
Abstract
Metadherin (MTDH) is a well-established oncogene in various cancers including Hepatocellular Carcinoma (HCC). However, the precise mechanism through which MTDH promotes cancer-related signaling pathways in HCC remains unknown. In this study, we identified DDX17 as a novel binding partner of MTDH. Furthermore, MTDH increased the protein level of DDX17 by inhibiting its ubiquitination. We confirmed that DDX17 was a novel oncogene, with dramatically upregulated expression in HCC tissues. The increased expression of DDX17 was closely associated with vascular invasion, TNM stage, BCLC stage, and poor prognosis. In vitro and in vivo tests demonstrated that DDX17, a downstream target of MTDH, played a crucial role in tumor initiation and progression. Mechanistically, DDX17 acted as a transcriptional regulator that interacted with Y-box binding protein 1 (YB1) in the nucleus, which in turn drove the binding of YB1 to its target epidermal growth factor receptor (EGFR) gene promoter to increase its transcription. This in turn increased expression of EGFR and the activation of the downstream MEK/pERK signaling pathway. Our results identify DDX17, stabilized by MTDH, as a powerful oncogene in HCC and suggest that the DDX17/YB1/EGFR axis contributes to tumorigenesis and metastasis of HCC.
Similar content being viewed by others


Introduction
Liver cancer is the sixth most common malignancy and the third leading cause of cancer-related death worldwide [1]. Hepatocellular carcinoma is the most common primary liver cancer, which is mainly related to HBV infection, HCV infection, alcohol abuse, and nonalcoholic fatty liver disease [1]. Despite substantial progress in HCC treatment, it still has a very low five-year survival owing to its high recurrence and metastasis rate [2]. Thus, it is necessary to further investigate molecular mechanisms underlying the initiation and progression of HCC to hopefully develop novel therapeutic strategies.
MTDH is originally identified as a HIV-inducible gene in human fetal astrocytes [3, 4]. The human MTDH gene is located on chromosome 8q22. It contains 12 exons/11 introns and encodes a protein of 582 amino acids [4]. It is widely distributed in the cell membrane, cytoplasm, nucleus and endoplasmic reticulum, therefore, suggesting that MTDH may have multiple physiological functions in different cellular contexts [4]. Numerous studies reported that MTDH played critical roles in the progression of various cancers. MTDH was a powerful oncogene that was overexpressed and correlated with tumor progression in HCC [5, 6], breast cancer [7, 8], gastric cancer [9], prostate cancer [10], and non-small cell lung cancer [11]. In breast cancer, MTDH expression was found to be highly correlated with tumor metastasis. MTDH acted as a transmembrane protein that promoted the metastasis from breast to lung via its lung homing domain [12]. Additionally, MTDH was also found to promote tumor formation and metastasis in a transgenic mouse model though binding to SND1 [8]. In HCC patients, high expression of MTDH was closely correlated with microvascular invasion, pathological satellites, TNM stage, and poor prognosis [13]. MTDH was reported to promote tumor growth [14], epithelial-mesenchymal transition [13], anoikis resistance [15], drug resistance [16], metastasis [17], and tumorigenesis [5, 18] in HCC. MTDH was also found to be involved in multiple oncogenic signaling pathways, including NF-κB [19], AKT [14], Wnt-β-catenin [20] and MAPK [4, 6]. Interaction of MTDH with NF-κB and CBP promoted NF-κB mediates transcription [19] and activated the Wnt-β-catenin pathway via upregulating LEF1 expression or indirectly activating ERK42/44 [6]. However, the direct mechanism through which MTDH activated the ERK pathway remains poorly understood. The identification of various interacting partners may help us understand the direct mechanism through which MTDH promotes tumor progression and reveal new therapeutic targets for the treatment of HCC.
DDX17, together with its ortholog DEAD-box helicase 5 (DDX5), belongs to the DEAD box family of RNA helicases, which participates in a range of different genetic processes, including transcription, RNA metabolism, and miRNA biogenesis [21, 22]. Recently, new evidence has shown that DDX17 may be involved in cancer progression. DDX17, as well as its ortholog DDX5, interacted with β-catenin to induce transcription of its target genes in colorectal cancer (CRC) [23]. Knocking down both of these RNA helicases (DDX17 and DDX5) significantly inhibited the proliferation of tumor cells in vitro [23]. In breast cancer, DDX17 was configured with ER-α and this interaction was crucial for the regulation of estrogen-responsive genes and estrogen-stimulated tumor cell growth [24]. Although a previous study reported that knockdown of DDX17 inhibited the migration and invasion of HCC cells [25], the precise role of DDX17 in tumor progression and tumorigenesis remained largely unknown.
The effects of MTDH on proliferation, apoptosis, metastasis, and tumorigenesis in HCC are relatively well understood, but the direct mechanisms mediating these effects are poorly defined. In order to obtain a better picture, we aimed to identify the direct target of MTDH that mediates these cancer-promoting effects. We discovered that MTDH interacted with DDX17 and increased its protein level by inhibiting its ubiquitination. We then validated the effects of DDX17 on HCC tumor proliferation, cell cycle, metastasis, and tumorigenesis. Moreover, we clarified the direct mechanism underling the cancer-promoting effect of DDX17. Finally, we demonstrated that DDX17 mediated the effect of MTDH on HCC progression via the EGFR-pERK signaling pathway and inferred its clinical significance.
Results
DDX17 is a novel MTDH interaction partner in HCC
To elucidate the specific molecular mechanism through which MTDH promoted HCC progression, MTDH interaction partners were identified using immunoaffinity purification combined with LC-MS/MS (Supplementary Figure 1A and Supplementary Table 1). After excluding proteins that overlap with the IgG group, a total of 24 proteins (unique peptides ≥ 4) were detected in the HA-MTDH group. To identify the top ten proteins, we removed non-specific proteins including ribosomal proteins and heat shock proteins. Finally, we focused on three proteins, i.e., SND1, DDX17 and PAPBC1. Since SND1 was previously reported as a binding partner of MTDH [8], we validated whether MTDH was bound to DDX17 and PAPBC1. PAPBC1 showed a weak binding affinity to MTDH (Supplementary Figure 1B). However, DDX17 with 6 matching peptides was identified as a potential interaction partner to MTDH. In the co-immunoprecipitation (CO-IP) assays, Flag-DDX17 was pulled down by HA-MTDH and vice versa (Fig. 1A) in HEK293T cells. Moreover, interaction between endogenous DDX17 and endogenous MTDH was confirmed by Co-IP in HLF cells and HCCLM3 cells (Fig. 1B, C). Immunofluorescence (IF) staining showed that MTDH and DDX17 mainly co-localized in the nucleus (Fig. 1D). Domain mapping analysis revealed that the amino terminal region of MTDH (aa: 1-291) was responsible for the binding between MTDH and DDX17 (Fig. 1E). Additionally, deletion of the helicase C-terminal domain (aa: 405-553) of DDX17 almost completely abolished the interaction between MTDH and DDX17 (Fig. 1F, G). However, DDX5, the ortholog of DDX17, did not interact with MTDH in HEK293T cells, and MTDH knockdown had little effect on the expression of DDX5 in HCCLM3 and HLF cells (Supplementary Figure 1C, D). These data suggested that DDX17 interacted with MTDH.

