
Abstract
Uncontrolled proliferation of intestinal epithelial cells caused by mutations in genes of the WNT/β-catenin pathway is associated with development of intestinal cancers. We previously reported that intestinal stromal cell-derived angiopoietin-like protein 2 (ANGPTL2) controls epithelial regeneration and intestinal immune responses. However, the role of tumor cell-derived ANGPTL2 in intestinal tumorigenesis remained unclear. Here, we show that tumor cell-derived ANGPTL2 promotes β-catenin-driven intestinal tumorigenesis. ANGPTL2 deficiency suppressed intestinal tumor development in an experimental mouse model of sporadic colon cancer. We also found that increased ANGPTL2 expression in colorectal cancer (CRC) cells augments β-catenin pathway signaling and promotes tumor cell proliferation. Relevant to mechanism, our findings suggest that tumor cell-derived ANGPTL2 upregulates expression of OB-cadherin, which then interacts with β-catenin, blocking destruction complex-independent proteasomal degradation of β-catenin proteins. Moreover, our observations support a model whereby ANGPTL2-induced OB-cadherin expression in CRC cells is accompanied by decreased cell surface integrin α5β1 expression. These findings overall provide novel insight into mechanisms of β-catenin-driven intestinal tumorigenesis.
Similar content being viewed by others


Introduction
Most sporadic human colorectal cancers (CRCs) arise as a consequence of mutations in components of the WNT/β-catenin signaling pathway and in the absence of overt inflammation. In the absence of WNT signaling, β-catenin is phosphorylated by the β-catenin destruction complex, which is composed of adenomatous polyposis coli (APC), glycogen synthase kinase 3 (GSK3), AXIN, and casein kinase 1α (CK1α), and undergoes proteasomal degradation [1]. However, dysregulation of WNT/β-catenin signaling due to alterations of the complex, such as mutations in APC or CTNNB1, which encodes β-catenin, leads to tumorigenesis [2, 3]. On the other hand, the β-catenin pathway is reportedly regulated by numerous stimuli, such as growth factors, extracellular matrix–integrin interaction, mechanical force, or changes in the adherens junction, in a manner independent of the destruction complex [4]. Those regulatory activities are, however, less understood.
Previously, we and others identified a family of secretory proteins structurally similar to angiopoietin and designated them angiopoietin-like proteins (ANGPTLs) [5, 6]. We later reported that one of those proteins, ANGPTL2, contributes to maintenance of tissue homeostasis [7]. Recently, we showed that intestinal stromal cell-derived ANGPTL2 maintains the stem cell niche to regulates epithelial regeneration and homeostasis [8]. On the other hand, excess ANGPTL2 signaling leads to chronic inflammation and irreversible tissue remodeling, promoting development and progression of various diseases, including some cancers [7, 9]. Furthermore, we showed that, in an autocrine manner, tumor cell-derived ANGPTL2 accelerates tumor cell migration and invasion in lung cancer, breast cancer, and osteosarcoma [10, 11]. These findings suggest that cell-autonomous ANGPTL2 signaling in tumor cells functions in tumor metastasis. However, it remains unclear whether tumor cell-derived ANGPTL2 is associated with the regulation of WNT/β-catenin signaling pathway and tumor cell proliferation.
Here, we report that tumor cell-derived ANGPTL2 contributes to β-catenin-driven intestinal tumorigenesis by suppressing destruction complex-independent degradation of β-catenin proteins in tumor cells. These findings provide novel insight into mechanisms responsible for intestinal tumorigenesis caused by dysfunction of β-catenin destruction complex.
Results
Human and mouse CRC cells show upregulated ANGPTL2 expression
To assess ANGPTL2 expression in CRC tumors, we examined ANGPTL2 expression in paraffin-embedded tumor samples and normal colonic mucosa from biopsies of patients with CRC. ANGPTL2 was undetectable in normal colonic epithelium but expressed in some cancer cells and stromal cells of primary CRC tumors (Fig. 1a). Furthermore, analysis of the Gene Expression database of Normal and Tumor tissues (GENT) showed that expression levels of ANGPTL2 mRNA were significantly increased in human CRC samples compared to nontumorous colon tissues (Fig. 1b).

a Representative images of ANGPTL2 immunostaining in surgical specimens of primary tumors from patients with colon cancer. Scale bar, 100 μm. b Comparison of ANGPTL2 transcript levels in normal (n = 397) and tumor (n = 3775) colon tissues based on the GENT database. Data are means ± SD. ***P < 0.001, Mann-Whitney test. Representative images of H&E staining (c) and immunohistochemical staining for β-catenin (d) and ANGPTL2 (e) in tumor and nontumor tissues from WT mice at week 24 of the AOM model. Angptl2−/− panels at right serve as a negative control for anti-ANGPTL2 antibody. Scale bar, 200 μm (top); 50 μm (middle and bottom).
To searched for the in vivo source of ANGPTL2 in mice CRC tumors, we took advantage of a sporadic colorectal tumorigenesis model, in which mice are repeatedly injected with the potent mutagen azoxymethane (AOM) to induce colon cancer. In this model, AOM induces several mutations, including in Ctnnb1, leading to accumulation of β-catenin protein [12]. Immunohistochemical analysis of β-catenin in normal colon epithelial cells revealed it was localized primarily at cell membrane (Fig. 1c, d). However, in tumor cells, β-catenin proteins accumulated in the cytoplasm and nuclei (Fig. 1c, d). Moreover, immunohistochemical analysis of ANGPTL2 in wild-type (WT) mice revealed that colonic stromal cells, but not colonic epithelial cells, abundantly expressed ANGPTL2 in nontumorous regions (Fig. 1e). We also observed upregulated ANGPTL2 expression in tumor cells (Fig. 1e). Furthermore, we examined Angptl2 mRNA expression in epithelial and stromal cells from normal and tumor tissues of WT mice. Based on qRT-PCR analysis, Angptl2 transcripts were nearly undetectable in normal intestinal epithelial cells (IECs) purified from normal colon, while tumor cells from WT mice of the AOM model abundantly expressed Angptl2 mRNA (Supplementary Fig. 1a). By contrast, qRT-PCR analysis revealed comparable levels of Angptl2 transcripts in normal intestinal subepithelial myofibroblasts (ISEMFs) from WT mice and tumor-associated fibroblasts (TAFs) from WT mice of the AOM model (Supplementary Fig. 1a). Taken together, these results indicate that ANGPTL2 expression is upregulated in both mouse and human intestinal tumor cells.
Angptl2-deficient mice show decreased β-catenin-dependent tumorigenesis in a colon cancer model
Interestingly, we did not observe robust β-catenin accumulation in dysplastic crypts that showed high expression of ANGPTL2 protein (Supplementary Fig 1b). Accordingly, we hypothesized that ANGPTL2 expression is induced prior to β-catenin accumulation in intestinal tumor cells and that ANGPTL2 contributes to β-catenin-dependent intestinal tumorigenesis. To investigate that possibility, we subjected both Angptl2-deficient (Angptl2−/−) mice, which show normal intestinal development in the absence of injury-inducing stimuli [8], and WT mice to the AOM model and assessed tumor development at 16 and 24 weeks after initial AOM injection (Fig. 2a and Supplementary Fig. 2a). Angptl2−/− mice showed a significantly lower incidence of colonic tumors (Fig. 2b–d and Supplementary Fig. 2b) and fewer tumors in colon relative to WT mice (Fig. 2e and Supplementary Fig. 2c). Moreover, Angptl2−/− mice showed a significant decrease in the size of tumors in the colon relative to WT mice at week 16 of the AOM model (Supplementary Fig. 2d). By week 24, average tumor size was comparable between genotypes (Fig. 2f), but Angptl2−/− mice exhibited a lower frequency of large tumors (≥3.5 mm) than did WT mice (Fig. 2g). Histopathologic analysis confirmed that the number of tumorous lesions in the colon of Angptl2−/− mice decreased compared with those seen in WT mice (Fig. 2h). Moreover, Angptl2−/− mice exhibited prolonged survival relative to WT mice (Fig. 2i). These results overall suggest that ANGPTL2 promotes development of colorectal cancers in this context.

