
Abstract
p110α is a catalytic subunit of phosphoinositide 3-kinase (PI3K), a major downstream effector of receptor tyrosine kinase ErbB2, that is amplified and overexpressed in 20–30% of breast cancers, 40% of which have an activating mutation in p110α. Despite the high frequency of PIK3CA gain-of-function mutations, their prognostic value is controversial. Here, we employ a knock-in transgenic strategy to restrict the expression of an activated form of ErbB2 and p110α kinase domain mutation (p110αHR) in the mammary epithelium. Physiological levels of transgene expression under the control of their endogenous promoters did not result in a major synergistic effect. However, tumors arising in ErbB2/p110αHR bi-genic strain metastasized to the lung with significantly reduced capacity compared to tumors expressing ErbB2 alone. The reduced metastasis was further associated with retention of the myoepithelial layer reminiscent of ductal carcinoma in situ (DCIS), a non-invasive stage of human breast cancer. Molecular and biochemical analyses revealed that these poorly metastatic tumors exhibited a significant decrease in phospho-myosin light chain 2 (MLC2) associated with cellular contractility and migration. Examination of human samples for MLC2 activity revealed a progressive increase in cellular contractility between non-invasive DCIS and invasive ductal carcinoma. Collectively, these data argue that p110αHR mutation attenuates metastatic behavior in the context of ErbB2-driven breast cancer.
Similar content being viewed by others

Introduction
Breast cancer accounts for ~15% of cancer-related deaths and over two million diagnoses every year, making it the most common cancer among women worldwide. In the clinic, breast cancer is still classified histologically into three classes: (1) HER2/ErbB2-positive (2) estrogen receptor (ER) or progesterone receptor (PR) positive, and (3) triple-negative [1, 2]. More recently, molecular classification of breast cancer has been developed allowing for the classification of additional subtypes at a transcriptomic level.
One important driving oncogene found to be mutated across all subtypes is phosphoinositide 3-kinase (PI3K), a lipid kinase whose activation leads to the phosphorylation of phosphatidylinositol 4,5-bisphosphate to phosphatidylinositol (3,4,5)-trisphosphate. PIP3 is an important phospholipid that allows for the activation of downstream signaling important in cell growth and survival [3,4,5]. The catalytic subunit of class I PI3K is comprised of four isoforms: p110α, p110β, p110γ, and p110δ. Of these isoforms, p110α and p110β are globally expressed while p110γ and p110δ are generally found in leukocytes [6,7,8]. ErbB2-positive tumors are characterized by the overexpression and/or amplification of this receptor and occur in 20–30% of breast cancers, forty percent of which display a mutation in PIK3CA, the gene encoding for the p110α catalytic subunit [9, 10]. Interestingly, mutations in p110α are clustered in three “hotspots”: two in the helical domain and one in the kinase domain. The helical domain mutations (E542K, E545K) render p110α independent of its regulatory subunit p85, and the kinase domain mutation (H1047R, p110αHR) is thought to make p110α independent of membrane tethering by rat sarcoma (Ras), both mechanisms leading to a gain-of-function phenotype [11,12,13]. However, the correlation between PIK3CA mutations and pathological parameters remains controversial among many studies, largely due to limited sample sizes, variable prognostic values among different hotspot mutations, and subtype heterogeneity [14]. Several studies have reported a good prognosis for breast cancer with p110αHR mutations [14,15,16,17] whereas other studies have indicated adverse patient outcomes, particularly due to its role in therapy resistance [18, 19].
To address the physiological role of p110αHR in the context of ErbB2-positive breast cancer, we generated a mouse model where p110αHR and NeuNT (activated rat ErbB2) are expressed in a heterozygous state and are driven by their endogenous promoters, mimicking how these mutations arise in breast cancer patients [20, 21]. Although combining both p110αHR and NeuNT alleles had little impact on tumor onset, we observed that mutant p110α was protective from lung metastasis. Analyses of mammary tumors and human breast cancer samples revealed that the mutant p110αHR allele promotes non-invasive pathology associated with ductal carcinoma in situ (DCIS), and impairment of myosin kinase light chain 2 (MLC2, MYL2) activity important for cell contractility and motility. Together these data argue that the p110αHR allele plays an anti-metastatic role in ErbB2-positive breast cancers.