A HEK293T cells were transfected with vectors expressing Flag-DDX17 and HA-MTDH, followed by immunoprecipitation and immunoblotting using antibodies against Flag and HA, respectively. B Immunoprecipitation analysis of DDX17 and MTDH in HLF cells. C Immunoprecipitation analysis of DDX17 and MTDH in HCCLM3 cells. D MTDH colocalized with DDX17 in the nucleus of HLF cells. Endogenous DDX17 and MTDH were stained by anti-DDX17 (green) and anti-MTDH (red) antibodies, respectively. Scale bar, 10 μm. E HEK293T cells were co-transfected with vectors expressing Flag-tagged full length MTDH or its truncation mutants and HA-DDX17. The cell lysates were immunoprecipitated with Flag antibody, followed by immunoblotting with the indicated antibodies. F HEK293T cells were co-transfected with vectors expressing Flag-tagged full length DDX17 or its truncation mutants and HA-MTDH. The cell lysates were immunoprecipitated with Flag antibody, followed by immunoblotting with the indicated antibodies. G HEK293T cells were co-transfected with vectors expressing Flag-tagged full length DDX17 or its truncation mutants and HA-MTDH. Immunoprecipitation analysis of Flag and HA in indicated cells.
MTDH increases the protein level of DDX17 by inhibiting its ubiquitination
We next investigated whether there was regulation between MTDH and DDX17. As shown in Fig. 2A, overexpression of MTDH increased the protein level of DDX17 in Huh7 cells, while MTDH knockdown decreased DDX17 expression in HCCLM3 cells. However, MTDH overexpression or knockdown had little effect on DDX17 mRNA expression (Fig. 2B). By contrast, DDX17 knockdown or overexpression had little effect on the mRNA and protein levels of MTDH (Supplementary Figure 2A, B). When HEK293T cells were transfected with different concentrations of plasmid expressed with MTDH, protein expression of DDX17 was increased, while the mRNA level was not changed (Fig. 2C, D). These results suggested that MTDH might increase the protein level of DDX17 at the post-transcriptional stage. Thus, we hypothesized that MTDH may stabilize DDX17 by inhibiting its degradation via the ubiquitin-proteasome pathway. Expression of DDX17 was markedly increased in MTDH-deficient HLF and HCCLM3 cells when the proteasome inhibitor MG132 was added (Fig. 2E). A cycloheximide (CHX) chase assay was, then, used to investigate the effect of MTDH on stability of DDX17. Overexpression of MTDH significantly increased the half-life of DDX17, while knockdown of MTDH destabilized expression of endogenous DDX17 (Fig. 2F, G). Furthermore, an in vitro deubiquitination assay showed that MTDH reduced the polyubiquitination of DDX17 (Fig. 2H). Consistently, knockdown of MTDH dramatically increased ubiquitination of DDX17 in HCCLM3 cells (Fig. 2I), whereas the ectopic expression of MTDH in Huh7 cells significantly decreased its polyubiquitination (Fig. 2J). These data suggested that MTDH increased the protein level of DDX17 by repressing its polyubiquitination.

A Immunoblotting analysis of DDX17, MTDH and GAPDH in MTDH-expressing or vector control Huh7 cells, or in shNC control and DDX17 deficient HCCLM3 cells. B RT-PCR analysis of MTDH and DDX17 in MTDH-overexpressing or vector-control Huh7 cells, or in shNC control and DDX17 deficient HCCLM3 cells. C HEK293T cells were transfected with the indicated concentrations of HA-MTDH for 48 h, after which immunoblotting analysis of DDX17, HA and GAPDH was performed. D HEK293T cells were transfected with the indicated concentrations of HA-MTDH for 48 h, after which RT-PCR analysis of DDX17 and MTDH was performed. E MTDH-deficient HLF and HCCLM3 cells and its shNC control were treated with or without 10 μM MG132, followed by immunoblotting analysis of DDX17 and GAPDH. F MTDH-expressing and vector control Huh7 cells were treated with CHX for the indicated time, followed by immunoblotting analysis of DDX17, MTDH and GAPDH. The lower panel shows the DDX17 protein levels quantified densitometrically using ImageJ software. G MTDH-deficient HLF and HCCLM3 cells and its shNC control were treated with CHX for indicated time, Immunoblotting analysis of DDX17, MTDH and GAPDH. The lower panel shows the DDX17 protein levels quantified densitometrically using ImageJ software. H HEK293T cells were co-transfected with vectors expressing Flag-DDX17, Myc-MTDH and HA-ub for 48 h, followed by treatment with 10 μM MG132 for 6 h. The cell lysates were immunoprecipitated with Flag antibody and then subjected to immunoblotting with the indicated antibodies. I MTDH-deficient HCCLM3 cells and the corresponding shNC control cells were treated with 10 μM MG132 for 6 h. The cell lysates were immunoprecipitated with DDX17 antibody and then subjected to immunoblotting with the indicated antibodies. J MTDH-overexpressing Huh7 cells and vector control cells were treated with 10 μM MG132 for 6 h. The cell lysates were immunoprecipitated with DDX17 antibody and then subjected to immunoblotting with the indicated antibodies. The data represent the means ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
DDX17 promotes the growth and metastasis of HCC
We next verified the physiological function of DDX17 in HCC. First, we detected the expression of DDX17 and MTDH in HCC cell lines (Supplementary Figure 3A). To explore the oncogenic role of DDX17 in HCC progression, endogenous DDX17 was reliably knocked down using lentivirus-expressed short hairpin RNAs in HLF and HCCLM3 cells (referred to as HLF-shDDX17#1-3 and HCCLM3-shDDX17#1-3, respectively) and exogenous DDX17 was overexpressed in Huh7 cells (referred to as Huh7-DDX17) (Fig. 3A). DDX17 knockdown or overexpression had no effect on the expression of its homologous protein DDX5 (Fig. 3A). Cell counting kit 8 (CCK8) and colony formation assays were performed to determine the effect of DDX17 on cell proliferation. The results showed that DDX17 knockdown inhibited the proliferation of HLF and HCCLM3 cells, whereas DDX17 overexpression enhanced the proliferation of Huh7 cells (Fig. 3B, C, and Supplementary Fig. 3B). The cell cycle assay showed that DDX17 knockdown increased the percentage of cells in the G2-M phase, but decreased it in the G0-G1 phase (Fig. 3D and Supplementary Figure 3C). By contrast, overexpression of DDX17 had the opposite effect on Huh7 cells (Fig. 3D and Supplementary Fig. 3C). Next, we tested the effect of DDX17 knockdown on HCC xenograft growth in vivo. HLF-shDDX17#1 or corresponding control cells were subcutaneously injected into the right flank of nude mice. The growth of xenograft tumors was significantly inhibited in the shDDX17-injected group compared to the shNC-injected control group, as reflected in the tumor growth curve, tumor volume and final tumor weigh (Fig. 3E). Moreover, IHC staining of the xenograft tumors showed that the expression of DDX17 and the proliferation marker, Ki-67, were significantly decreased in the shDDX17 group compared to the shNC group (Fig. 3F). These results suggested that DDX17 promoted HCC cell proliferation in vitro and in vivo.

