ovarian cancer cells after knockdown or overexpression of RUNX1-IT1. The result showed that the expression of E-cadherin and NFKBIA were significantly upregulated, while the nuclear protein p65 and N-cadherin were significantly downregulated upon RUNX1-IT1 knockdown (Supplementary Fig. S5B). Similar results were observed in the RUNX1-IT1 overexpression groups (Fig. 6C), suggesting that RUNX1-IT1 induces the transition from an epithelial phenotype to a mesenchymal phenotype and activates the NF-κB signaling pathway in ovarian cancer cells.

A GSEA analysis of RUNX1-IT1 knockdown and control OVCA429 cells. B Relative expression of selected targets after knockdown of RUNX1-IT1 in OVCA429 cells. C Western blot analysis of N-cadherin, E-cadherin, NFKBIA, nuclear protein p65 and cytoplasm protein p65 in 3AO and SKOV3 cells with RUNX1-IT1 overexpression followed by HDAC1 or STAT1 knockdown as indicated. D The ROS levels were detected by flow cytometry in 3AO and SKOV3 cells overexpressing RUNX1-IT1 untreated or treated with 5 mM NAC (5 mM, 24 h) (bottom) and quantified by the fluorescence intensities (top). E Transwell assays to detect the invasion and migration ability of SKOV3 cells overexpressing RUNX1-IT1 untreated or treated with 100 μM NAC for 24 h. Representative images and quantitative analysis are shown in left and right. F The ROS levels were quantified by the fluorescence intensities in 3AO and SKOV3 cells treated as indicated. G Transwell assays to detect the invasion and migration ability of SKOV3 cells treated as indicated. Representative images and quantitative analysis are shown in left and right. The data are presented as the mean ± SD of three independent experiments. Two-tailed t test, *p < 0.05, **p < 0.01, ***p < 0.001.
Another important question concerns the role of GPX1, a downstream target gene directly regulated by the RUNX1-IT1-STAT1-NuRD complex, in the promotion of ovarian cancer metastasis by RUNX1-IT1. GPX1 plays a critical role in regulating redox homeostasis in cells by controlling intracellular levels of reactive oxygen species (ROS). We examined intracellular ROS levels following alterations in RUNX1-IT1 expression. The results showed that upregulation of RUNX1-IT1 significantly promoted ROS production, while downregulation of RUNX1-IT1 led to a clear decrease in ROS levels (Fig. 6D and Supplementary Fig. S5C, D). Furthermore, elevated levels of ROS or increased metastasis resulting from RUNX1-IT1 overexpression can both be reversed by administration of the ROS scavenger N-acetyl-l-cysteine (NAC), indicating that alterations in ROS levels underlie the influence of RUNX1-IT1 on ovarian cancer metastasis (Fig. 6D, E and Supplementary Fig. S5E).
Previous studies have indicated that the accumulation of reactive oxygen species (ROS) can activate the NF-κB signaling pathway [28, 29]. Furthermore, NF-κB has been demonstrated to play a crucial role in regulating the EMT process, which is considered a critical step in metastasis [30,31,32]. Building on these findings, we propose that the RUNX1-IT1-STAT1-NuRD complex activates the NF-κB signaling pathway by increasing intracellular ROS levels, thereby promoting EMT and metastasis. To test this hypothesis, we initially assessed ROS levels and NF-κB signaling in ovarian cancer cells overexpressing RUNX1-IT1. The results revealed that upregulation of RUNX1-IT1 led to elevated ROS production and activation of the NF-κB signaling pathway, and this effect was reversed by knocking down STAT1 or HDAC1 (Fig. 6C, F). Subsequently, we examined the metastatic capacity and EMT markers in cells overexpressing RUNX1-IT1 and found that its upregulation promoted metastasis and the EMT process in ovarian cancer cells, a trend that was reversed upon knockdown of STAT1 or HDAC1 (Fig. 6C, G and Supplementary Fig. S5G). Overall, these findings suggest that the RUNX1-IT1-STAT1-NuRD complex regulates ROS production to activate the NF-κB signaling pathway and promote metastasis in ovarian cancer cells.