Results
The p110αHR allele results in lower ErbB2 expression and impaired metastatic progression
Using a publicly available dataset (BRCA, TCGA, Firehose Legacy) [22, 23] we found that breast cancer patients with ErbB2-positive breast cancer by IHC have a wide range of mutations in p110α (PIK3CA). A large majority occurred at His 1047 in the kinase domain leading to a change to Arg or Leu (Fig. S1a). To further explore this clinical observation, we generated physiologically relevant mouse models to accurately recapitulate PIK3CA mutated ErbB2-positive breast cancer. Unlike overexpression systems, both knock-in mouse models are maintained in a heterozygous state and are driven by their endogenous promoters to mimic as closely as possible what occurs in the clinic. Wild-type exon 20 of the Pik3ca gene, which is flanked with loxP sites, is immediately followed by a mutated exon 20. In the presence of Cre recombinase expression driven by a mammary specific promoter, the mouse mammary tumor virus (MMTV) promoter, the loxP sites recombine and excise the wild-type exon, allowing for the expression of the mutated exon 20. In the NeuNT model, a neomycin stop cassette is inserted before NeuNT cDNA in the first exon of ErbB2 and is flanked by loxP sites. Expression of Cre recombinase will allow for excision of the neomycin cassette, thereby enabling NeuNT expression under its endogenous promoter regulation. Thus, every cell expressing Cre will result in the expression of p110αHR and/or NeuNT alleles (Fig. S1b–d).
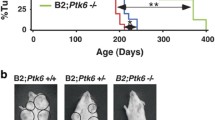
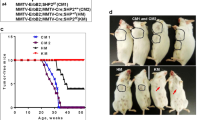
Although tumor onsets of bi-genic mice were comparable to animals with activation of p110α alone (averaged onset of 211 days vs. 237 days), the onset is significantly accelerated relative to the NeuNT monogenic strain (377 days) (Fig. 1a). In addition, there was an increase in tumor number per animal and a trend towards increased tumor burden in bi-genic mice (Fig. 1b). Corroborating this trend, we report higher levels of a proliferative marker Ki67 in tumors expressing p110αHR (Fig. S2a) but did not observe a difference in the presence of blood vessels stained by CD31 (Fig. S2b) or cell death assessed by TUNEL (Fig. S2c). Histological analyses of mammary tumors revealed considerable inter- and intra-tumoral heterogeneity (Fig. S3a–c). The NeuNT tumors mainly exhibited adenocarcinoma and Neu type histopathology. By contrast, the p110αHR tumors displayed adenosquamous and biphasic pathology. Interestingly, the majority of bi-genic tumors are classified as adenosquamous and Neu type tumors which were a combination of the most common pathologies from each monogenic strain (Fig. S3d-f).

a Tumor onset curve and table indicating tumor penetrance, T50, and average onset. P values were calculated between the NeuNT cohort and each of the other two cohorts. b Quantification of tumor number and total tumor burden for each genotype. c Immunoblot analysis for ErbB2 expression levels. d Quantification of ErbB2 signal, normalized to α-tubulin. Error bar SEM. e Representative H&E images of lung metastasis. Scale bar 400 μm. f Percentage of animals that developed lung metastasis.
Biochemical analyses revealed a reduction in ErbB2 protein levels in bi-genic tumors (Fig. 1c, d) in comparison to monogenic NeuNT tumors. There was no difference in copy number of NeuNT or ErbB2 allele between the two groups of tumors by quantitative PCR method (Fig. S4a). However, mRNA levels of NeuNT were significantly lower in the bi-genic tumors (Fig. S4b). Consistent with the bi-genic murine model, inquiry of a public dataset for ErbB2+ breast cancer revealed a lower trend in ErbB2 protein levels in tumors harboring a p110αHR mutation (Fig. S4c). Although bi-genic animals possessed a comparable tumor burden to monogenic mice when size was adjusted, no animal developed distant metastasis to the lungs whereas 27.3% of NeuNT mice developed lung metastasis (Fig. 1e, f). Together, our data indicate a modest synergistic effect between ErbB2 activation and p110αHR expression to promote larger tumor burdens, but surprisingly the p110αHR mutation decreases the incidence of lung metastasis.