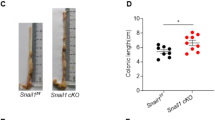
a Schematic illustrating experimental design of the AOM model. i.p., intraperitoneal injection. b Representative endoscopy images of distal colonic tumors (arrows) at week 24 of the AOM model. c Gross appearance of colon of WT and Angptl2−/− mice at week 24 of the AOM model. Arrows indicate a tumor. Scale bar, 1 cm. d Tumor incidence in colon of WT (n = 19) and Angptl2−/− (n = 16) mice at week 24 of the AOM model. ***P < 0.001, Fisher’s exact test. e Comparison of tumor number in WT (n = 19) and Angptl2−/− (n = 16) mice at week 24 of the AOM model. Data are means ± SD. ***P < 0.001, Mann-Whitney test. Maximum tumor diameter (f) and number of large tumors (g) in WT (n = 41 tumors from 19 mice) and Angptl2−/− (n = 4 tumors from 16 mice) mice at week 24 of the AOM model. Data are means ± SD. ns, not significant (P > 0.05); **P < 0.01, unpaired t-test (f) or Mann-Whitney test (g). h H&E-stained images of colon tissues from representative WT or Angptl2−/− mice at week 24 of the AOM model. Arrowheads indicate tumors. p, proximal; d, distal. Scale bar, 2 mm. i Kaplan-Meier survival curves of WT (n = 25) and Angptl2−/− (n = 17) mice of the AOM model. *P < 0.05, log-rank test. j (left) Representative immunoblotting of β-catenin in tumor tissues from WT (n = 8) or Angptl2−/− (n = 3) mice at week 24 of the AOM model. HSC70 served as a loading control. The asterisk indicates a non-specific band. (right) Quantification of normalized β-catenin expression. Data are means ± SD. **P < 0.01, unpaired t-test. k, l qRT-PCR analysis of transcripts of indicated genes in colonic tumors from WT (n = 8) and Angptl2−/− (n = 3) mice. WT levels were set to 1. Data are means ± SD. ns, not significant (P > 0.05); *P < 0.05, unpaired t-test or Mann-Whitney test.
Next, we evaluated β-catenin protein accumulation and observed reduced β-catenin levels in colonic tumors from Angptl2−/− compared to WT mice (Fig. 2j). Tumors from Angptl2−/− mice also showed significantly lower transcript levels of β-catenin target genes, such as Ccnd1 and Ccnd2, and modest but not significant decreases in Myc and Axin2 relative to WT mice (Fig. 2k). We also observed significantly lower Mki67 expression in tumors from Angptl2−/− relative to WT mice (Fig. 2l). Overall, these results suggest that in this mouse model, ANGPTL2 contributes to development of colorectal tumorigenesis by regulating the β-catenin pathway.
ANGPTL2 deficiency decreases tumor growth and β-catenin signaling in CRC cells
To assess ANGPTL2 function in CRC progression, we deleted ANGPTL2 in the human colorectal cancer line SW480 using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 genome-editing technology and verified lack of ANGPTL2 protein expression in multiple independent ANGPTL2 KO clones (ANGPTL2 KO SW480) (Fig. 3a, b). ANGPTL2 KO SW480 tumor cells showed significantly decreased clonogenic capacity relative to control SW480 cells (Fig. 3c). We then used a xenograft model to investigate whether ANGPTL2 contributes to tumor progression in vivo. To establish the model, we injected ANGPTL2 KO or control SW480 cells into the back flanks of nude mice and monitored volume of resultant tumors over the next 4 weeks. Based on that analysis, mice injected with ANGPTL2 KO SW480 cells showed significantly decreased tumor growth relative to mice injected with control cells (Fig. 3d). Interestingly, immunoblot analysis revealed decreased β-catenin protein levels in ANGPTL2 KO relative to control SW480 cells (Fig. 3e). Accordingly, immunoblot and qRT-PCR analysis revealed that respective protein and transcript levels of β-catenin target genes, such as c-Myc (encoded by MYC), Cyclin D2 (encoded by CCND2), and Axin2 (encoded by AXIN2), significantly decreased ANGPTL2 KO relative to control SW480 cells (Fig. 3e, f). These results support the idea that tumor cell-derived ANGPTL2 promotes β-catenin protein accumulation in CRC cells, accelerating their proliferation.

a Representative immunoblotting for ANGPTL2 in control SW480 cells and in three lines (#1, #2, and #3) of ANGPTL2 KO SW480 cells. HSC70 served as a loading control. b Representative immunofluorescent staining for ANGPTL2 (red) in ANGPTL2 KO SW480 lines (#1, #2, and #3) and control cells. Nuclei are counterstained with DAPI (blue). c (left) Clonogenic analysis of indicated groups at day 12 based on crystal violet staining. (right) Quantification of data. Shown are means ± SD; n = 4 for each group. ***P < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test. d Comparison of tumor volumes in nude BALB/c mice injected with either ANGPTL2 KO SW480 line #1 or control cells. Data are means ± SD; n = 10 tumors per group. ***P < 0.001, two-way ANOVA test. e (left) Representative immunoblotting of ANGPTL2 KO SW480 lines (#1, #2, and #3) or control cells. HSC70 served as a loading control. Numbers below panels indicate normalized protein expression. (right) Quantification of data. Control cell levels were set to 1. Shown are means ± SD; n = 3 for each group. ***P < 0.001; **P < 0.01; *P < 0.05, one-way ANOVA test followed by Tukey’s multiple comparison test. f Comparison of MYC, CCND2, and AXIN2 transcript levels in ANGPTL2 KO lines (#1, #2, and #3) or control SW480 cells. Control cell levels were set to 1. Data are means ± SD; n = 4 for each group. ***P < 0.001; **P < 0.01, one-way ANOVA test followed by Tukey’s multiple comparison test.
ANGPTL2 expressed in CRC tumor cells enhances their growth and β-catenin signaling
Given the increased ANGPTL2 expression seen in colonic tumor cells in the AOM model (Fig. 1), we asked whether ANGPTL2 induction in those cells contributes to β-catenin-driven intestinal tumorigenesis. To do so, we established two independent cell lines overexpressing (OE) ANGPTL2 in the human colorectal cancer cell line SW480 (ANGPTL2-OE SW480) (Fig. 4a). ELISA analyses confirmed that relative to control cells, ANGPTL2-OE SW480 cells showed enhanced ANGPTL2 protein secretion into culture medium (Fig. 4b). ANGPTL2 overexpression significantly increased clonogenic capacity of tumor cells of both lines relative to cells transduced with control vector (Fig. 4c), suggesting that tumor cell-derived ANGPTL2 promotes CRC cell proliferation. Furthermore, mice subcutaneously injected with ANGPTL2-OE SW480 cells showed a significant increase in tumor volume relative to mice injected with SW480 control cells (Fig. 4d). Interestingly, immunoblot analysis revealed that β-catenin protein levels increased in ANGPTL2-OE relative to control SW480 cells (Fig. 4e). ANGPTL2-OE SW480 cells also showed increased levels of β-catenin protein in both cytosolic and nuclear fractions relative to control cells (Fig. 4f). Accordingly, qRT-PCR analysis revealed that transcript levels of β-catenin target genes, such as MYC, CCND2, and AXIN2, significantly increased in ANGPTL2-OE relative to control SW480 cells (Fig. 4g).