RUNX1-IT1 presents a promising therapeutic target for ovarian cancer
Finally, we evaluated the mRNA expression of GPX1 and HDAC1 in 38 pairs of primary and metastatic ovarian tumors. The results showed that GPX1 expression was significantly lower, while HDAC1 expression was significantly higher in metastatic ovarian cancer tissues compared to primary tumors (Fig. 7A). Furthermore, analysis of ovarian cancer patients using Kaplan–Meier Plotter revealed that patients with high levels of HDAC1 had a shorter overall survival time and progression-free survival time than patients with low HDAC1 levels (Fig. 7B). Considering the crucial role of RUNX1-IT1 in the progression of ovarian cancer, we further investigated the potential utility of RUNX1-IT1 as a therapeutic target for ovarian cancer. Luciferase-labeled OVCA429 cells were injected into the peritoneal cavity of nude mice to simulate ovarian cancer metastasis. The mice were then randomly divided into four groups and treated with in vivo optimized ASO-Ctrl, ASO-RUNX1-IT1, Entinostat (a class I HDAC1 inhibitor) alone or a combination of Entinostat and ASO-RUNX1-IT1 (Fig. 7C). We observed that the mice treated with ASO-RUNX1-IT1 or Entinostat alone exhibited weaker luciferase signals of metastasis compared to the control group, whereas the combined treatment showed a better effect than single treatment alone (Fig. 7D, E). In addition, treatment with ASO-RUNX1-IT1 or Entinostat significantly reduced the number of metastatic nodules, and the combination treatment group had the lowest number of metastatic tumors (Fig. 7F, G). Taken together, these results suggest that targeting RUNX1-IT1 and HDAC1 may effectively suppress the progression of ovarian cancer and serve as potential therapeutic targets for ovarian cancer.

A Expression of GPX1 and HDAC1 in 38 paired primary and metastasis tumors of ovarian cancer. Paired t test. B Kaplan–Meier survival analysis of the relationship between HDAC1 expression and overall survival time or progression free survival time in ovarian cancer patients. C Timeline of treatment with antisense oligonucleotides (ASO)-Ctrl, ASO-RUNX1-IT1, Entinostat, and the combination of ASO-RUNX1-IT1 and Entinostat. D, E Representative images and quantification of the bioluminescence radiance of metastatic nodules in the abdomens of mice with different treatment. The OVCA429 cells labeled with luciferase were implanted into the peritoneal cavity of nude mice. F Representative images of macroscopic observations of metastatic lesions and range in nude mice. G The number of metastatic nodules of each group with different treatment. The data are presented as the mean ± SD of 5 mice per group. Two-tailed t test, *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Metastasis is a leading cause of cancer-related deaths. Emerging evidence suggests that lncRNAs play a critical role in tumor metastasis [33]. In this study, we identified the metastasis-associated lncRNA RUNX1-IT1 in ovarian cancer by profiling lncRNA expression in primary and metastatic tumors. We found that RUNX1-IT1 is significantly associated with ovarian cancer progression and reduced patient survival. Knockdown of RUNX1-IT1 significantly inhibited proliferation, invasion and migration, as well as in vivo metastasis. Mechanistically, RUNX1-IT1 suppresses the transcription of GPX1 by promoting the deacetylation of the GPX1 promoter through the NuRD complex. Importantly, we demonstrated that targeting RUNX1-IT1 or HDACs significantly suppressed tumor metastasis in mice. Our results provide insight into the molecular mechanisms underlying the oncogenic role of the aberrantly upregulated RUNX1-IT1 in ovarian cancer progression and offer a potential target for ovarian cancer treatment.
RUNX1-IT1 is transcribed from the intron of RUNX1, an important transcription factor that is closely associated with tumor proliferation, metastasis, and chemoresistance [34,35,36,37]. Interestingly, the function of RUNX1-IT1 appears to be diverse in different types of cancers. For example, in hepatocellular carcinoma (HCC), RUNX1-IT1 suppresses proliferation and promotes apoptosis by regulating the miR-632/GSK-3β/Wnt axis [38]. However, another study on pancreatic cancer revealed that RUNX1-IT1 exhibits characteristics of promoting tumor proliferation and invasion, and high levels of RUNX1-IT1 are associated with poor prognosis in pancreatic cancer patients [39]. In this study, we found that the oncogenic effect of RUNX1-IT1 is mediated by HDAC1, which is a member of the NuRD complex that participates in the epigenetic regulation of downstream targets.
NuRD is a multi-subunit protein complex that inhibits gene expression by participating in nucleosome remodeling and histone deacetylation [40, 41]. Previous studies have shown that lncRNAs can recruit histone-modifying enzymes or transcription factors to activate oncogenes or inhibit tumor suppressors, thereby promoting tumor proliferation and invasion [42,43,44,45]. Based on RNA pulldown assays, we observed that RUNX1-IT1 interacts with the NuRD complex and STAT1. Knockdown of either HDAC1 or STAT1 led to the upregulation of downstream tumor suppressor genes. This suggests that the RUNX1-IT1/STAT1/NuRD complex axis activates transcriptional repression mediated by histone deacetylation. HDAC1 is considered as one of the most promising therapeutic targets among the subunits of the NuRD complex. Recent studies have shown that HDAC1 plays a crucial role in cancer metastasis, including nasopharyngeal, lung, and pancreatic cancers [46, 47]. For instance, DNTTIP1 recruits HDAC1 to the DUSP2 promoter and suppresses its expression, leading to activation of ERK signaling and upregulation of MMP2, which promotes metastasis in nasopharyngeal carcinoma [48]. Due to its significant role in cancer progression and metastasis, Targeting HDACs has become a highly promising approach for cancer treatment, and there have been promising developments in the development of HDAC inhibitors as therapeutic agents. Besides Vorinostat, which has already been approved for clinical use, several other drugs, such as Entinostat, Pracinostat, Abexinostat, and Panobinostat, are currently undergoing phase III clinical trials.