A The efficiency of knockdown or overexpression of DDX17 in the indicated cells was validated by immunoblotting, respectively. B The indicated cells were subjected to the CCK8 assay. C The indicated cells were subjected to the colony formation assay. D The indicated cells were subjected to the cell cycle assay. E Subcutaneous tumors composed of HLF-shDDX17#1 cells and control cells were shown in the left panel. The tumor volume growth curve was shown in the middle panel. The tumor weight was shown in the right panel. F IHC staining for DDX17 and Ki-67 expression in xenograft tumors of different groups (left panel). Statistical comparison of the DDX17 IHC scores of the indicated groups (right panel). G The indicated cells were subjected to transwell migration and invasion assays. H Orthotopic liver tumors derived from HLF-DDX17#1 and the shNC control cells were showed in the left panel. HE staining for orthotopic tumors in the liver from the indicated groups was shown in middle panel. Statistical comparisons of the tumor numbers in the indicated groups were shown in right panel. The data represent the means ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
We next investigated the effect of DDX17 on the mobility and metastasis of HCC cells. The transwell migration and invasion assay showed that knockdown of DDX17 significantly impaired the ability of cell migration and invasion compared to its shNC control, while the ectopic expression of DDX17 enhanced the migration and invasion ability compared to the vector control (Fig. 3G and Supplementary Fig. 3D). We further examined the role of DDX17 in HCC metastasis using an in situ intrahepatic metastasis model and ectopic tumor metastasis model. HLF-shDDX17 cells and the shNC control cells were injected into the left outer lobe of the liver. The metastatic foci in the liver were significantly decreased in the shDDX17 group compared to the shNC group (Fig. 3H). Next, nude mice were injected with HCCLM3-shNC, HCCLM3-shDDX17#1 and HCCLM3-shDDX17#2 cells, as well as HLF-shNC, HLF-shDDX17#1 and HLF-shDDX17#2 cells into the tail vein, respectively. The mice were sacrificed after 8 weeks. The number of lung metastatic nodules was counted using H&E staining. The results showed that DDX17 knockdown in either HCCLM3 or HLF cells reduced the number of metastatic foci and decreased the metastasis rate in the lungs compared to the shNC control (Supplementary Figs. 4A–H). Taken together, the results confirmed that DDX17 promoted the mobility and metastasis of HCC cells in vitro and in vivo.
DDX17 promoted the tumorigenesis of HCC
To investigate the possible role of DDX17 in HCC development, DDX17+/+ mice (DDX17fl/fl mice) and conditional liver DDX17 knockout mice (DDX17fl/fl-Alb-Cre mice, referred to as DDX17−/− mice for sake of brevity) were injected with 80 mg/kg of the hepatic carcinogen diethylnitrosamine (DEN) at 4 weeks of age and fed a high-fat diet (HFD) (Fig. 4A). DDX17 expression in the liver was effectively diminished in DDX17−/− mice compared to DDX17+/+ mice (Fig. 4B). The mice were sacrificed after 52 weeks following DEN treatment. DDX17-/- mice exhibited decreased HCC tumorigenesis with fewer tumors and smaller tumor diameters (Fig. 4C, D). Additionally, the expression of the cell proliferation marker Ki67 was lower in DDX17−/− mice than in the control, which further supported the idea that DDX17 contributes to HCC tumorigenesis (Fig. 4E). Furthermore, the number of lung metastatic foci was significantly decreased in DDX17−/− mice compared to DDX17+/+ mice (Fig. 4E). Subsequently, we tested the effect of DDX17 knockout on the classical signaling pathways in mice, and found that the tissues from DDX17−/− mice showed less phosphorylation in ERK, compared to pAKT, pP38 and pJNK (Fig. 4F). Taken together, these results suggested that DDX17 promoted HCC tumorigenesis in vivo.

A Schematic representation of the DEN-HFD-induced HCC model. For week-old mice were intraperitoneally injected with DEN (80 mg/kg) and 2 weeks later were placed on a high-fat diet (HFD, 60% fat) for 52 weeks (n = 10). B Immunoblotting analysis of liver tissues from DDX17+/+ and DDX17−/− mice. C. Gross morphology (top) of livers of the DDX17+/+ and DDX17−/− mice described in A. Scale bars, 0.5 cm. D Quantification of total tumor number, number of tumors bigger than 3 mm, and maximal tumor diameters. E Ki67 staining of liver tumors from DDX17+/+ and DDX17−/− mice (left panel). H&E staining of sections for lung metastases was performed (left panel). Statistical comparisons of the indicated groups were showed in the right panel. F. Immunoblotting analysis of the indicated proteins in the liver tumors derived from DDX17+/+ and DDX17−/− mice (left panel). The levels of the indicated proteins were quantified densitometrically using ImageJ software. The data represent the means ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
RNA sequencing identify EGFR as a downstream effector of DDX17
We next explored the specific mechanism through which DDX17 contributed to the tumorigenesis and progression of HCC. Since DDX17 acted as a transcription cofactor, we conducted mRNA sequencing to identify its downstream target genes in HLF-shNC versus HLF-shD1 cells and HCCLM3-shNC versus HCCLM3-shD1 cells (Fig. 5A and supplementary Figure 5A). Compared with the respective controls, a total of 214 genes were up- and 640 genes were downregulated in HCCLM3-shD1 cells, whereas 402 genes were up- and 553 genes were downregulated in HLF-shD1 cells (p < 0.05 and Foldchange>2.0) (supplementary Fig. 5B). A Venn diagram showed that 138 genes were downregulated and 25 genes were upregulated in both DDX17-silenced cell lines compared to the respective control cells (supplementary Figure 5C, D, Supplementary Table 2). Among the 138 downregulated genes, EGFR was selected for further study for three reasons. First, it was a well-established and powerful oncogene that contributed to tumor initiation and progression. Second, EGFR was enriched in multiple signaling pathways such as pathways in cancer, focal adhesion, MAPK signaling pathway and regulation of actin cytoskeleton according to KEGG pathway analysis (Fig. 5B and supplementary Fig. 5E). Third, as the most important downstream modulator of EGFR, pERK expression was significantly decreased in DDX17−/− mice compared to DDX17+/+ mice.