a Representative immunoblotting for ANGPTL2 in two lines (#1 and #2) of ANGPTL2-OE SW480 and control cells. HSC70 served as a loading control. b ANGPTL2 protein concentrations in culture medium of ANGPTL2-OE SW480 lines #1 and #2 or control cells. Data are means ± SD; n = 4 for each group. ***P < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test. c (left) Clonogenic analysis of indicated groups at day 12 based on crystal violet staining. (right) Quantification of data. Shown are means ± SD; n = 4 for each group. ***P < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test. d Comparison of tumor volumes in nude BALB/c mice injected with either the ANGPTL2-OE SW480 line #1 (n = 10) or control SW480 cells (n = 8). Data are means ± SD. ***P < 0.001, two-way ANOVA test. e Representative immunoblotting for β-catenin in ANGPTL2-OE SW480 lines #1 and #2 and control cells. HSC70 served as a loading control. Numbers below panels represent normalized expression of proteins. f (left) Representative immunoblotting for β-catenin in ANGPTL2-OE SW480 lines #1 and #2 and control cells. HDAC2 served as a loading control for the nuclear fraction. GAPDH served as a loading control for the cytosolic fraction. (right) Shown are relative β-catenin/GAPDH (cytosolic) and β-catenin/HDAC2 (nuclear) levels. Data are means ± SD; n = 3 for each group. ***P < 0.001; *P < 0.05, one-way ANOVA test followed by Tukey’s multiple comparison test. g Comparison of MYC, CCND2, and AXIN2 transcript levels in ANGPTL2-OE SW480 lines #1 and #2 and control cells. Control cell levels were set to 1. Data are means ± SD; n = 4 for each group. ***P < 0.001; **P < 0.01, one-way ANOVA test followed by Tukey’s multiple comparison test. h Representative immunoblotting for ANGPTL2 in ANGPTL2-OE or control HCT116 cells. HSC70 served as a loading control. i ANGPTL2 protein concentrations in the culture medium of ANGPTL2-OE HCT116 or control cells. Data are means ± SD; n = 4 for each group. *P < 0.05, Mann-Whitney test. j (left) Clonogenic analysis of indicated groups at day 10 based on crystal violet staining. (right) Quantification of data. Shown are means ± SD; n = 3 for each group. *P < 0.05, unpaired t-test. k Representative immunoblotting for β-catenin in ANGPTL2-OE or control HCT116 cells. HSC70 served as a loading control. Numbers below panels represent normalized protein expression. l Comparison of MYC, CCND2, and AXIN2 transcript levels in ANGPTL2-OE or control HCT116 cells. Control cell levels were set to 1. Data are means ± SD; n = 6 for each group. ***P < 0.001; *P < 0.05, unpaired t-test.
To further assess the relationship between ANGPTL2 expression and β-catenin accumulation in CRC cells, we overexpressed ANGPTL2 in a different human colorectal cancer line, namely, HCT116 cells (ANGPTL2-OE HCT116) (Fig. 4h, i). As in SW480 cells, clonogenic capacity of ANGPTL2-OE HCT116 cells was significantly increased relative to control cells (Fig. 4j), and ANGPTL2-OE HCT116 cells showed relatively increased levels of β-catenin protein (Fig. 4k). Moreover, qRT-PCR analysis revealed significantly increased levels of MYC, CCND2 and AXIN2 transcripts in ANGPTL2-OE relative to control cells (Fig. 4l). Overall, these results support our observations that tumor cell-derived ANGPTL2 promotes β-catenin protein accumulation in CRC cells and consequent upregulation of β-catenin target genes, accelerating tumor cell proliferation.
ANGPTL2 suppresses proteasomal degradation of β-catenin independent of the destruction complex
To determine mechanisms underlying accumulation of β-catenin protein in tumor cells overexpressing ANGPTL2, we assessed stability of endogenous β-catenin proteins in ANGPTL2-OE SW480 cells cultured in the presence of the protein synthesis inhibitor cycloheximide. Cycloheximide chase analysis revealed that in control SW480 cells, most β-catenin protein was degraded within 12 h, whereas >50% of initial β-catenin protein remained in ANGPTL2-OE SW480 cells (Fig. 5a). β-catenin protein phosphorylated at Ser33, Ser37, and Thr41 by GSK-3, a component protein of β-catenin destruction complex, is ubiquitinated and undergoes degradation [13]. Interestingly, Angptl2-deficient mice show decreased tumorigenesis in the AOM model, in which the GSK-3β phosphorylation consensus motif of β-catenin is reportedly mutated [12]. Therefore, we hypothesized that ANGPTL2 blocks β-catenin protein degradation independent of GSK-3β. SW480 cells harbor mutant APC (Fig. 5b), which leads to inactivation of the β-catenin destruction complex [14,15,16]. Polyubiquitination of phospho-β-catenin was readily detectable in HEK293 cells cultured in the presence of the proteasome inhibitor MG132, based on immunoblot analysis, while phospho-β-catenin was not ubiquitinated in SW480 cells cultured in the presence or absence of MG132 (Fig. 5b). These findings suggest that the destruction complex is inactivated in SW480 cells. Indeed, β-catenin degradation was not inhibited by LiCl or the GSK-3 inhibitor CHIR99021 in cycloheximide-treated SW480 cells (Fig. 5c, d). Nevertheless, culture of cycloheximide-treated control SW480 cells with MG132 suppressed β-catenin protein degradation (Fig. 5e). Immunoblot analysis also revealed that polyubiquitination of phospho-β-catenin was not detectable in either ANGPTL2-OE or control SW480 cells in the presence of MG132, and ratios of phosphorylated (S33/S37/T41) to total β-catenin protein were comparable in ANGPTL2-OE SW480 and control cells (Fig. 5f). These findings suggest overall that ANGPTL2 does not contribute to β-catenin phosphorylation or to ubiquitination of phospho-β-catenin. On the other hand, the increase in β-catenin protein levels seen relative to controls in ANGPTL2-OE SW480 cells was minimal in the presence of MG132 (Fig. 5g). Overall, these results suggest that β-catenin undergoes proteasomal degradation in SW480 cells independent of the GSK-3-containing destruction complex and that ANGPTL2 suppresses this activity.

a (left) Representative immunoblotting for β-catenin in ANGPTL2-OE SW480 lines #1 and #2 and control cells incubated with cycloheximide (CHX, 100 μg/ml) for indicated times. HSC70 served as a loading control. (right) Quantification of the percentage of β-catenin remaining at indicated incubation times. Data are means ± SD; n = 3 for each group. **P < 0.01; *P < 0.05, two-way ANOVA test. b Representative immunoblotting of indicated proteins in HEK293 and SW480 cells incubated with or without MG132 (20 μM) for 4 h. HSC70 served as a loading control. c Representative immunoblotting for β-catenin in control SW480 cells incubated with cycloheximide (100 μg/ml) and/or LiCl (20 mM) for 12 h. HSC70 served as a loading control. Numbers below panels represent normalized expression of proteins. d Representative immunoblotting for β-catenin in control SW480 cells incubated with cycloheximide (100 μg/ml) and/or CHIR99021 (3 μM) for 12 h. HSC70 served as a loading control. Numbers below panels represent normalized protein expression. e (left) Representative immunoblotting for β-catenin in control SW480 cells incubated with cycloheximide (100 μg/ml) in the absence or presence of the proteasome inhibitor MG132 (20 μM) for indicated times. HSC70 served as a loading control. (right) Quantification of the percentage of β-catenin remaining at indicated incubation times. Data are means ± SD; n = 3 for each group. **P < 0.01, two-way ANOVA test. f (left) Representative immunoblotting for β-catenin and phosphorylated (p)-β-catenin in ANGPTL2-OE SW480 lines #1 and #2 and control cells incubated 12 h with MG132 (20 μM). HSC70 served as a loading control. (right) Shown are relative levels of p-β-catenin/β-catenin. Data are means ± SD; n = 3 for each group. ns, not significant (P > 0.05), one-way ANOVA test followed by Tukey’s multiple comparison test. g Representative immunoblotting for β-catenin in ANGPTL2-OE SW480 lines #1 and #2 and control cells incubated with or without MG132 (20 μM) for 12 h. HSC70 served as a loading control. Numbers below panels represent normalized expression of proteins. Controls each condition (either with or without MG132) were set to 1.
Tumor cells overexpressing ANGPTL2 show OB-cadherin upregulation and β-catenin stabilization
Next to determine how ANGPTL2 suppresses destruction complex-independent β-catenin degradation in tumor cells, we searched for proteins that interact with β-catenin in colon cancer cells. To do so, we immunoprecipitated β-catenin proteins from whole cell lysates of ANGPTL2-OE SW480 or control cells and identified co-immunoprecipitated proteins by mass spectrometry (Fig. 6a, b). That analysis revealed that Cadherin-11 (also known as OB-cadherin) preferentially interacts with β-catenin in ANGPTL2-OE SW480 relative to control cells (Fig. 6b and Supplementary Fig. 3a). Immunoblot analysis also showed upregulated OB-cadherin expression in ANGPTL2-OE relative to control SW480 cells, and that OB-cadherin was co-immunoprecipitated with β-catenin in ANGPTL2-OE SW480 cells in the presence or absence of MG132 (Fig. 6c and Supplementary Fig. 3b).