GPX1 is one of the most abundant antioxidant enzymes that regulates the levels of reactive oxygen species, thus maintaining cellular redox balance [49]. Several studies have suggested that GPX1 inhibits tumor metastasis and enhances chemosensitivity, while others have shown that GPX1 promotes tumor metastasis [50, 51]. This complicated effect may depend not only on GPX1 itself, but also on the dichotomous roles of reactive oxygen species regulated by GPX1 in tumor cells. In addition, NF-κB is abnormally activated in various types of cancers, including ovarian cancer, and is closely associated with tumor progression and recurrence [52, 53]. ROS is considered one of the key factors responsible for activating NF-κB signaling [54], which is associated with the degradation of IκBα [55]. Our study found that RUNX1-IT1 activates the NF-κB signaling pathway. Knockdown of RUNX1-IT1 reduced HDAC1-mediated GPX1 transcriptional repression, resulting in a decrease in ROS-mediated NF-κB activation. Therefore, we investigated whether therapeutic strategies targeting RUNX1-IT1 or HDACs could alleviate ovarian cancer progression.
The concept of antisense oligonucleotide therapy has been proposed for decades [56] and represents a promising lncRNA-directed therapeutic strategy that is still under active investigation [57]. In the present study, ASOs targeting RUNX1-IT1 or HDAC inhibitor Entinostat treatment achieved a significant repression effect on tumor burden in mouse xenograft models. Furthermore, the combination of these two reagents had greater benefits than each treatment alone. Since Entinostat has already been approved for the treatment of locally advanced or metastatic HR-positive, HER2-negative breast cancer, exploring its efficacy in ovarian cancer is necessary.
We acknowledge the limitations of this study. While multiple ovarian cancer datasets have shown that high expression of RUNX1-IT1 is associated with poor prognosis, this conclusion still needs to be validated in larger cohorts to confirm its reliability. Moreover, as RUNX1-IT1 has distinct functions in different tumors, it is crucial to accurately distinguish cancer types when discussing its potential clinical applications. In addition, the mechanism underlying the abnormal high expression of RUNX1-IT1 in ovarian cancer metastasis remains to be elucidated and warrants further investigation. This study has identified NF-kB as a downstream pathway of RUNX1-IT1 involved in mediating metastasis and the EMT process in ovarian cancer. While the critical role of NF-kB in EMT regulation has been demonstrated [30], further investigation is necessary to understand the specific mechanisms responsible for its promotion of EMT.
In summary, we have identified RUNX1-IT1 as a metastasis-related lncRNA in ovarian cancer. Mechanistically, RUNX1-IT1 acts as a molecular scaffold linking STAT1 and the NuRD complex, co-localizes to the GPX1 promoter, and epigenetically suppresses GPX1 transcription. This downregulation of GPX1 results in the accumulation of ROS, ultimately leading to the activation of NF-κB (Fig. 8). Treatment with ASOs targeting RUNX1-IT1 and HDAC inhibitors showed that the combination of the two reagents was effective in mitigating peritoneal metastasis in mice, providing new therapeutic options for the development of novel therapies for ovarian cancer.

RUNX1-IT1 suppresses GPX1 expression through the STAT1/NuRD complex, leading to downregulation of GPX1. This downregulation induces an increase in ROS levels and activates the NF-κB signaling, which ultimately contributes to the promotion of tumor growth and metastasis of ovarian cancer.
Materials and methods
Patient samples
The ovarian cancer specimens used in this study were obtained from the Cancer Hospital of the Chinese Academy of Medical Sciences. All samples were surgically resected, immediately frozen in liquid nitrogen, and stored at −80 °C until use. This study was approved by the Ethics Committee of the Cancer Hospital, Chinese Academy of Medical Sciences, and informed consents were obtained from all patients.
Cell line and cell culture
The ovarian cancer cell lines (SKOV3, ES-2, OVCA429, TOV-21G, and OVTOKO) were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). IOSE80 and A2780 cells were purchased from the National Experimental Cell Resource Sharing Platform (Beijing, China). 3AO cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured following their instructions.