A Heatmap showing the differentially expressed genes modulated by DDX17 knockdown (left panel), Scatterplot showing the differentially expressed genes modulated by DDX17 knockdown in HLF cells compared to HLF-shNC cells (right panel). B KEGG pathway analysis of the mRNA sequencing data. C RT-PCR analysis of DDX17 and EGFR in the indicated cells. D The EGFR promoter activity was measured in the indicated cells. E Immunoblotting analysis of EGFR, pERK, ERK, DDX17 and GAPDH in the indicated cells. F Immunoblotting analysis of EGFR and GAPDH in liver tumors derived from the indicated transgenic mice. G HLF DDX17-deficient or HCCLM3 DDX17-deficient cells, together with corresponding shNC control cells, were treated with 10 μg/mL EGF for the indicated time periods. Then, immunoblotting analysis for the indicated proteins was performed. H HLF DDX17-deficient or HCCLM3 DDX17-deficient cells, together with corresponding shNC control cells, were treated with 10 μg/mL EGF for the indicated time period. Then, these cells were subjected to transwell migration and invasion assays (left panel). Statistical comparisons of the indicated groups are shown in the right panel. The data represent the means ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
RT-PCR and immunoblotting analysis were performed to validate the mRNA sequencing results. The mRNA and protein expression of EGFR were significantly decreased after knockdown of DDX17 in HLF and HCCLM3 cells (Fig. 5C, E), whereas their expression were markedly increased by ectopic DDX17 expression in Huh7 cells (Supplementary Fig. 6A, C). Consistent with the results in transgenic mice, the phosphorylation of ERK downstream of EGFR was also influenced by DDX17 in HCC cell lines (Fig. 5E and Supplementary Fig. 6C). Moreover, the transcriptional activity of the EGFR promoter was decreased or enhanced in response to DDX17 knockdown or overexpression, respectively (Fig. 5D and Supplementary Fig. 6B). In vivo, EGFR expression was obviously downregulated in DDX17−/− mice compared to DDX17+/+ mice (Fig. 5F). Additionally, we assessed the expression of DDX17 and EGFR in HCC tissue samples, and the results showed that DDX17 expression was positively correlated with EGFR expression in HCC (cohort I) (Supplementary Fig. 6D, E). Additionally, TCGA analysis confirmed these results (Supplementary Figure 6F). We subsequently treated DDX17-deficient cells and control cells with EGF for the indicated duration and detected the phosphorylation sites of EGFR at Y1068, Y1173 and Y845 which assisted in the binding of downstream anchor proteins and promoted the activation of downstream pathways. And results showed that the phosphorylation sites of EGFR at Y1068, Y1173 and Y845 were significantly reduced in DDX17 deficient cells compared to control cells after EGF treatment (Fig. 5G). Consistently, DDX17 knockdown impaired the EGF-induced migration and invasion of HLF cells compared to the HLF-shNC control (Fig. 5H). Taken together, these results revealed that EGFR was a vital downstream modulator of DDX17.
DDX17 interacts with YB1 and enhances the transcription of EGFR
The underlying mechanism of EGFR regulated by DDX17 was further investigated. DDX17 played an important role in transcription regulation [21, 22] and the above results showed that DDX17 upregulated transcription of EGFR. Hence, we hypothesized that DDX17 may enhance EGFR expression by binding to an unknown transcription factor (TF). We performed CO-IP followed by LC-MS/MS analysis to detect the possible TFs binding to DDX17, and the three TFs Y-box binding protein 1(YB1), Y-box binding protein 3 (YB3) and zinc finger protein 326 (ZNF326) were found to bind DDX17 (Supplementary Fig. 7A and Supplementary Table 3). With 23 detected peptides, YB1 showed stronger binding to DDX17 than the other two identified interaction partners (Supplementary Fig. 7A and B). Moreover, YB1 was reported to regulate EGFR transcription in breast cancer [26]. We therefore hypothesized that DDX17 might interact with YB1 to induce EGFR transcription in HCC. To validate this hypothesis, we first investigated whether there was an interaction between DDX17 and YB1. HEK293T cells were co-transfected with vectors expressing Flag-DDX17 and HA-YB1, followed by a Co-IP assay, which showed that ectopically expressed DDX17 and YB1 interact with each other (Fig. 6A). Additionally, DDX5 was also found to interact with YB1 in HEK293T cells. However, DDX5 knockdown had little effect on EGFR expression (Supplementary Fig. 7C, D). The interaction between endogenous DDX17 and YB1 was also detected in HLF and HCCLM3 cells (Fig. 6B). IF staining showed the co-localization of endogenous DDX17 and YB1 in the nucleus (Fig. 6C). To determine which domain was required for interaction, Co-IP assays were performed. The results revealed that YB1 mainly bound to the C-terminal region of DDX17 (aa: 553-729) (Fig. 6D), and the C-terminal domain (CTD) of YB1 was responsible for the binding to DDX17 (Fig. 6E).