a Representative images of silver staining of anti-β-catenin immunoprecipitates from ANGPTL2-OE SW480 lines #1 and #2 and control cells incubated 12 h with MG132 (20 μM). b Results of anti-β-catenin IP/MS experiments showing log2 ratio of normalized protein abundance in ANGPTL2-OE SW480 cells (lines #1 and #2) to abundance seen in control cells. n = 1 for each group. The X-axis indicates the log2 ratio of normalized protein abundance in the ANGPTL2-OE SW480 line #1 relative to control cells; the Y-axis indicates the log2 ratio of normalized protein abundance in the ANGPTL2-OE SW480 line #2 relative to control cells. c Representative immunoblotting for β-catenin and OB-cadherin in input samples and anti-β-catenin immunoprecipitates from ANGPTL2-OE SW480 lines #1 and #2 and control cells incubated 12 h with MG132 (20 μM). HSC70 served as a loading control. d Representative immunoblotting for OB-cadherin and β-catenin in ANGPTL2-OE SW480 lines #1 and #2 transfected with two different siCDH11 constructs (#1 or #2) or control siRNA. HSC70 served as a loading control. e Representative immunoblotting for β-catenin and OB-cadherin in input samples and anti-β-catenin immunoprecipitates from ANGPTL2-OE SW480 cells (line #1 only) transfected with siCDH11 constructs #1 or #2 or control siRNA and then incubated 12 h more in MG132 (20 μM). HSC70 served as a loading control. f (upper) Representative immunoblotting for β-catenin in ANGPTL2-OE SW480 cells (line #1 only) transfected with siCDH11 construct #2 or control siRNA and incubated with cycloheximide (100 μg/ml) for indicated times. HSC70 served as a loading control. (lower) Shown is the percentage of β-catenin remaining at indicated incubation times. Data are means ± SD; n = 3 for each group. *P < 0.05, two-way ANOVA test. g (upper) Representative immunoblotting for β-catenin in ANGPTL2-OE SW480 cells (line #1 only) transfected with siCDH11 construct #2 and incubated with cycloheximide (100 μg/ml) in the absence or presence of the proteasome inhibitor MG132 (20 μM) for indicated times. HSC70 served as a loading control. (lower) Shown is the percentage of β-catenin remaining at indicated incubation times. Data are means ± SD; n = 3 for each group. *P < 0.05, two-way ANOVA test.
We next asked whether reduced OB-cadherin expression seen in ANGPTL2-overexpressing CRC cells underlies decreased β-catenin accumulation by transfecting ANGPTL2-OE SW480 cells with either CDH11-specific or control scramble siRNA (Fig. 6d, e). OB-cadherin knockdown in ANGPTL2-OE SW480 cells decreased levels of β-catenin proteins (Fig. 6d). Furthermore, cycloheximide chase assays revealed β-catenin protein to be less stable in ANGPTL2-OE SW480 cells transfected with CDH11-specific siRNA relative to control siRNA-transduced cells (Fig. 6f), an effect blocked by MG132 treatment (Fig. 6g). We next asked whether OB-cadherin/β-catenin interaction promotes β-catenin translocation from the plasma membrane to the cytoplasm. Although cytosolic β-catenin protein levels increased in ANGPTL2-OE SW480 relative to control cells (Fig. 4f), the ratio of cytosolic to plasma membrane-bound β-catenin protein was comparable in both groups (Supplementary Fig. 3c). Taken together, these results suggest that ANGPTL2-induced OB-cadherin is required to suppress proteasomal degradation of β-catenin proteins in tumor cells.
ANGPTL2 induction of CDH11 expression is accompanied by decreased cell surface expression of integrin α5β1
We next asked how ANGPTL2 induces OB-cadherin expression in CRC cells. qRT-PCR analysis revealed markedly increased CDH11 transcript levels in ANGPTL2-OE SW480 relative to control cells (Fig. 7a). To further assess a potential correlation between ANGPTL2 and CDH11 expression in CRC cells, we analyzed gene expression data from the Cancer Cell Line Encyclopedia (CCLE) and found a positive correlation between ANGPTL2 and CDH11 mRNA expression in CRC lines (Fig. 7b), suggesting that ANGPTL2 may transcriptionally regulate CDH11 in CRC.

a Comparison of CDH11 transcript levels in ANGPTL2-OE SW480 lines #1 and #2 and control cells. Levels in control cells were set to 1. Data are means ± SD; n = 4 for each group. ***P < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test. b Correlation of ANGPTL2 and CDH11 mRNA levels in CRC cell lines from the CCLE database (n = 58). Pearson’s correlation coefficient (r) was calculated for each correlation. c (left) Representative histograms of cell surface expression of integrin α5β1 in ANGPTL2-OE SW480 lines #1 and #2 and control cells. (right) Shown is quantification of integrin α5β1 Mean Fluorescence Intensity (MFI). d Comparison of levels of ITGA5 (left) and ITGB1 (right) transcripts in ANGPTL2-OE SW480 lines #1 and #2 and control cells. Levels in control cells were set to 1. Data are means ± SD; n = 4 for each group. ns, not significant (P > 0.05), one-way ANOVA test followed by Tukey’s multiple comparison test. e Representative images of immunofluorescence for integrin α5 (red) in tumor and adjacent nontumor tissues from WT mice at week 24 of the AOM model. Nuclei are counterstained with DAPI (white). Scale bar, 50 μm. f Representative histograms of cell surface expression of integrin α5β1 in SW480 control cells transfected with two different siITGA5 constructs (#1 and #2) or control siRNA. g Comparison of CDH11 transcript levels in SW480 control cells transfected with two different siITGA5 constructs (#1 and #2) or control siRNA. Levels in control siRNA were set to 1. Data are means ± SD; n = 3 for each group. ***P < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test. h Model of proposed molecular mechanism underlying ANGPTL2-mediated β-catenin-driven colorectal tumorigenesis. In CRC cells, tumor cell-derived ANGPTL2 signaling upregulates OB-cadherin, which blocks proteasomal degradation of β-catenin, thereby augmenting β-catenin signaling and promoting cell proliferation.
We next investigated molecular mechanisms underlying upregulation of CDH11 by ANGPTL2. Because ANGPTL2 binds to integrin α5β1 [7, 11, 17], we asked whether integrin α5β1 plays a role in ANGPTL2-dependent CDH11 induction in CRC cells, by first assessing cell surface expression of integrin α5β1 in these cells. Interestingly, flow cytometry analysis revealed a significant decrease in cell surface integrin α5β1 protein in ANGPTL2-OE SW480 relative to control SW480 cells (Fig. 7c), while integrin α5 (ITGA5) and β1 (ITGB1) transcript levels were comparable between groups (Fig. 7d). Consistent with this, immunofluorescence analysis revealed integrin α5 protein primarily localized at cell-to-cell borders in normal colon epithelial cells, while levels of cell surface integrin α5 were lower in adjacent tumor cells in the AOM model (Fig. 7e). To investigate mechanisms underlying ANGPTL2 regulation of cell surface integrin α5β1 expression, we examined integrin α5 protein levels in cytosolic and plasma membrane fractions of ANGPTL2-OE SW480 and control cells. Based on immunoblot analysis, ANGPTL2-OE SW480 cells showed relatively increased levels of early endosome antigen 1 (EEA1), which localizes to early endosomes and is essential for endosome trafficking, and integrin α5 protein in cytosolic fractions relative to control cells (Supplementary Fig. 4a), suggesting that ANGPTL2 enhances ligand-induced integrin α5 internalization. OB-cadherin, a type 2 cadherin, is a biomarker for mesenchymal phenotypes [18], and integrin α5 at the lateral membrane is required for epithelialization [19, 20]. Thus, we asked whether decreased levels of cell surface integrin α5β1 contribute to ANGPTL2-dependent CDH11 induction in tumor cells. To do so, we transfected SW480 cells with either ITGA5-specific or control scramble siRNA and assessed CDH11 levels (Fig. 7f). CDH11 transcript levels significantly increased in integrin α5-knockdown compared to control cells (Fig. 7g). To further assess potential integrin α5β1 function in tumor progression in vivo, we deleted integrin α5 in ANGPTL2-OE SW480 cells using the CRISPR/Cas9 system (Supplementary Fig. 4b) and then injected either integrin α5 KO or control ANGPTL2-OE SW480 cells into the back flanks of nude mice. We observed no differences in tumor volume between groups (Supplementary Fig. 4c). However, relative to control cells, ANGPTL2-overexpressing SW480 cells already show decreased surface expression of integrin α5β1 (Fig. 7c) plus increased CHD11 expression (Fig. 7a) and tumor growth in vivo (Fig. 4d); thus, integrin α5 KO in those cells may not be sufficient to alter tumor progression in vivo.