Virus production and cell infection
Virus production and cell infection were performed as previously described [9]. The shRNA and sgRNA sequences are listed in Supplementary Table S1.
FISH and immunofluorescence (IF) staining
RNA FISH was used to detect the subcellular localization of RUNX1-IT1 with Ribo fluorescent in situ hybridization kit (C10910, RiboBio, Guangzhou, China). Cy3-labeled probes targeting RUNX1-IT1 was used to perform in situ hybridization according to the manufacturer’s instructions. IF staining was performed as previously described [58].
CCK8
Cell proliferation was measured using a Cell Counting. Kit 8 (Dojindo, Japan). Briefly, 2 × 103 ovarian cancer cells were suspended in 100 μL of medium and seeded into a 96-well plates. The absorbance at 450 nm was measured using a microplate reader (BioTek, VT, USA) every 24 h for 4 consecutive days.
Transwell
The invasion and migration ability of ovarian cancer cells were measured by Transwell chambers (3422; Corning, NY, USA). For migration assays, 3 × 104 transfected cells were suspended in 100 μL of medium and seeded into upper chambers, and 650 μL medium containing 10% FBS were added into lower chambers. After 24 h of incubation at 37 °C with 5% CO2, the remaining cells in the upper layer of the membrane were gently wiped up with cotton swab. The cells that passed through the membrane were stained with a methanol solution containing 0.5% crystal violet for 30 min. Representative images were photographed by microscopy, and the number of colored cells was counted for statistical analysis. For the invasion assay, serum-free medium and Matrigel (Corning) were premixed at a ratio of 25:1, and 100 μl of the mixture was added to the upper chamber and incubated at 37 °C for 2 h before seeding the cells.
Wound healing
For wound healing assay, transfected cells were seeded into a 6-well plate and grown until the confluence reached 90% and scratched using a 10 μl tip. The wound formation was photographed by microscopy at 0 h and 24 h. The size of the wound was measured using Image J software.
RNA extraction and real-time quantitative polymerase chain reaction (RT‒qPCR)
Total RNA was extracted from tumor tissues or cells with TRIzol reagent (15596018; Invitrogen, CA, USA). Briefly, 1 μg of total RNA was used for reverse transcription with Quant script RT Kit (KR103, Tiangen Biotech, Beijing, China) or lnRcute lncRNA First-Strand cDNA Kit (KR202; Tiangen Biotech, Beijing, China). PowerUp SYBR Green Master Mix (A25918; Applied Biosystems, CA, USA) were used to determine RNA level via StepOnePlus Real-Time PCR system (Applied Biosystems), and the relative quantification (2−ΔΔCt) method was used for fold-change calculation. The RT-qPCR primers are shown in Supplementary Table S2.
Western blotting and antibodies
Total proteins were lysed with RIPA buffer containing protease inhibitor cocktail (Roche, Basel, Switzerland) and quantified using the PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). The proteins were separated by electrophoresis using 10% SDS polyacrylamide gels and then transferred onto PVDF membranes (Millipore, Billerica, MA, USA). After blocking with 5% skimmed milk at room temperature for 1 h, the membranes were incubated with the appropriate primary antibody overnight at 4 °C. Subsequently, the membranes were washed 3 times with TBST and incubated with HRP-labeled secondary antibodies for 2 h at room temperature. Finally, the membranes were visualized using enhanced chemiluminescence (ECL) horseradish peroxidase (HRP) substrate (Thermo Fisher Scientific). Following antibodies were used for Western blot analysis: HDAC1 (#34589, Cell Signaling Technology, 1:1000), GAPDH (G9545, Sigma-Aldrich, 1:10000), RBAP46 (#6882, Cell Signaling Technology, 1:1000), HDAC2 (#57156, Cell Signaling Technology, 1:1000), MTA1 (#5674, Cell Signaling Technology,1:1000), GATAD2B (ab76925, Abcam, 1:2000), NF-κB p65 (#8242, Cell Signaling Technology, 1:1000), RUNX1 (A20836, Abclonal, 1:500), GPX1 (ab108429, Abcam, 1:1000), STAT1 (#14994, Cell Signaling Technology, 1:1000), IκBα (A11397, Abclonal, 1:500), Ecadherin (#3195, Cell Signaling Technology, 1:1000), Ncadherin (#13116, Cell Signaling Technology, 1:1000), H3 (#4499, Cell Signaling Technology, 1:2000).