A HEK293T cells were transfected with vectors expressing Flag-DDX17 and HA-YB1, followed by immunoprecipitation and immunoblotting with HA and Flag antibodies, respectively. B Immunoprecipitation analysis with DDX17 or YB1 in HCCLM3 and HLF cells. C DDX17 colocalized with YB1 in the nucleus of HCCLM3 and HLF cells. Endogenous DDX17 and MTDH were stained with anti-DDX17 (green) and anti-YB1 (red) antibodies, respectively. Scar bar, 20 μm. D HEK293T cells were co-transfected with vectors expressing Flag-tagged full-length DDX17 or its truncation mutants and Flag-DDX17. The cell lysates were immunoprecipitated with Flag antibody, followed by immunoblotting with the indicated antibodies. E HEK293T cells were co-transfected with vectors expressing HA-tagged full length YB1 or its truncation mutants and Flag-DDX17. The cell lysates were immunoprecipitated with Flag antibody, followed by immunoblotting with the indicated antibodies. F HEK293T cells were transfected with the indicated plasmids, and the luciferase activity of the EGFR promoter construct was measured. G In DDX17-expressing Huh7 cells, DDX17 directly interacted with the EGFR promoter at all four YB1-binding regions. The data represent the means ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Since DDX17 acted as a transcription coactivator, we hypothesized that DDX17 interacted with YB1 to activate the transcription of EGFR. To determine whether DDX17 cooperated with YB1 to enhance the transcription of EGFR, we transfected HEK293T cells with constructs expressing DDX17, YB1, or DDX17 and YB1, along with an EGFR promoter reporter construct, and then measured the luciferase reporter activity. We found that ectopic co-expression of DDX17 with YB1 synergistically enhanced the promoter activity of EGFR (Fig. 6F). A previous study showed that YB1 upregulated EGFR transcription by binding to its promoter at four binding sites (named EGFR1b, EGFR2a, EGFR2b and EGFR3) [26]. To confirm this, we performed a chromatin immunoprecipitation (CHIP) assay in Huh7-DDX17 cells using a Flag antibody. We observed that Flag-DDX17 was enriched in the same region of the EGFR promoter as that of the YB1 binding sites, especially the binding site EGFR2a and EGFR2b (Fig. 6G). These results suggested that DDX17 interacted with YB1 and enhanced the transcription activity of YB1 on the EGFR promoter.
In addition, we intended to confirm whether DDX17 regulated other YB1 downstream genes in addition to EGFR. CCNA2, MET and MDR1, known to be regulated by YB1 [27], were chosen for validation. The mRNA expression of these genes was significantly decreased by DDX17 knockdown in HCCLM3 cells (Supplementary Fig. 8A), whereas DDX17 overexpression had the opposite effect in Huh7 cells (Supplementary Fig. 8B). Luciferase reporter assays showed that the transcription activity of the CCNA2, MDR1 and MET promoters was inhibited by DDX17 knockdown (Supplementary Fig. 8C). Moreover, the protein levels of these factors were also reduced after DDX17 knockdown in HCCLM3 cells (Supplementary Fig. 8D). Furthermore, TCGA data analysis revealed that DDX17 expression was positively correlated with CCNA2 and MET (Supplementary Fig. 8E). These results suggest that DDX17 may also regulate multiple downstream genes of YB1 by enhancing its transcriptional activity.
Consequently, we investigated whether YB1 mediated the DDX17-induced upregulation of EGFR expression and tumor progression in vitro. DDX17 knockdown had little effect on the expression of YB1 in HLF and HCCLM3 cells (Supplementary Fig. 9A). We knocked down YB1 using siRNA in Huh7 cells, in the presence of DDX17 overexpression. YB1 silencing dramatically reduced the mRNA and protein expression of EGFR induced by DDX17 overexpression (Supplementary Fig. 9B, C). Moreover, pERK, the downstream effector of EGFR, was also reduced after YB1 knockdown in Huh7-DDX17 cells (Supplementary Fig. 9C). Furthermore, YB1 silencing dramatically impaired the proliferation, migration and invasion capacity of Huh7 cells, which was induced by DDX17 overexpression (Supplementary Fig. 9D, E). Taken together, these results suggested that YB1 was essential for DDX17-induced EGFR upregulation and oncogenic function.
DDX17 partially mediates the effect of MTDH on HCC progression via the EGFR-pERK pathway
After showing that DDX17 acted as a downstream effector of MTDH, we assessed whether MTDH-induced tumor progression was mediated by increased DDX17 expression, by overexpressing DDX17 in HLF and HCCLM3 cells with a knockdown of endogenous MTDH (designated as HLF-shMTDH/DDX17 and HCCLM3-shMTDH/DDX17, respectively). We observed that MTDH significantly inhibited the proliferation, migration and invasion of HLF and HCCLM3 cells, and this effect was restored by DDX17 overexpression (Fig. 7A–C). Moreover, an in vivo metastasis assay revealed that overexpression of DDX17 in HLF-shMTDH cells significantly promoted ectopic lung metastasis compared to the vector control in HLF-shMTDH cells (Fig. 7D). Furthermore, immunoblotting analysis revealed that DDX17 overexpression rescued the inhibition of the EGFR-pERK signaling pathway due to MTDH knockdown (Fig. 7E). These data indicates that DDX17 at least partially mediates the cancer-promoting effect of MTDH via the EGFR-pERK signaling pathway.

A MTDH-deficient HLF and HCCLM3 cells were transfected with a vector expressing DDX17 or an empty control. The indicated cells were subjected to the CCK8 assay. B MTDH-deficient HLF and HCCLM3 cells were transfected with a vector expressing DDX17 or an empty control. The indicated cells were subjected to a colony formation assay. Representative images are shown in the left panel and statistical comparisons of the indicated groups are shown in the right panel. C MTDH-deficient HLF and HCCLM3 cells were transfected with a vector expressing DDX17 or an empty control. The indicated cells were subjected to the transwell migration and invasion assay. Representative images are shown in the left panel and statistical comparisons of the indicated groups are shown in the right panel. D MTDH-deficient HLF cells were transfected with a vector expressing DDX17 or an empty control and injected into nude mice for the ectopic lung metastasis assay. Representative images were showed in the left panel, HE staining of the liver tissues was showed in the middle panel, and statistical comparisons of the lung tumor number in the indicated groups were showed in the right panel. E MTDH-deficient HLF and HCCLM3 cells were transfected with a vector expressing DDX17 or an empty control. Immunoblotting analysis for the indicated proteins was performed. F Knockdown DDX17 sensitized HCC cells to sorafenib treatment. Representative images of subcutaneous tumors in HLF-shNC and HLF-shDDX17 cells treated with or without sorafenib. G Statistical comparisons of tumor weigh and tumor volume from indicated groups were shown. The data represent the means ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
EGFR signaling pathway is critical to the development and progression of HCC and Targeting EGFR is able to overcome sorafenib resistance [28]. Hence, we investigated whether targeting DDX17 may enhance the inhibitory effect of sorafenib in HCC. We inoculated the shNC and shDDX17 HLF in the right flank of nude mice, respectively. When tumors became palpable at day 14, mice were treated with or without sorafenib for another 20 days. Results showed combined sorafenib treatment and DDX17 knockdown inhibited tumor growth by about 75% in terms of tumor volume and tumor weigh compared to sorafenib treatment alone (Fig. 7F, G). These findings suggest that targeting DDX17 sensitizes HCC cells to sorafenib treatment possibly by down-regulating EGFR expression.