ANGPTL2 expression in human primary tumor tissues correlates with β-catenin expression
Finally, we assessed a potential correlation between ANGPTL2, β-catenin, and integrin α5 expression levels in primary tumors from patients with CRC. Immunohistochemical analysis of tumor tissues from these patients revealed relatively lower β-catenin expression but higher integrin α5 expression in tissues expressing low ANGPTL2 protein (Supplementary Fig. 5a–c, patients #1 and #2). By contrast, CRC cells expressing abundant ANGPTL2 protein showed high β-catenin and low integrin α5 expression (Supplementary Fig. 5a–c, patients #3 and #4).
Discussion
Here, we demonstrate that ANGPTL2 deficiency inhibits tumor development in an experimental mouse model of sporadic CRC. Moreover, induction of ANGPTL2 expression in tumor cells led to accumulation of β-catenin proteins in CRC cells and enhanced tumor cell proliferation. We also found that ANGPTL2 overexpression blocks destruction complex-independent proteasomal degradation of β-catenin in tumor cells. Mechanistically, our findings suggest that ANGPTL2 enhances expression of OB-cadherin, which interacts with β-catenin, resulting in inhibition of β-catenin degradation in CRC cells. Furthermore, we showed that ANGPTL2 induces OB-cadherin expression in CRC cells likely by decreasing cell surface expression of integrin α5β1. Based on these results we propose that ANGPTL2 promotes β-catenin-driven colorectal tumorigenesis by accelerating β-catenin accumulation in tumor cells via the integrin α5β1/OB-cadherin pathway (Fig. 7h).
WNT/β-catenin signaling controls epithelial proliferation, intestinal homeostasis, and maintenance of intestinal stem cells [21, 22]. However, aberrant activation of β-catenin leads to development of adenomas and CRC [23, 24]. Loss of APC, which encodes a component of the β-catenin destruction complex, in intestinal epithelial cells drives formation of adenomas via increased WNT/β-catenin signaling [25]. This mutation is found in approximately 30%–70% of sporadic adenomas and sporadic CRCs. In the AOM model, the GSK-3β phosphorylation consensus motif of β-catenin is reportedly mutated [12], suggesting that β-catenin degradation in AOM-induced tumor cells does not depend solely on the destruction complex. We demonstrate here that ANGPTL2 is required for AOM-induced tumor development and that ANGPTL2 overexpression causes β-catenin accumulation in SW480 cells, which harbor a typically truncated form of APC, resulting in enhanced cell proliferation. These results support the idea that ANGPTL2 controls β-catenin stability independent of the destruction complex, thereby promoting development of AOM-induced tumors. Importantly, we demonstrated that knockdown of OB-cadherin decreases levels of β-catenin protein in ANGPTL2-OE SW480 cells. Thus, even if the β-catenin destruction complex becomes nonfunctional in CRC cells, aberrant activation of β-catenin signaling, which promotes intestinal tumorigenesis, could be suppressed by targeting ANGPTL2 or OB-cadherin.
OB-cadherin, which was initially identified in mouse osteoblasts [26], is also expressed in breast, prostate, gastric, and colon cancers [27,28,29,30,31]. OB-cadherin functions in tumor cell invasion and migration and cancer progression [32, 33]. OB-cadherin expression is also reportedly associated with enhanced migration and invasion of CRC cells [34]. Furthermore, recent studies demonstrate that inhibiting OB-cadherin inactivates WNT/β-catenin signaling [35, 36]. Our study indicates that OB-cadherin interacts with β-catenin in ANGPTL2-OE cells and that OB-cadherin loss via knockdown promotes proteasomal degradation of β-catenin, suggesting that β-catenin/OB-cadherin interaction may protect β-catenin proteins from degradation and activate β-catenin pathway.
We also report that integrin α5β1 expression on tumor cells decreases following ANGPTL2 overexpression and that integrin α5 knockdown induces OB-cadherin mRNA expression in tumor cells. OB-cadherin is a marker of the epithelial–mesenchymal transition (EMT) [37], and integrin α5 expression positively correlates with epithelial phenotypes in CRC cells [38]. Because ANGPTL2 signaling enhances cancer cell metastasis via EMT activation [17, 39], ANGPTL2 may induce OB-cadherin expression by activating the EMT, although further studies are required to confirm this possibility.
Here, we demonstrated that tumor cell-derived ANGPTL2 activates WNT/β-catenin signaling. On the other hand, we previously reported that Angptl2-deficient mice show impaired intestinal epithelial regeneration after dextran sulfate sodium (DSS)-induced injury [8]. Mechanistically, in that study we found that stromal cell-derived ANGPTL2 maintained the intestinal stem cell niche by modulating signaling between bone morphogenetic protein (BMP) and β-catenin and showed de-repression of Bmp2 and Bmp7 expression in ANGPTL2-deficient stromal cells. In humans, perturbed β-catenin/BMP signaling is associated with juvenile polyposis syndrome (JPS), familial adenomatous polyposis (FAP), and colorectal cancer [40,41,42]. Thus, stromal cell-derived ANGPTL2-mediated suppression of BMP expression may also function in intestinal tumorigenesis.
Complementing this study, we previously showed that ANGPTL2 signaling in tumor cells has tumor-promoting activity [10, 11, 39, 43, 44]. However, we recently demonstrated that tumor stroma-derived ANGPTL2 exhibits anti-tumor activities in a AOM/DSS mouse model of colitis-associated colon cancer, in a mouse kidney cancer model, and in murine syngeneic tumor models [45, 46]. These studies suggest a dual role for ANGPTL2 in oncogenesis functioning either in tumor promotion or suppression, depending on context, although mechanisms underlying context-dependency remain unclear. One possibility is that ANGPTL2 may have anti-tumor activity in highly immunogenic tumor microenvironments characterized by T cell infiltration and molecular signatures indicative of immune activation. Interestingly, in our recent analysis of anti-tumor ANGPTL2 activity in mouse kidney cancer, and murine syngeneic and AOM/DSS models, we observed abundant T cell infiltration of tumors [45, 46]. By contrast, in the AOM model, which is an inflammation-independent sporadic colorectal tumorigenesis model, ANGPTL2 showed tumor-promoting activities, supporting the idea that ANGPTL2 function in cancer pathology depends on the nature of the tumor microenvironment, as well as the type of cell expressing ANGPTL2.
In conclusion, we propose that tumor cell-derived ANGPTL2 accelerates tumor cell proliferation by enhancing OB-cadherin expression, in turn suppressing proteasomal degradation of β-catenin and promoting β-catenin-driven intestinal tumorigenesis. Our findings represent a novel mechanism underlying destruction complex-independent regulation of β-catenin signaling in CRC cells and suggest that the ANGPTL2–OB-cadherin axis could be targeted by novel therapeutics.
Materials and methods
Animals
Angptl2−/− mice on a C57BL/6 N background were described previously [9]. Angptl2+/− mice were backcrossed to a C57BL/6 strain for 10 or more generations. Age-and sex-matched littermate mice were used for experiments. Nude BALB/c mice were purchased from CLEA (Japan). Sample sizes were based on the literature. Data collection and analysis were conducted in a randomized and blinded manner. Mice showing clear signs of abnormal sickness were excluded from the study. All experimental procedures were approved by the Ethics Review Committee for Animal Experimentation of Kumamoto University.
Induction of sporadic colon tumorigenesis
Mice received intraperitoneal injections of 10 mg/kg AOM (Wako) once a week for 6 weeks. Tumor development was assessed at 16 and 24 weeks after the initial AOM injection. Colonoscopy was performed using an endoscope system (AVS), air pump (Tetra), and video capture box (IO-DATA).