Immunoprecipitation (IP)
For co-IP assays, Cell Lysis Buffer for Western and IP (P0013; Beyotime Biotechnology, Shanghai, China) were used for protein extraction. Equal amounts of lysate were incubated with corresponding antibodies or rabbit IgG overnight at 4 °C, followed by the addition of Protein A/G Magnetic Beads and further incubation at 4 °C for 8 h. The beads were washed three times with cell lysis buffer and the immunoprecipitated protein complexes were subjected to western blot analysis.
RNA-seq
RNA purification, reverse transcription, library construction and sequencing were performed in Sequanta Technologies Co., Ltd at Shanghai according to the manufacturer’s instructions. The mRNA-focused sequencing libraries from 1 µg total RNA were prepared using Illumina VAHTS® Universal V6 RNA-seq Library Prep Kit (Vazyme). PolyA mRNA was purified from total RNA using oligo-dT-attached magnetic beads and then fragmented by fragmentation buffer. Taking these short fragments as templates, first strand cDNA was synthesized using reverse transcriptase and random primers. Then in the process of second strand cDNA synthesis, RNA template was removed and a replacement strand, incorporating dUTP in place of dTTP to generate ds cDNA, was synthesized. AMPure XP (Beckmen) beads were then used to purify the ds cDNA from the reaction mix. Next, the ds cDNA was subjected to end-repair, phosphorylation and ‘A’ base addition according to Illumina’s library construction protocol. In the following process, Illumina sequencing adapters were added to both size of the ds cDNA fragments. Before PCR amplification, the second strand containing U was digested using UDG enzyme. After PCR amplification for DNA enrichment, the AMPure XP Beads were used to clean up the target fragments of 200–300 bp. After library construction, Qubit 3.0 fluorometer dsDNA HS Assay (Thermo Fisher Scientific) was used to quantify concentration of the resulting sequencing libraries, while the size distribution was analyzed using Agilent BioAnalyzer (Agilent). Sequencing was performed using an Illumina Novaseq 6000 following Illumina-provided protocols for 2 × 150 paired-end sequencing in Sequanta Technologies Co., Ltd. P adj < 0.05 and |FC | >= 2 were used as criteria for differentially expressed genes. The detailed information of differentially expressed genes were listed in Supplementary Table S7.
ISH
The RNA ISH was performed using RNA in situ hybridization kit (Servicebio Technology, Wuhan, China) to detect RUNX1-IT1 expression in ovarian cancer patients according to the manufacturer’s instructions. All the tumor tissues were surgically resected from patients at the Cancer Hospital, Chinese Academy of Medical Sciences. Written informed consents were obtained from all patients. Detailed clinical information were shown in Supplementary Table S6. The H-scores were calculated according to the percentage of positive cells and the staining intensity by two independent observers. H-score = ∑pi×i, where pi represents the percentage of positive cells (0–100%) and i represents the staining intensity (0: negative, 1: weak, 2: medium, and 3: strong). The probes targeting RUNX1-IT1 designed and synthesized by Servicebio Technology (Wuhan, China) were shown in Supplementary Table S3.
RNA immunoprecipitation (RIP) assay
RIP assays were conducted with the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (17–700, Sigma-Aldrich, USA) according to the manufacturer’s instructions. Immunoprecipitated RNA was purified and analyzed by relative quantification by RT‒qPCR.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation was performed using the ChIP kit (#9003, Cell Signaling Technology) according to the manufacturer’s instructions. Chromatin was crosslinked and fragmented by sonication and precipitated with antibodies against HDAC1 (#34589, Cell Signaling Technology) or H3Ac (06–599, Sigma Aldrich), while IgG (#2729, Cell Signaling Technology) was used as a negative control. The immunoprecipitated DNA were analyzed by RT-qPCR. ChIP-reChIP is executed in basically the same way as ChIP. In the first round of ChIP, Immunoprecipitated chromatin were eluted and incubated with 10 mM DTT at 37 °C for 30 min and diluted 20 times with ChIP dilution buffer. Diluted chromatin was subsequently used for a second round of ChIP. The primers used for ChIP-qPCR are listed in Supplementary Table S4.
Detection of ROS
The cellular ROS were measured by flow cytometry. Briefly, indicated cells were harvested and washed with PBS, followed by incubation with 5 μM H2DCFDA (D6883, Sigma-Aldrich, USA) at 37 °C for 30 min in the dark. After washing with PBS, the labeled cells were resuspended in 200 μl of PBS and analyzed using a flow cytometer. The ROS scavenger, N-acetyl-l-cysteine (NAC) (HY-B0215) were purchased from MedChemExpress (MCE, Monmouth Junction, MA, USA).