Tumors with a p110αHR mutation maintain a non-invasive pathology
Breast tumors naturally progress in a stepwise manner, arising from a normal mammary duct with a basement membrane, a layer of myoepithelial cells (K14+), and a layer of luminal cells (K8/18+), to ductal hyperplasia, apical ductal hyperplasia and then DCIS. DCIS lesions often maintain the proper architecture with intact myoepithelial layer and luminal cells in the correct compartments. Eventually, selected DCIS lesions progress further to invasive ductal carcinoma (IDC), where the gradual loss of K14+ myoepithelial cells promotes local dissemination and distant metastases of cancer cells [24]. To further explore whether the diminished metastatic capacity of bi-genic tumors was due to retention of the myoepithelial barrier, we stained tumors for myoepithelial (K14) and luminal (K8/18) markers and found that the distribution of these cells is highly distinct between the two genotypes. The NeuNT tumors display K14+ cells that are also K8/18+ and disperse throughout the tumor nest whereas bi-genic tumors show K14+/K8/18− myoepithelial cells strictly outlining tumor areas and adjacent to surrounding stroma (Fig. 2a, b). This well-differentiated myoepithelial organization is very reminiscent of non-invasive DCIS stage, arguing for a potential role of p110αHR mutation in the myoepithelium which functions as a physical barrier to metastasis.

a Immunohistochemical staining of tumor differentiation markers, luminal keratin K8/18, and myoepithelial keratin K14. Normal duct shows typical keratin localization. Scale bar 50 μm. b Quantification of K8/18+ and K14+ cells. Error bar SEM. P values were calculated for each category of K14+ cells between the two genotypes while p values are insignificant for K8/18 analysis. c Representative images of tumors stained for E-cadherin and a cell membrane marker, Epcam. Scale bar 25 μm. d Membrane area was first generated based on Epcam signal using Imaris. Membrane signal of E-cadherin is determined as the area of colocalization (percentage of total area) and Pearson correlation coefficient. Error bar SEM. e Immunoblot analysis of E-cadherin levels.
Consistent with the non-invasive nature of bi-genic tumors, immunohistochemical analyses of E-cadherin and its association with a membrane marker Epcam (Epithelial cellular adhesion molecule) revealed a distinct distribution of E-Cadherin at adherens junction along the cell membrane, indicating stable cell-cell contact (Fig. 2c, d). In contrast, invasive NeuNT tumors exhibit diffuse cytoplasmic E-cadherin with reduced colocalization with the membrane Epcam signal. This different localization of E-cadherin is not due to an alteration in E-cadherin expression levels (Fig. 2e). Collectively, these analyses argue that the p110αHR mutation in the context of activated ErbB2 signaling results in retention of the myoepithelial cell layer and strong cell-cell adhesion, which in part account for the non-invasive behavior of these cells.
Phosphorylation of MLC2 correlates with metastatic potential in ErbB2-positive tumors
To further understand the pathways associated with each genotype, we utilized microarray analysis using total RNA extracted from bi-genic and NeuNT monogenic tumors. Due to the heterogeneous nature of our tumors, the number of genes that were found to be significantly (p < 0.05, fold change >1.5) upregulated or downregulated was 68 and 73 respectively (Fig. S5a). Despite the limited number of differential genes, using the EnrichR Jensen Compartments classification, one significantly upregulated pathway in our p110αHR/NeuNT/Cre tumors was found to associate with the myosin complex and activity (Fig. S5b, c). Consistent with murine results, analyses of a TCGA dataset of ErbB2-positive tumors harboring a p110αHR mutation revealed top enriched pathways such as calcium and RhoA signaling, including genes in myosin regulation (Fig. S5d). One integral protein of these pathways, MLC2, is phosphorylated and activated by kinases such as MLCK or ROCK in a Ca2+/calmodulin-dependent manner [25]. MLC2 regulates actin cytoskeleton dynamics and contractility important to the migratory capacity of metastatic cancer cells [25]. To explore whether phosphorylation of MLC2 was involved in the difference in metastatic behaviors due to the p110αHR allele, we performed immunohistochemical and immunoblot analyses using antibodies for phosphorylated MLC2 (Ser19) and total MLC2. Although the total levels of MLC2 were unchanged, MLC2 phosphorylation was significantly lower in bi-genic tumors (Fig. 3a–d). Consistent with murine data, analyses of TCGA mass spectrometry data from ErbB2-positive breast cancer revealed that the H1047R mutation also was associated with lower levels of MLC proteins (Fig. S5e).