Isolation of colonic epithelial cells and fibroblasts
To isolate tumor cells, tissues from 15 pooled tumor specimens from WT mice (n = 10) of the AOM model were washed twice with cold PBS and incubated in 5 mM EDTA in PBS for 90 min at 4 °C on a tube roller. After incubation, small pieces were vigorously shaken, passed through a 100-μm cell strainer and centrifuged at 300 g for 5 min. Collected cells were used for qRT-PCR. Remaining tissue was then incubated 2 h in 20 ml RPMI 1640 medium (Wako) containing 10 mg dispase and 10 mg collagenase D in a shaking 37 °C incubator. Digested tissue was treated with red blood cell lysis buffer (ACK buffer; 0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA) for 5 min, washed and then passed through a 100-μm cell strainer. TAFs were isolated by MACS using a tumor-associated fibroblast isolation kit (Miltenyi Biotec). Mouse normal colonic crypts or ISEMFs were isolated as described [8].
Histology, immunohistochemistry, immunofluorescence
For histological analysis, colonic tissue samples were fixed with 15% neutral buffered formalin, embedded in paraffin, sectioned with a microtome, and stained with hematoxylin and eosin (H&E).
For immunohistochemistry, after antigen retrieval, endogenous peroxidase was blocked using 3% H2O2 for 10 min. Samples were then blocked with 5% serum for 20 min at room temperature and incubated with primary antibodies overnight at 4 °C. Appropriate secondary antibodies were applied for 60 min at room temperature, and 0.02% DAB solution was used for detection and visualization of staining. Slides were counterstained with hematoxylin and mounted. Antibodies used were as follows: anti-β-catenin (1:50, BD Biosciences, 610154), anti-integrin α5 (1:100, abcam, ab150361). Immunohistochemical staining for human ANGPTL2 was performed as described [10]. Immunohistochemical staining for mouse ANGPTL2 was performed as described [47].
For immunofluorescence, after antigen retrieval, samples were blocked with 5% serum for 20 min at room temperature and incubated with primary antibodies overnight at 4 °C, then incubated with antibodies conjugated Alexa Flour 594 for 60 min at room temperature. Nuclei were stained with DAPI for 20 min. Antibodies used were as follows: anti-integrin α5 (1:00, abcam, ab150361).
Clinical samples
We obtained paraffin-embedded tumor samples from biopsy specimens of primary tumors from patients with unresectable CRC at Kumamoto University Hospital (Kumamoto, Japan). This study was approved by the Ethics Committees of Kumamoto University. Written informed consent was obtained from each subject.
Cell culture
The human CRC cell line SW480, purchased from ATCC, was cultured in Leibovitz’s medium (Invitrogen) supplemented with 10% FCS at 37 °C in a humidified 5% CO2 atmosphere. The human CRC line HCT116 (purchased from ATCC) was cultured in McCoy’s 5a medium (Invitrogen) supplemented with 10% FCS at 37 °C in a humidified 5% CO2 atmosphere.
ANGPTL2- or integrin α5-deficient cells were generated using the Guide-it CRISPR/Cas9 system (Takara Bio) according to the manufacturer’s instructions. Cells were transfected with expression plasmids encoding Cas9 and ZsGreen1, and with single-guide RNAs (sgRNAs). The latter were 5′-CGACCAGAGACACGACCCCG-3′ for human ANGPTL2 #1; 5′-TGCGACCAGAGACACGACCC-3′ for human ANGPTL2 #2; 5′-GGTGTAGGTGCACTTGTCCT-3′ for human ANGPTL2 #3; and 5′- TCATGGATCGGACCCCTGACGGG-3′ for human integrin α5. A Guide-it Control Anneals Oligos served as control. After transfection, ZsGreen1+ cells were isolated by cell sorting (using a SH800S sorter, Sony Biotechnology). ANGPTL2 KO was confirmed based on lack of ANGPTL2 protein expression in established lines.
To create stably transfected cells, SW480 or HCT116 cells were transfected with ANGPTL2 or control vectors [10] using Lipofectamine 2000 or 3000 (Invitrogen), according to the manufacturer’s protocol.
For transient knockdown of CDH11 or ITGA5, SW480 cells were transfected with siRNA (Trilencer-27 human siRNA; OriGene) using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer’s protocol. As a control, we used Trilencer-27 Universal Scrambled Negative Control (OriGene).
Immunostaining
Cells were first fixed 10 min in 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100 in PBS for 15 min at room temperature. Non-specific binding was minimized by blocking with 5% normal goat serum. Cells were incubated with anti-human ANGPTL2 antibodies [10] and then with Alexa 594-conjugated anti-rabbit antibodies. Nuclei were counterstained with DAPI.
Quantitation of ANGPTL2 protein by ELISA
Cells were grown to confluency, and then medium was collected to quantify ANGPTL2 protein by ELISA. ANGPTL2 concentrations in the medium were estimated using an ANGPTL2 Assay Kit (IBL) based on the manufacturer’s instructions.
Clonogenic assay
Two thousand cells were seeded into 6-well plates. Growth medium was refreshed every three days. After 10–12 days, cells were fixed with 4% paraformaldehyde and stained 30 min with 0.5% crystal violet in 25% methanol and images of colonies were acquired manually. Stained cells were then solubilized in 1% SDS, and the OD595 values were determined
Xenograft tumor model
ANGPTL2 KO and control SW480 cells (4 × 106) in 100 μL PBS were injected subcutaneously into both back flanks of 5-wk-old nude mice. Likewise, ANGPTL2-OE and control SW480 cells (1 × 106) in 50 μL PBS were injected using the same protocol. Tumor volume (V) was measured with calipers (V = width2 × height × 0.523).
Immunoblot analysis
Solubilized proteins were subjected to SDS-PAGE, and proteins were electro-transferred to nitrocellulose membranes. Immunoblotting was performed with antibodies against ANGPTL2 (1:1000, R&D Systems, BAF1444), β-catenin (1:1000, BD Biosciences, 610154), c-Myc (1:1000, Cell Signaling Technology, #5605), Cyclin D2 (1:1000, Cell Signaling Technology, #3741), Axin2 (1:1000, Cell Signaling Technology, #2151), HDAC2 (1:1000, Cell Signaling Technology, #57156), GAPDH (1:1000, Cell Signaling Technology, #5174), APC (1:1000, Santa Cruz Biotechnology, #sc53165), phospho-β-catenin (1:1000, Cell Signaling Technology, #9561), OB-cadherin (1:1000, Cell Signaling Technology, #4442), Integrin α5 (1:1000, abcam, ab150361), EEA1 (1:1000, Cell Signaling Technology, #3288), Na/K-ATPase (1:1000, Cell Signaling Technology, #3010), or HSC70 (1:4000, Santa Cruz Biotechnology, #sc7298). Immunodetection was carried out using an ECL kit (GE Healthcare) according to the manufacturer’s protocol.
Cytosolic/nuclear/plasma membrane fractionation
Cytosolic/nuclear fractionation was performed as described [48]. Cells were lysed in ‘cytosolic buffer’ (0.33 M Sucrose, 10 mM HEPES, 1 mM MgCl2, 0.1% Triton X100) for 10 min. The lysate was centrifuged at 2400 rpm for 5 min, and the resulting supernatant was used as cytosolic fraction. The nuclear fraction was obtained by dissolving pellets in ‘membrane buffer’ (0.25 M Sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA) for 25 min on ice to remove membrane-bound β-catenin. The lysate was then centrifuged at 3000 rpm for 5 min, and the pellet was considered the nuclear fraction. Plasma membrane fractions were prepared using a Trident Membrane Protein Extraction Kit (GeneTex) according to the manufacturer’s protocol.
Immunoprecipitation
Cells were lysed with lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM EDTA, protease inhibitor (Roche), 10 mM NaF, 1 mM Na3VO4, and 10 mM Na3P2O7. Lysates were centrifuged to remove debris, and supernatants were incubated for 120 min at 4 °C with Dynabeads protein G (Invitrogen)-conjugated anti-β-catenin antibody (5 μg, BD Biosciences, 610154) or control IgG (R&D Systems, MAB002). Resulting precipitates were washed and eluted with SDS sample buffer. Purified proteins were fractionated by SDS-PAGE and silver-stained (Wako).