Animal experiments
Female BALB/c nude mice aged 6 weeks (GemPharmatech, Nanjing, China) were randomly grouped and intraperitoneally injected with luciferase labeled ovarian cancer cells (2 × 106). Four weeks after injection, the tumors were monitored by bioluminescence imaging with a IVIS LUMINA XRMS system. For tumor treatment evaluation, mice were randomly divided into four groups and treated as follows: (a) 10 nM ASO-Ctrl by intraperitoneal injection every 3 days for a total 16 days; (b) 10 nM ASO-RUNX1-IT1 by intraperitoneal injection every 3 days for a total 16 days; (c) 20 mg/kg Entinostat by oral gavage once daily. (d) combination of ASO-RUNX1-IT1 and Entinostat. In vivo optimized ASOs were synthesized by RiBobio (Guangzhou, China). Entinostat (HY-12163) were purchased from MedChemExpress (MCE, Monmouth Junction, MA, USA). The animal experimental procedures were approved by the Animal Care and Use Committee of the Chinese Academy of Medical Sciences Cancer Hospital.
RNA pull-down assays and mass spectrometry analyses
RNA pulldown assays were performed with Pierce™ Magnetic RNA–Protein Pull-Down Kit (20164; Thermo Fisher Scientific) according to the manufacturer’s instructions. The sense and antisense strands of RUNX1-IT1 were in vitro transcribed using a TranscriptAid T7 High Yield Transcription Kit (K0441; Thermo Fisher Scientific) and purified with GeneJET RNA Purification Kit (K0731; Thermo Fisher Scientific). The RNA binding proteins were separated by electrophoresis following silver staining (24600; Thermo Fisher Scientific). Specific bands were excised for mass spectrometry (Beijing Qinglian Biotech Co., Ltd., Beijing, China) and searched in the human proteomics repository.
Chromatin isolation by RNA purification
The ChIRP assay were performed with Magna ChIRP™ Chromatin Isolation by RNA Purification Kit (Millipore) according to the manufacturer’s instructions. 3′-end biotin -labeled probes against RUNX1-IT1 and LacZ were designed and synthesized by RiboBio (Guangzhou, China). A total of 2 × 107 cells were crosslinked and sonicated to create 100–500 bp DNA fragments. After incubation with probes and chromatin at 37 °C for 4 h, the mixture was subjected to streptavidin magnetic beads purification. The immunoprecipitated Chromatin were eluted for RT-qPCR analysis. The probes used for ChIRP assay are listed in Supplementary Table S5.
Statistical analyses
Two-tailed Student’s t test or paired t test were used for comparisons between two groups. Overall survival curves were established using the Kaplan‒Meier method, and the significance of differences was calculated using the log-rank test. All data were representative of at least three independent experiments and presented as the mean ± SD. Statistical analyses were performed using GraphPad Prism 7.0, and p < 0.05 was considered statistically significant.
Data availability
The data generated in this study are available within the article and its supplementary information files. The raw data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE224495 and GSE245778.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Peres LC, Cushing-Haugen KL, Kobel M, Harris HR, Berchuck A, Rossing MA, et al. Invasive epithelial ovarian cancer survival by histotype and disease stage. J Natl Cancer Inst. 2019;111:60–8.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33.
Rosendahl M, Hogdall CK, Mosgaard BJ. Restaging and survival analysis of 4036 ovarian cancer patients according to the 2013 FIGO classification for ovarian, fallopian tube, and primary peritoneal cancer. Int J Gynecol Cancer. 2016;26:680–7.
Goodall GJ, Wickramasinghe VO. RNA in cancer. Nat Rev Cancer. 2021;21:22–36.
Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500.
Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–307.
Yang X, Zhang S, He C, Xue P, Zhang L, He Z, et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol Cancer. 2020;19:46.
Nan Y, Luo Q, Wu X, Liu S, Zhao P, Chang W, et al. DLGAP1-AS2-mediated phosphatidic acid synthesis activates YAP signaling and confers chemoresistance in squamous cell carcinoma. Cancer Res. 2022;82:2887–903.
Park MK, Zhang L, Min KW, Cho JH, Yeh CC, Moon H, et al. NEAT1 is essential for metabolic changes that promote breast cancer growth and metastasis. Cell Metab. 2021;33:2380–97 e9.
Li RH, Tian T, Ge QW, He XY, Shi CY, Li JH, et al. A phosphatidic acid-binding lncRNA SNHG9 facilitates LATS1 liquid-liquid phase separation to promote oncogenic YAP signaling. Cell Res. 2021;31:1088–105.
Elguindy MM, Mendell JT. NORAD-induced Pumilio phase separation is required for genome stability. Nature. 2021;595:303–8.
Zhao L, Ji G, Le X, Wang C, Xu L, Feng M, et al. Long noncoding RNA LINC00092 acts in cancer-associated fibroblasts to drive glycolysis and progression of ovarian cancer. Cancer Res. 2017;77:1369–82.