The above results showed that both YB1 and DDX17 regulated EGFR expression in HCC cells. Then we wanted to study whether targeting both YB1 and DDX17 further enhanced the sensitivity of HCC cells to sorafenib. HLF cells with shDDX17, shYB1 and a double knock down of YB1/DDx17 were inoculated in the right flank of nude mice. Then the mice were treated with sorafenib for 20 days. We found that the tumors with shYB1 and a double-knockdown of YB1/DDx17 were markedly reduced by sorafenib treatment compare to shDDX17 group (Supplementary Fig. 10A–C). Knockdown of YB1 and double-knockdown of YB1 and DDX17 had almost no significant difference in the downregulation of EGFR (Supplementary Fig. 10D).
This showed that the regulation of EGFR by DDX17 almost no longer existed in the absence of YB1. Moreover, the regulation of EGFR by YB1 was even partly due to the fact that it also downregulated the expression of DDX17 (Supplementary Fig. 10D).
DDX17 is upregulated and positively correlate with MTDH in HCC
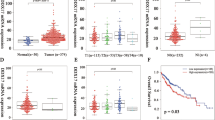
Finally, we measured the expression of MTDH and DDX17 in a cohort of 122 paired HCC tissues and adjacent non-tumor tissues (cohort II). IHC analysis showed that MTDH and DDX17 were both upregulated in HCC tissues compared to adjacent non-tumor tissues (p < 0.001) (Fig. 8A and Supplementary Fig. 11A). Additionally, immunoblotting analysis was performed to measure DDX17 expression in a cohort of 92 paired HCC tissues and adjacent non-tumor tissues (cohort I). The results showed that DDX17 expression was much higher in HCC tissues than adjacent non-tumor tissues (Fig. 8B and Supplementary Fig. 6D). TCGA analysis also revealed that MTDH and DDX17 were high expression in HCC tissues compared to normal liver tissues (Supplementary Fig. 11B, C). In addition, MTDH expression was positively correlated with DDX17 expression in HCC tissues (Fig. 8C, D).

A DDX17 expression in tumors and adjacent normal tissues of HCC patients as detected by immunohistochemistry in cohort II. Scale bar, 300 μm (upper panel), 50 μm (lower panel). Statistical comparisons of IHC scores in the indicated groups were showed in the right panel. B DDX17 expression in tumors and adjacent normal tissues of HCC patients was detected by immunoblotting in cohort I. C Representative images of IHC staining for MTDH and DDX17 in HCC tissues. Scale bar, 300 μm (upper panel), 30 μm (lower panel). D Correlations of IHC data for high or low expression of MTDH relative to the level of DDX17. E Kaplan-Meier analyses of HCC patients with tumors with high or low expression levels of MTDH or DDX17. F A schematic diagram illustrating the proposed molecular mechanisms through which the MTDH-DDX17-EGFR axis promotes the initiation and progression of HCC. The data represent the means ± SEM from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
To further investigate the clinicopathological correlates and prognostic value of DDX17 in HCC, we performed a tissue microarray-based IHC study of DDX17 in 122 HCC tissue samples with comparable clinicopathological features and follow-up data (12 of these patients were lost in follow-up). These patients were categorized according to their immunostaining scores into high (score 2–3) or low (score 0–1) DDX17 expression groups (Supplementary Fig. 11D). High expression of DDX17 was closely associated with vascular invasion (p = 0.008), TNM stage (p = 0.039) and BCLC stage (p = 0.030) (Supplementary Table 4). Additionally, our results revealed that elevated expression of MTDH or DDX17 in clinical HCC specimens was correlated with poorer OS and disease-free survival (DFS) (Fig. 8E, cohort II). Additionally, multivariate Cox regression analysis revealed that DDX17 was an independent risk factor for OS (Hazard ratio [HR] = 2.675, 95% Confidence interval [Cl], 1.230–5.815; p = 0.013) and DFS (HR = 1.917, 95% Cl, 1.091–3.368, p = 0.024) in HCC patients (Supplementary Table 5). These findings indicated that increased expression of DDX17 was significantly associated with a poor prognosis in HCC patients.
Discussion
In the present study, we identified DDX17 as a novel partner of MTDH in HCC. The helicase C-terminal domain (aa: 405-553) of DDX17 was mainly responsible for the binding to MTDH. Furthermore, MTDH increased the protein levels of DDX17 by inhibiting its ubiquitination. Subsequently, the enhanced expression of DDX17 resulted in its binding to YB1 via the C-terminal region of DDX17 (aa: 553-729), which in turn drove the binding of YB1 to the EGFR gene promoter to increase its transcription (Fig. 8F).
Accumulated evidence from functional studies has established MTDH as a key regulator of tumor proliferation [29], metastasis [12], angiogenesis [30], EMT [13], and anoikis resistance [15] in various cancers. It is convincing that MTDH may regulate its downstream genes by interacting with diverse binding partners. For example, MTDH was found to interact with p65 in the nucleus to facilitate NF-κB-mediated transcription [19]. In the cytoplasm, MTDH associates with CBP and SND1 to increase their protein levels [8, 31]. In the present study, we identified DDX17 as a novel binding partner of MTDH in HCC. Moreover, MTDH could increase the protein levels of DDX17 in the nucleus by preventing its polyubiquitination and degradation. The prevention of ubiquitin-mediated degradation of DDX17 by MTDH may also involve an E3 ubiquitin ligase, which needs to be further investigated in the future.
DDX17 acts as a transcriptional co-regulator which either promotes or suppresses gene transcription by binding to certain TFs. Thus, we conducted mRNA sequencing to identify the possible downstream effectors of DDX17 in HCC. KEGG enrichment analysis of the mRNA sequencing results indicated that DDX17 was involved in multiple pathways related to cancer progression and metastasis, including the KEGG categories pathways in cancer, focal adhesion, MAPK signaling pathway, and regulation of actin cytoskeleton, in both HLF and HCCLM3 cells. EGFR, which is involved in all these four signaling pathways, was found to be regulated by DDX17 in HCC cells. EGFR is a member of the ErbB family of receptor tyrosine kinases (RTKs) and is overexpressed in different types of human cancers, including HCC [32]. EGFR dysregulation can activate multiple signaling pathways, including Ras-Raf-MEK-ERK, PI3K-AKT, STAT and Src [32, 33]. The aberrant activation of EGFR signaling promotes cell proliferation, apoptosis resistance, metastasis, angiogenesis, drug resistance and tumorigenesis in multiple cancers [34, 35]. In the present study, we confirmed that DDX17 upregulated the mRNA and protein expression of EGFR. Moreover, we confirmed that DDX17 expression was positively correlated with EGFR expression in clinical HCC tissue samples. Furthermore, as a downstream effector of EGFR, pERK was activated by DDX17 in this context, while pAKT was not. This suggested that the EGFR-pERK axis possibly mediated the effect of DDX17 in HCC, but not the EGFR-pAKT pathway.