In-gel digestion/Mass spectrometry
Extracted proteins were separated by SDS-PAGE followed by negative staining. Total proteins in gels were reduced by treatment with 10 mM dithiothreitol for 30 min, and alkylated using 50 mM iodoacetamide for 30 min. After overnight trypsin digestion, peptides were extracted from gel pieces and purified with SDB-XC StageTip [49, 50]. NanoLC-MS/MS was conducted using a Triple TOF 5600 (SCIEX) with an Ultimate 3000 RSLCnano (Thermo Fisher Scientific). An Acclaim PepMap RSLC (2 mm, 25 cm × 75 mm) column was used for peptide separation. The mobile phase consisted of (A) 0.1% formic acid in water and (B) 0.1% formic acid in acetonitrile. A linear gradient of 2% B to 25% B in 60 min, 50% B to 90% B in 15 min and 90% B for 5 min was employed. MS data were subjected to a search against the Uniprot Human database with Protein Pilot V.4.5 (AB Sciex). Peak areas of peptides were calculated by PeakView (AB Sciex).
Total RNA extraction and real-time quantitative RT-PCR
Total RNA was isolated from cells and tissues using TRIzol regent (Invitrogen). DNase-treated RNA was reversed-transcribed with a PrimeScript RT regent Kit (Takara Bio). PCR products were analyzed using a Thermal Cycler Dice Real Time System (Takara Bio). PCR primer sequences (forward and reverse, respectively) were 5′-CCTGGTGCTCCATGAGGAGAC-3′ and 5′-CAGACTCTGACCTTTTGCCAGG-3′ for human MYC; 5′-GAGAAGCTGTCTCTGATCCGCA-3′ and 5′-CTTCCAGTTGCGATCATCGACG-3′ for human CCND2; 5′-ACTGCCCACACGATAAGGAG-3′ and 5′-CTGGCTATGTCTTTGGACCA-3′ for human AXIN2; 5′-TGGCAGCAAGTATCCAATGG-3′ and 5′-TTTGGTTACGTGGTAGGCAC-3′ for human CDH11; 5′-AGACATTCGATCCCTCTACAACT-3′ and 5′-AATCGGCCAAACTCATCATGG-3′ for human ITGA5; 5′-CCTACTTCTGCACGATGTGATG-3′ and 5′-CCTTTGCTACGGTTGGTTACATT-3′ for human ITGB1; 5′-TTTGCGAGTACTCAACACCAACATC-3′ and 5′- GAGCATATCTTCGGCCCACAC -3′ for human 18 S. Relative transcript abundance was normalized to that of 18 S mRNA.
Flow cytometry
Cells were suspended in MACS buffer (Miltenyi Biotec), stained with anti-integrin α5β1 (Millipore, MAB1999) or control IgG (R&D Systems, MAB002), and then incubated with appropriate secondary antibodies. Viable cells were identified as unstained with 7-AAD (Beckman Coulter). Stained cells were analyzed by BD FACSVerse (BD Biosciences). Data analysis was undertaken using FlowJo software (TreeStar).
Statistical analysis
Statistical analyses were performed using GraphPad prism 9 software (GraphPad Software). Statistical parameters and methods are reported in respective figures and figure legends. Results with p-values < 0.05 were considered significant (P < 0.05 = *; P < 0.01 = **; P < 0.001 = ***). Comparisons between two groups were performed using an unpaired two-tailed t-test when variance was similar between groups, or the Mann-Whitney U test. Comparisons between three or more groups were performed using one-way ANOVA with Tukey’s multiple comparison test. For comparisons with two or more independent variable factors, we used two-way ANOVA followed by Sidak’s multiple comparison test. Survival rate was analyzed by log-rank test. For comparison of tumor incidence, we used Fisher’s exact test.
References
Kimelman D, Xu W. β-Catenin destruction complex: Insights and questions from a structural perspective. Oncogene. 2006;25:7482–91.
Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and β-catenin signalling: Diseases and therapies. Nat Rev Genet. 2004;5:691–701.
Reya T, Clevers H. Wnt signalling in stem cells and cancer. 434, 843–850 (2005). Nature. 2005;434:843–50.
Kumawat K, Koopmans T, Gosens R. Β-Catenin As a Regulator and Therapeutic Target for Asthmatic Airway Remodeling. Expert Opin Ther Targets. 2014;18:1023–34.
Hato T, Tabata M, Oike Y. The Role of Angiopoietin-Like Proteins in Angiogenesis and Metabolism. Trends Cardiovasc. Med. 2008;18. https://doi.org/10.1016/j.tcm.2007.10.003.
Oike Y, Yasunaga K, Ito Y, Matsumoto S ichiro, Maekawa H, Morisada T, et al. Angiopoietin-related growth factor (AGF) promotes epidermal proliferation, remodeling, and regeneration. Proc Natl Acad Sci USA 2003;100. https://doi.org/10.1073/pnas.1531901100.
Kadomatsu T, Endo M, Miyata K, Oike Y. Diverse roles of ANGPTL2 in physiology and pathophysiology. Trends Endocrinol Metab. 2014;25:245–54.
Horiguchi H, Endo M, Kawane K, Kadomatsu T, Terada K, Morinaga J, et al. ANGPTL2 expression in the intestinal stem cell niche controls epithelial regeneration and homeostasis. EMBO J. 2017;36:409–24.
Tabata M, Kadomatsu T, Fukuhara S, Miyata K, Ito Y, Endo M, et al. Angiopoietin-like Protein 2 Promotes Chronic Adipose Tissue Inflammation and Obesity-Related Systemic Insulin Resistance. Cell Metab. 2009;10:178–88.
Endo M, Nakano M, Kadomatsu T, Fukuhara S, Kuroda H, Mikami S, et al. Tumor Cell-Derived Angiopoietin-like Protein ANGPTL2 Is a Critical Driver of Metastasis. Cancer Res. 2012;72:1784–94.
Odagiri H, Kadomatsu T, Endo M, Masuda T, Morioka MS, Fukuhara S, et al. The Secreted Protein ANGPTL2 Promotes Metastasis of Osteosarcoma Cells Through Integrin 5 1, p38 MAPK, and Matrix Metalloproteinases. Sci Signal. 2014;7:ra7.
Takahashi M, Fukuda K, Sugimura T, Wakabayashi K. Β-Catenin Is Frequently Mutated and Demonstrates Altered Cellular Location in Azoxymethane-Induced Rat Colon Tumors. Cancer Res. 1998;58:42–46.
Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–54.
Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991;253. https://doi.org/10.1126/science.1651563.
Su YY, Fu C, Ishikawa S, Stella A, Kojima M, Shitoh K, et al. APC Is Essential for Targeting Phosphorylated β-Catenin to the SCFβ-TrCP Ubiquitin Ligase. Mol Cell. 2008;32. https://doi.org/10.1016/j.molcel.2008.10.023.
Li VSW, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, et al. Wnt Signaling through Inhibition of β-Catenin Degradation in an Intact Axin1 Complex. Cell. 2012;149. https://doi.org/10.1016/j.cell.2012.05.002.
Osumi H, Horiguchi H, Kadomatsu T, Tashiro K, Morinaga J, Takahashi T, et al. Tumor cell-derived angiopoietin-like protein 2 establishes a preference for glycolytic metabolism in lung cancer cells. Cancer Sci. 2020;111:1241–53.
Hoffmann I, Balling R. Cloning and expression analysis of a novel mesodermally expressed cadherin. Dev Biol. 1995;169:337–46.
Koshida S, Kishimoto Y, Ustumi H, Shimizu T, Furutani-Seiki M, Kondoh H, et al. Integrinα5-dependent fibronectin accumulation for maintenance of somite boundaries in zebrafish embryos. Dev Cell. 2005;8. https://doi.org/10.1016/j.devcel.2005.03.006.
Starchenko A, Graves-Deal R, Yang YP, Li C, Zent R, Singh B, et al. Clustering of integrin α5 at the lateral membrane restores epithelial polarity in invasive colorectal cancer cells. Mol Biol Cell. 2017;28. https://doi.org/10.1091/mbc.E16-12-0852.