Wang X, Li X, Lin F, Sun H, Lin Y, Wang Z, et al. The lnc-CTSLP8 upregulates CTSL1 as a competitive endogenous RNA and promotes ovarian cancer metastasis. J Exp Clin Cancer Res. 2021;40:151.
Mitra R, Chen X, Greenawalt EJ, Maulik U, Jiang W, Zhao Z, et al. Decoding critical long non-coding RNA in ovarian cancer epithelial-to-mesenchymal transition. Nat Commun. 2017;8:1604.
Wu DD, Chen X, Sun KX, Wang LL, Chen S, Zhao Y. Role of the lncRNA ABHD11-AS(1) in the tumorigenesis and progression of epithelial ovarian cancer through targeted regulation of RhoC. Mol Cancer. 2017;16:138.
Liang H, Zhao X, Wang C, Sun J, Chen Y, Wang G, et al. Systematic analyses reveal long non-coding RNA (PTAF)-mediated promotion of EMT and invasion-metastasis in serous ovarian cancer. Mol Cancer. 2018;17:96.
Zhao P, Wang Y, Yu X, Nan Y, Liu S, Li B, et al. Long noncoding RNA LOC646029 functions as a ceRNA to suppress ovarian cancer progression through the miR-627-3p/SPRED1 axis. Front Med. 2023. https://doi.org/10.1007/s11684-023-1004-z.
Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41:e74.
Guo JC, Fang SS, Wu Y, Zhang JH, Chen Y, Liu J, et al. CNIT: a fast and accurate web tool for identifying protein-coding and long non-coding transcripts based on intrinsic sequence composition. Nucleic Acids Res. 2019;47:W516–W22.
Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood. 2017;129:2070–82.
Mill CP, Fiskus W, DiNardo CD, Birdwell C, Davis JA, Kadia TM, et al. Effective therapy for AML with RUNX1 mutation by cotreatment with inhibitors of protein translation and BCL2. Blood. 2022;139:907–21.
Hou Y, Zhang Q, Pang W, Hou L, Liang Y, Han X, et al. YTHDC1-mediated augmentation of miR-30d in repressing pancreatic tumorigenesis via attenuation of RUNX1-induced transcriptional activation of Warburg effect. Cell Death Differ. 2021;28:3105–24.
Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–8.
Zhao Y, Wang H, Zhou J, Shao Q. Glutathione peroxidase GPX1 and its dichotomous roles in cancer. Cancers (Basel). 2022;14:2560.
Tian X, Guan W, Zhang L, Sun W, Zhou D, Lin Q, et al. Physical interaction of STAT1 isoforms with TGF-beta receptors leads to functional crosstalk between two signaling pathways in epithelial ovarian cancer. J Exp Clin Cancer Res. 2018;37:103.
Greenwood C, Metodieva G, Al-Janabi K, Lausen B, Alldridge L, Leng L, et al. Stat1 and CD74 overexpression is co-dependent and linked to increased invasion and lymph node metastasis in triple-negative breast cancer. J Proteom. 2012;75:3031–40.
Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. 2016;12:999–1014.
Meyer M, Schreck R, Baeuerle PA. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993;12:2005–15.
Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–81.
Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711–24.
Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, et al. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26:7445–56.
Liu SJ, Dang HX, Lim DA, Feng FY, Maher CA. Long noncoding RNAs in cancer metastasis. Nat Rev Cancer. 2021;21:446–60.
Li Q, Lai Q, He C, Fang Y, Yan Q, Zhang Y, et al. RUNX1 promotes tumour metastasis by activating the Wnt/beta-catenin signalling pathway and EMT in colorectal cancer. J Exp Clin Cancer Res. 2019;38:334.
Liu S, Xie F, Gan L, Peng T, Xu X, Guo S, et al. Integration of transcriptome and cistrome analysis identifies RUNX1-target genes involved in pancreatic cancer proliferation. Genomics. 2020;112:5343–55.
Hong M, He J, Li D, Chu Y, Pu J, Tong Q, et al. Runt-related transcription factor 1 promotes apoptosis and inhibits neuroblastoma progression in vitro and in vivo. J Exp Clin Cancer Res. 2020;39:52.
Zhao K, Cui X, Wang Q, Fang C, Tan Y, Wang Y, et al. RUNX1 contributes to the mesenchymal subtype of glioblastoma in a TGFbeta pathway-dependent manner. Cell Death Dis. 2019;10:877.
Sun L, Wang L, Chen T, Shi Y, Yao B, Liu Z, et al. LncRNA RUNX1-IT1 which is downregulated by hypoxia-driven histone deacetylase 3 represses proliferation and cancer stem-like properties in hepatocellular carcinoma cells. Cell Death Dis. 2020;11:95.