There is emerging evidence that DDX17/DDX5 acts as a transcriptional regulator that can mediate either the activation or suppression of gene expression [22, 36]. DDX17, together with its ortholog DDX5, was found to interact with ERα, β-catenin, and MyoD to enhance their transcriptional activity [37]. However, DDX17 also played an important role in transcription regulation independent of DDX5. Lambert et al. revealed that DDX17 enhanced REST-mediated transcriptional suppression by interacting with it [38]. Thus, we proposed that DDX17 possibly regulated EGFR expression through the same mechanism. In the study, we identified three TFs by screening the interaction partners of DDX17. Among them, YB1 came to our attention because it was known to regulate the transcription of EGFR as a TF. YB1 was a well-established oncogene which regulated various genetic processes, including transcription, pre-mRNA splicing and mRNA translation [39]. Since YB1 was able to bind to four putative YB-1-responsive elements (YRE) in the EGFR promoter to enhance its mRNA expression [26], we proposed that DDX17 may up-regulated the expression of EGFR by binding to YB1. Our results demonstrated that DDX17 could be recruited to the EGFR promoter to induce its transcription by binding to YB1. Sook Shin et al. showed that DDX17 increased the transcriptional activity of β-catenin by interacting with a domain spanning approximately 200 amino acids at its C-terminal domain [23]. Correspondingly, we also found that DDX17 associated with YB1 via its C-terminal domain in our context. These evidences suggest that the C-terminal domain of DDX17 may be essential for regulating the transcriptional activity of TFs. Notably, YB1 can both promote and inhibit transcription, upregulating a subset of genes including MET, MDR1, MVP, PDGFB, cyclin A2, and smad7, while downregulating many targeted genes such as P21, P53, MMP12, MMP13 and VEGF [27]. In our study, we found that DDX17 also promoted the transcription of MET, MDR1 and CCNA2 in addition to EGFR. This suggested that DDX17 may be recruited to the promoters of multiple YB1-targeted genes to regulate their expression. Despite this, the specific mechanism through which DDX17 regulates the transcriptional activity of YB1 remains to be elucidated. A previous study reported that DDX17 deficiency may inhibit the binding of REST to the promoters of its targeted genes [38], suggesting that a similar mechanism might exist in our study. However, we could not verify this conclusion due to a lack of appropriate YB1 CHIP antibodies. Taken together, these results suggest that DDX17 interacts with YB1 and recruits it to the promoter on its target genes to enhance their transcription.
EGFR is highly expressed in 60-80% of human HCC cases, while EGFR mutations appear to be rare [40]. Sorafenib was a first-line systemic treatment for advanced HCC, while EGFR overexpression contributed to drug resistance of EGFR-positive cells to sorafenib [41, 42]. Our results revealed that DDX17 knockdown markedly reduced EGFR expression in HCC cells. Furthermore, DDX17 was positively correlated with EGFR expression in HCC tissues. Notably, DDX17 enhanced the EGF-induced EGFR signaling pathway, suggesting that DDX17 strengthened EGFR signaling in HCC, making tumor resistant to sorafenib treatment. In addition, we found that DDX17 also downregulated the expression of MDR1, which was strongly related with multidrug resistance. Thus, targeting DDX17 may abrogate the DDX17-induced resistance to sorafenib or other chemotherapeutic drugs.
In conclusion, our results demonstrate that MTDH interacts with and stabilizes DDX17 by inhibiting its ubiquitination. We identify DDX17 as a powerful oncogene using in vivo and in vitro experiments. DDX17 interacts with YB1, which in turn binds to the EGFR-promoter to activate its transcription and downstream EGFR-pERK signaling pathway in HCC. Our findings suggest that the DDX17/YB1/EGFR axis contributes to HCC metastasis and could be a potential therapeutic target for the treatment of HCC.
Materials and methods
Available in supplementary information
Data availability
The datasets used and analysed during the current study are available from the corresponding author upon reasonable request.
References
Craig AJ, von Felden J, Garcia-Lezana T, Sarcognato S, Villanueva A. Tumour evolution in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2020;17:139–52.
Chen S, Cao Q, Wen W, Wang H. Targeted therapy for hepatocellular carcinoma: Challenges and opportunities. Cancer Lett. 2019;460:1–9.
Su ZZ, Kang DC, Chen Y, Pekarskaya O, Chao W, Volsky DJ, et al. Identification and cloning of human astrocyte genes displaying elevated expression after infection with HIV-1 or exposure to HIV-1 envelope glycoprotein by rapid subtraction hybridization, RaSH. Oncogene. 2002;21:3592–602.
Zhu HD, Liao JZ, He XX, Li PY. The emerging role of astrocyte-elevated gene-1 in hepatocellular carcinoma (Review). Oncol Rep. 2015;34:539–46.
Robertson CL, Srivastava J, Siddiq A, Gredler R, Emdad L, Rajasekaran D, et al. Genetic deletion of AEG-1 prevents hepatocarcinogenesis. Cancer Res. 2014;74:6184–93.
Yoo BK, Emdad L, Su ZZ, Villanueva A, Chiang DY, Mukhopadhyay ND, et al. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J Clin Investig. 2009;119:465–77.
Hu G, Chong RA, Yang Q, Wei Y, Blanco MA, Li F, et al. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell. 2009;15:9–20.
Wan L, Lu X, Yuan S, Wei Y, Guo F, Shen M, et al. MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer Cell. 2014;26:92–105.
Li G, Wang Z, Ye J, Zhang X, Wu H, Peng J, et al. Uncontrolled inflammation induced by AEG-1 promotes gastric cancer and poor prognosis. Cancer Res. 2014;74:5541–52.
Wang Z, Wei YB, Gao YL, Yan B, Yang JR, Guo Q. Metadherin in prostate, bladder, and kidney cancer: A systematic review. Mol Clin Oncol. 2014;2:1139–44.
Yao Y, Gu X, Liu H, Wu G, Yuan D, Yang X, et al. Metadherin regulates proliferation and metastasis via actin cytoskeletal remodelling in non-small cell lung cancer. Br J Cancer. 2014;111:355–64.