Sancho E, Batlle E, Clevers H. Live and let die in the intestinal epithelium. Curr Opin Cell Biol. 2003;15:763–70.
Kuhnert F, Davis CR, Wang HT, Chu P, Lee M, Yuan J, et al. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc Natl Acad Sci USA. 2004;101:266–71.
Miyoshi Y, Ando H, Nagase H, Nishisho I, Horii A, Miki Y, et al. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc Natl Acad Sci USA. 1992;89:4452–6.
Kinzler KW, Vogelstein B Lessons from hereditary colorectal cancer. Cell. 1996;87. https://doi.org/10.1016/S0092-8674(00)81333-1.
Barker N, Ridgway RA, Van Es JH, Van De Wetering M, Begthel H, Van Den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–11.
Okazaki M, Takeshita S, Kawai S, Kikuno R, Tsujimura A, Kudo A, et al. Molecular cloning and characterization of OB-cadherin, a new member of cadherin family expressed in osteoblasts. J Biol Chem. 1994;269:12092–8.
Shibata T, Ochiai A, Gotoh M, Machinami R, Hirohashi S. Simultaneous expression of cadherin-11 in signet-ring cell carcinoma and stromal cells of diffuse-type gastric cancer. Cancer Lett. 1996;99:147–53.
Feltes CM, Kudo A, Blaschuk O, Byers SW. An alternatively spliced cadherin-11 enhances human breast cancer cell invasion. Cancer Res. 2002;62:6688–97.
Tomita K, Van Bokhoven A, Van Leenders GJLH, Ruijter ETG, Jansen CFJ, Bussemakers MJG, et al. Cadherin switching in human prostate cancer progression. Cancer Res. 2000;60:3650–4.
Pishvaian MJ, Feltes CM, Thompson P, Bussemakers MJ, Schalken JA, Byers SW. Cadherin-11 is expressed in invasive breast cancer cell lines. Cancer Res. 1999;59:947–52.
Munro SB, Turner JM, Farookhi R, Blaschuk OW, Jothy S. E-cadherin and ob-cadherin mrna levels in normal human colon and colon carcinoma. Exp Mol Pathol. 1995;62:118–22.
Kaur H, Phillips-Mason PJ, Burden-Gulley SM, Kerstetter-Fogle AE, Basilion JP, Sloan AE, et al. Cadherin-11, a marker of the mesenchymal phenotype, regulates glioblastoma cell migration and survival in vivo. Mol Cancer Res. 2012;10. https://doi.org/10.1158/1541-7786.MCR-11-0457.
Chu K, Cheng CJ, Ye X, Lee YC, Zurita AJ, Chen DT, et al. Cadherin-11 promotes the metastasis of prostate cancer cells to bone. Mol Cancer Res. 2008;6. https://doi.org/10.1158/1541-7786.MCR-08-0077.
Zhu Q, Wang Z, Zhou L, Ren Y, Gong Y, Qin W, et al. The role of cadherin-11 in microcystin-LR-induced migration and invasion in colorectal carcinoma cells. Oncol Lett. 2018;15:1417–22.
Satriyo P, Bamodu O, Chen J-H, Aryandono T, Haryana S, Yeh C-T, et al. Cadherin 11 Inhibition Downregulates β-catenin, Deactivates the Canonical WNT Signalling Pathway and Suppresses the Cancer Stem Cell-Like Phenotype of Triple Negative Breast Cancer. J Clin Med. 2019;8:148.
Chen JH, Huang WC, Bamodu OA, Chang PMH, Chao TY, Huang TH. Monospecific antibody targeting of CDH11 inhibits epithelial-to-mesenchymal transition and represses cancer stem cell-like phenotype by up-regulating miR-335 in metastatic breast cancer, in vitro and in vivo. BMC Cancer. 2019;19:1–13.
Zeisberg M, Neilson EG Biomarkers for epithelial-mesenchymal transitions. J Clin Investig. 2009;119. https://doi.org/10.1172/JCI36183.
Halbleib JM, Sääf AM, Brown PO, Nelson WJ. Transcriptional modulation of genes encoding structural characteristics of differentiating enterocytes during development of a polarized epithelium in vitro. Mol Biol Cell. 2007;18. https://doi.org/10.1091/mbc.E07-04-0308.
Aoi J, Endo M, Kadomatsu T, Miyata K, Nakano M, Horiguchi H, et al. Angiopoietin-like protein 2 is an important facilitator of inflammatory carcinogenesis and metastasis. Cancer Res. 2011;71:7502–12.
Waite KA, Eng C From developmental disorder to heritable cancer: It’s all in the BMP/TGF-β family. Nat Rev Genet. 2003;4. https://doi.org/10.1038/nrg1178.
Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Petersen GM, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet. 2001;28. https://doi.org/10.1038/88919.
Van Es JH, Giles RH, Clevers HC. The many faces of the tumor suppressor gene APC. Exp Cell Res. 2001; 264. https://doi.org/10.1006/excr.2000.5142.
Masuda T, Endo M, Yamamoto Y, Odagiri H, Kadomatsu T, Nakamura T, et al. ANGPTL2 increases bone metastasis of breast cancer cells through enhancing CXCR4 signaling. Sci Rep. 2015;5:9170.
Horiguchi H, Endo M, Miyamoto Y, Sakamoto Y, Odagiri H, Masuda T, et al. Angiopoietin-like protein 2 renders colorectal cancer cells resistant to chemotherapy by activating spleen tyrosine kinase-phosphoinositide 3-kinase-dependent anti-apoptotic signaling. Cancer Sci. 2014;105:1550–9.
Horiguchi H, Kadomatsu T, Kurahashi R, Hara C, Miyata K, Baba M, et al. Dual functions of angiopoietin-like protein 2 signaling in tumor progression and anti-tumor immunity. Genes Dev. 2019;33:1641–56.
Horiguchi H, Kadomatsu T, Miyata K, Terada K, Sato M, Torigoe D, et al. Stroma-derived ANGPTL2 establishes an anti-tumor microenvironment during intestinal tumorigenesis. Oncogene. 2021;40:55–67.
Motokawa I, Endo M, Terada K, Horiguchi H, Miyata K, Kadomatsu T, et al. Interstitial pneumonia induced by bleomycin treatment is exacerbated in Angptl2-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2016;311:L704–L713.
Oudhoff MJ, Braam MJS, Freeman SA, Wong D, Rattray DG, Wang J, et al. SETD7 Controls Intestinal Regeneration and Tumorigenesis by Regulating Wnt/β-Catenin and Hippo/YAP Signaling. Dev Cell. 2016;37:47–57.
Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2. https://doi.org/10.1038/nprot.2007.261.
Rappsilber J, Ishihama Y, Mann M Stop And Go Extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem. 2003;75. https://doi.org/10.1021/ac026117i.
Acknowledgements
We thank Kiyoka Tabu, Noriko Shirai, and Sayomi Iwaki for technical assistance. This work was supported by the Scientific Research Fund of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (grant 21K07101 to TK and grant 21K15508 to HH), the Takeda Science Foundation (HH, TK), and Tasaki Memorial Research Grant for 2021 (HH).
Author information
Authors and Affiliations
Contributions
HH designed the study, performed and analyzed most of experiments, and wrote the paper. TK designed the study and wrote the paper. SYumoto and HB collected human data. TMasuda and SO performed proteomics. KM provided Angptl2 mutant mice. SYamamura, MS, and JM discussed the data. TMoroishi designed and supervised the study. YO coordinated, designed, and supervised the study, and wrote the paper. All authors discussed the data and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Horiguchi, H., Kadomatsu, T., Yumoto, S. et al. Tumor cell-derived ANGPTL2 promotes β-catenin-driven intestinal tumorigenesis. Oncogene 41, 4028–4041 (2022). https://doi.org/10.1038/s41388-022-02405-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-022-02405-8
- Springer Nature Limited
This article is cited by
-
Tumor stroma-derived ANGPTL2 potentiates immune checkpoint inhibitor efficacy
Cancer Gene Therapy (2024)
-
ANGPTL2 promotes immune checkpoint inhibitor-related murine autoimmune myocarditis
Communications Biology (2023)