Liu S, Zhang J, Yin L, Wang X, Zheng Y, Zhang Y, et al. The lncRNA RUNX1-IT1 regulates C-FOS transcription by interacting with RUNX1 in the process of pancreatic cancer proliferation, migration and invasion. Cell Death Dis. 2020;11:412.
Yin X, Teng X, Ma T, Yang T, Zhang J, Huo M, et al. RUNX2 recruits the NuRD(MTA1)/CRL4B complex to promote breast cancer progression and bone metastasis. Cell Death Differ. 2022;29:2203–17.
Deng P, Wang Z, Chen J, Liu S, Yao X, Liu S, et al. RAD21 amplification epigenetically suppresses interferon signaling to promote immune evasion in ovarian cancer. J Clin Invest. 2022;132:e159628.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66.
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–93.
Sun M, Nie F, Wang Y, Zhang Z, Hou J, He D, et al. LncRNA HOXA11-AS promotes proliferation and invasion of gastric cancer by scaffolding the chromatin modification factors PRC2, LSD1, and DNMT1. Cancer Res. 2016;76:6299–310.
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6.
von Burstin J, Eser S, Paul MC, Seidler B, Brandl M, Messer M, et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology. 2009;137:361–71.
Lin CW, Wang LK, Wang SP, Chang YL, Wu YY, Chen HY, et al. Daxx inhibits hypoxia-induced lung cancer cell metastasis by suppressing the HIF-1alpha/HDAC1/Slug axis. Nat Commun. 2016;7:13867.
Ding S, Gao Y, Lv D, Tao Y, Liu S, Chen C, et al. DNTTIP1 promotes nasopharyngeal carcinoma metastasis via recruiting HDAC1 to DUSP2 promoter and activating ERK signaling pathway. EBioMedicine. 2022;81:104100.
Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–303.
Meng Q, Shi S, Liang C, Liang D, Hua J, Zhang B, et al. Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3beta/Snail signaling. Oncogene. 2018;37:5843–57.
Lee E, Choi A, Jun Y, Kim N, Yook JI, Kim SY, et al. Glutathione peroxidase-1 regulates adhesion and metastasis of triple-negative breast cancer cells via FAK signaling. Redox Biol. 2020;29:101391.
Yang W, Liu L, Li C, Luo N, Chen R, Li L, et al. TRIM52 plays an oncogenic role in ovarian cancer associated with NF-kB pathway. Cell Death Dis. 2018;9:908.
Liang H, Yu T, Han Y, Jiang H, Wang C, You T, et al. LncRNA PTAR promotes EMT and invasion-metastasis in serous ovarian cancer by competitively binding miR-101-3p to regulate ZEB1 expression. Mol Cancer. 2018;17:119.
Gloire G, Piette J. Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid Redox Signal. 2009;11:2209–22.
Schoonbroodt S, Ferreira V, Best-Belpomme M, Boelaert JR, Legrand-Poels S, Korner M, et al. Crucial role of the amino-terminal tyrosine residue 42 and the carboxyl-terminal PEST domain of I kappa B alpha in NF-kappa B activation by an oxidative stress. J Immunol. 2000;164:4292–300.
Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA. 1978;75:280–4.
Lin C, Yang L. Long noncoding RNA in cancer: wiring signaling circuitry. Trends Cell Biol. 2018;28:287–301.
Wu X, Luo Q, Zhao P, Chang W, Wang Y, Shu T, et al. JOSD1 inhibits mitochondrial apoptotic signalling to drive acquired chemoresistance in gynaecological cancer by stabilizing MCL1. Cell Death Differ. 2020;27:55–70.
Funding
This research was funded by the National Key R&D Program of China (2021YFC2501000, 2020YFA0803300), the National Natural Science Foundation of China (82030089, 82188102), the CAMS Innovation Fund for Medical Sciences (2021–1-I2M-018, 2021-I2M-1-067, 2022-I2M-2-001).
Author information
Authors and Affiliations
Contributions
XY and PZ contributed equally to this work. ZL and ZC conceived the ideas and supervised the research. XY and PZ performed most of the experiments, analyzed the data and drafted the manuscript with assistance from ZL and ZC. WG performed the bioinformatics analyses. QL, XW, YW, and BL collected clinical samples. SL and YN assisted with animal studies. All authors reviewed the manuscript and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yu, X., Zhao, P., Luo, Q. et al. RUNX1-IT1 acts as a scaffold of STAT1 and NuRD complex to promote ROS-mediated NF-κB activation and ovarian cancer progression. Oncogene 43, 420–433 (2024). https://doi.org/10.1038/s41388-023-02910-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-023-02910-4
- Springer Nature Limited