Brown DM, Ruoslahti E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell. 2004;5:365–74.
Zhu K, Dai Z, Pan Q, Wang Z, Yang GH, Yu L, et al. Metadherin promotes hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin Cancer Res: Off J Am Assoc Cancer Res. 2011;17:7294–302.
Li WF, Ou Q, Dai H, Liu CA. Lentiviral-mediated short hairpin RNA knockdown of MTDH inhibits cell growth and induces apoptosis by regulating the PTEN/AKT pathway in hepatocellular carcinoma. Int J Mol Sci. 2015;16:19419–32.
Zhu HD, Liu L, Deng H, Li ZB, Sheng JQ, He XX, et al. Astrocyte elevated gene 1 (AEG-1) promotes anoikis resistance and metastasis by inducing autophagy in hepatocellular carcinoma. J Cell Physiol. 2020;235:5084–95.
Manna D, Sarkar D. Multifunctional role of Astrocyte Elevated Gene-1 (AEG-1) in Cancer: Focus on drug resistance. Cancers. 2021;13:1792.
Zhu K, Peng Y, Hu J, Zhan H, Yang L, Gao Q, et al. Metadherin-PRMT5 complex enhances the metastasis of hepatocellular carcinoma through the WNT-β-catenin signaling pathway. Carcinogenesis. 2020;41:130–8.
Srivastava J, Siddiq A, Gredler R, Shen XN, Rajasekaran D, Robertson CL, et al. Astrocyte elevated gene-1 and c-Myc cooperate to promote hepatocarcinogenesis in mice. Hepatology. 2015;61:915–29.
Sarkar D, Park ES, Emdad L, Lee SG, Su ZZ, Fisher PB. Molecular basis of nuclear factor-kappaB activation by astrocyte elevated gene-1. Cancer Res. 2008;68:1478–84.
He W, He S, Wang Z, Shen H, Fang W, Zhang Y, et al. Astrocyte elevated gene-1(AEG-1) induces epithelial-mesenchymal transition in lung cancer through activating Wnt/β-catenin signaling. BMC Cancer. 2015;15:107.
Fuller-Pace FV. The DEAD box proteins DDX5 (p68) and DDX17 (p72): multi-tasking transcriptional regulators. Biochimica et Biophysica acta. 2013;1829:756–63.
Xing Z, Ma WK, Tran EJ. The DDX5/Dbp2 subfamily of DEAD-box RNA helicases. Wiley Interdiscip Rev RNA. 2019;10:e1519.
Shin S, Rossow KL, Grande JP, Janknecht R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res. 2007;67:7572–8.
Wortham NC, Ahamed E, Nicol SM, Thomas RS, Periyasamy M, Jiang J, et al. The DEAD-box protein p72 regulates ERalpha-/oestrogen-dependent transcription and cell growth, and is associated with improved survival in ERalpha-positive breast cancer. Oncogene. 2009;28:4053–64.
Xue Y, Jia X, Li C, Zhang K, Li L, Wu J, et al. DDX17 promotes hepatocellular carcinoma progression via inhibiting Klf4 transcriptional activity. Cell death Dis. 2019;10:814.
Wu J, Lee C, Yokom D, Jiang H, Cheang MC, Yorida E, et al. Disruption of the Y-box binding protein-1 results in suppression of the epidermal growth factor receptor and HER-2. Cancer Res. 2006;66:4872–9.
Eliseeva IA, Kim ER, Guryanov SG, Ovchinnikov LP, Lyabin DN. Y-box-binding protein 1 (YB-1) and its functions. Biochem Biokhimiia. 2011;76:1402–33.
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 2020;5:87.
Li J, Yang L, Song L, Xiong H, Wang L, Yan X, et al. Astrocyte elevated gene-1 is a proliferation promoter in breast cancer via suppressing transcriptional factor FOXO1. Oncogene. 2009;28:3188–96.
Emdad L, Lee SG, Su ZZ, Jeon HY, Boukerche H, Sarkar D, et al. Astrocyte elevated gene-1 (AEG-1) functions as an oncogene and regulates angiogenesis. Proc Natl Acad Sci USA. 2009;106:21300–5.
Liang Y, Hu J, Li J, Liu Y, Yu J, Zhuang X, et al. Epigenetic activation of TWIST1 by MTDH promotes cancer stem-like cell traits in breast cancer. Cancer Res. 2015;75:3672–80.
Berasain C, Avila MA. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol. 2014;49:9–23.
Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12:3–20.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Uribe ML, Marrocco I, Yarden Y. EGFR in cancer: signaling mechanisms, drugs, and acquired resistance. Cancers. 2021;13:2748.
Fuller-Pace FV, Moore HC. RNA helicases p68 and p72: multifunctional proteins with important implications for cancer development. Future Oncol. 2011;7:239–51.
Giraud G, Terrone S, Bourgeois CF. Functions of DEAD box RNA helicases DDX5 and DDX17 in chromatin organization and transcriptional regulation. BMB Rep. 2018;51:613–22.
Lambert MP, Terrone S, Giraud G, Benoit-Pilven C, Cluet D, Combaret V, et al. The RNA helicase DDX17 controls the transcriptional activity of REST and the expression of proneural microRNAs in neuronal differentiation. Nucleic Acids Res. 2018;46:7686–7700.
Lyabin DN, Eliseeva IA, Ovchinnikov LP. YB-1 protein: functions and regulation. Wiley Interdiscip Rev RNA. 2014;5:95–110.
Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989–5005.
Ezzoukhry Z, Louandre C, Trécherel E, Godin C, Chauffert B, Dupont S, et al. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int J cancer. 2012;131:2961–9.
Niu L, Liu L, Yang S, Ren J, Lai PBS, Chen GG. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochimica et biophysica acta Rev cancer. 2017;1868:564–70.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81874189 to Bi-xiang Zhang, No. 82003003 to Jin Chen).
Author information
Authors and Affiliations
Contributions
CX, ZB, and CY conceived and designed the study. CJ, DH, and LQ performed the experiments. ND, DP, and MJ collected the clinical specimens and data. GQ, XL, and ZX performed the statistical analysis. CJ drafted the manuscript. LH, ZB, and CX contributed to the critical revision of the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jin, C., Han-hua, D., Qiu-meng, L. et al. MTDH-stabilized DDX17 promotes tumor initiation and progression through interacting with YB1 to induce EGFR transcription in Hepatocellular Carcinoma. Oncogene 42, 169–183 (2023). https://doi.org/10.1038/s41388-022-02545-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-022-02545-x
- Springer Nature Limited