a Representative images of IHC for total MLC2 and activated MLC2 (Ser19 phosphorylation). b Quantification of total MLC2 and p-MLC2 levels from images in Fig. 3a. P values were calculated using Student’s t-test for each category of p-MLC2-positive cells (low, medium, and high). There is no significant difference in total MLC2 levels. c Immunoblot analyses of total MLC2 and activated MLC2. β-actin is a loading control. d Quantification of total MLC2 and activated MLC2 levels, normalized to β-actin levels.
Although using public datasets to confirm the human relevance of a MLC2 signature is important, we were also interested in validating the results by IHC analyses of a panel of human breast cancer samples. The tumor cores comprising either DCIS or IDC were stained for ErbB2, total MLC2, phosphorylated MLC2, K8/18, and K14. Consistent with murine studies, we observed a significant increasing trend in MLC2 phosphorylation associated with progression into the invasive stage of IDC in both ErbB2-positive and ErbB2-negative cases (Fig. 4a, b). However, the levels of K8/18 or K14 were not significantly different between DCIS and IDC samples (Fig. 4c). Together, these results implicate this contractile signaling pathway in governing metastatic behavior of both murine and human ErbB2-positive breast cancer cells.

a A panel of human breast cancer samples are classified based on pathological stage of DCIS (ERBB2+ n = 2, ERBB2− n = 10) or IDC (ERBB2+ n = 28, ERBB2− n = 91) as well as ERBB2 positivity. Immunohistochemical analyses for markers including i. ERBB2 ii. p-MLC2 (Ser19) iii. K8/18 (luminal epithelial marker), iv. merge, v. total MLC2 (brown) and K14 (myoepithelial marker, green). b Quantification of p-MLC2 levels normalized to the area of total MLC2 positivity. c Quantification of a number of K14+ cells.
Discussion
Tumor classification has always been an efficient way to determine which patients receive what therapy based on one or few markers. However, this forgoes the issue of tumor heterogeneity and specific mutational status. Here we show that in the context of ErbB2 expression, a common mutation in the p110α subunit of PI3K results in suppression of ErbB2 metastatic phenotype. Originally this physiological p110αHR knock-in model was generated and characterized in the context of ovarian cancer where it was found that expression alone induced ovarian hyperplasia but PTEN loss was necessary to cause the formation of tumors [21]. In the context of mammary tumorigenesis, transgenic models utilizing various promoters to drive p110αHR mutation expression developed low histological grade tumors that rarely metastasize [26]. However, loss of p53 function has been shown to synergize with p110αHR to promote highly invasive tumors with increased metastatic potential [27]. Consistent with these studies, we found that mammary epithelial expression of this mutation results in poorly metastatic tumors, even in the context of ErbB2-positive cancer (Fig. 1e, f). One possible explanation for the decreased metastatic potential is the reduced levels of NeuNT oncogene despite similar levels of NeuNT gene amplification (Fig. 1c, d, S4a, b). The suppression of ErbB2/MAPK signaling axis may in turn impact the migratory and invasive properties of these bi-genic tumor cells. Consistent with this hypothesis, inhibition of this signaling has been shown to inhibit ErbB2-mediated metastatic progression [28].
Another possible explanation for the reduced metastatic potential is that, unlike the parental NeuNT tumors, the bi-genic tumors maintain a non-invasive DCIS pathology associated with intact myoepithelial layers (Fig. 2a, b). Indeed, mammary tumors driven by p110αHR alone acquire predominantly adenomyoepithelioma and adenosquamous carcinoma pathology, both of which exhibit intact K14+ or K5+ myoepithelial layers with rare distant metastasis [27, 29]. Our results are in line with the extensive literature supporting the repressive role of myoepithelial cells in metastatic dissemination of human breast cancer cells. These observations argue that the H1047R mutation is a key determinant of tumor differentiation that dictates metastatic behaviors in the context of ErbB2-mediated tumorigenesis.
Another commonality between human and mouse DCIS is the correlation between MLC2 activity and invasive progression. Indeed, ErbB2-negative and ErbB2-positive DCIS exhibit no or much lower phosphorylation levels of MLC2 whereas invasive breast cancer possesses much higher activity of this key contractile protein (Figs. 3–4). Although the molecular mechanism underlying this phenomenon remains to be determined, other studies have argued for a role of calcium/RhoA-mediated myosin signaling and activity in metastatic processes [25]. Indeed, a study transfecting benign rat mammary tumors with a calcium ion-binding protein p9Ka resulted in increased metastasis [30]. This supports our observation that activation of myosin signaling correlates with metastatic potential.
The clinical implication of PIK3CA mutations and other pathological parameters remain inconsistent among many clinical studies, without a clear consensus. Unlike helical domain mutations, the kinase domain mutation H1047R is found to be generally associated with good prognosis, including lymph node negativity [31] and prolonged disease-free survival [14]. In the particular context of ErbB2-positive breast cancer, the H1047R mutation presents a favorable prognostic value with low tumor grade and better metastasis-free survival when ErbB2-positive cancer patients were not given ErbB2-targeted therapies [15]. However, the H1047R and other PI3K mutations are also shown to confer resistance to ER and ErbB2-targeted therapies (trastuzumab and lapatinib), ultimately leading to poor response for these patients [18, 19]. It is possible that p110αHR determines key malignant and metastatic properties of breast cancer, including less invasive behaviors that contribute to better metastasis-free survival. However, like other PI3K mutations, this mutation also confers resistance to therapies, attributing to worse outcomes in those patients. It is also possible that under the selective pressure of ErbB2 therapies, this mutation can engage new functions that exacerbate the metastatic burden. These observations clearly illustrate the distinct roles of the H1047R mutation that seem opposing in different contexts of metastasis or therapy. Given the clinical importance of this observation, our mouse model will be instrumental to the development of therapies more adapted to patients with a p110αHR mutation.
Materials and methods
Animal husbandry
All strains were maintained on an FVB/N background. The strains used were: p110αHR [21], NeuNT [20], MMTV-Cre [20]. Animals were housed at the Rosalind and Goodman Cancer Research Institute (GCRI) animal facility in accordance with McGill University animal ethics guidelines. Only nulliparous female animals were used in this study.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Simond AM, Muller WJ. In vivo modeling of the EGFR family in breast cancer progression and therapeutic approaches. Adv Cancer Res. 2020;147:189–228.
Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000;100:603–6.
Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–75.
Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253:239–54.
Carpenter CL, Duckworth BC, Auger KR, Cohen B, Schaffhausen BS, Cantley LC. Purification and characterization of phosphoinositide 3-kinase from rat liver. J Biol Chem. 1990;265:19704–11.
Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272:19236–41.
Hiles ID, Otsu M, Volinia S, Fry MJ, Gout I, Dhand R, et al. Phosphatidylinositol 3-kinase: structure and expression of the 110 kd catalytic subunit. Cell. 1992;70:419–29.
Andrulis IL, Bull SB, Blackstein ME, Sutherland D, Mak C, Sidlofsky S, et al. neu/erbB-2 amplification identifies a poor-prognosis group of women with node-negative breast cancer. Toronto Breast Cancer Study Group. J Clin Oncol. 1998;16:1340–9.
Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Disco. 2014;13:140–56.
Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–8.
Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239–42.
Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27:5486–96.
Barbareschi M, Buttitta F, Felicioni L, Cotrupi S, Barassi F, Del Grammastro M, et al. Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas. Clin Cancer Res. 2007;13:6064–9.
Cizkova M, Susini A, Vacher S, Cizeron-Clairac G, Andrieu C, Driouch K, et al. PIK3CA mutation impact on survival in breast cancer patients and in ERalpha, PR and ERBB2-based subgroups. Breast Cancer Res. 2012;14:R28.
Dumont AG, Dumont SN, Trent JC. The favorable impact of PIK3CA mutations on survival: an analysis of 2587 patients with breast cancer. Chin J Cancer. 2012;31:327–34.
Shimoi T, Hamada A, Yamagishi M, Hirai M, Yoshida M, Nishikawa T, et al. PIK3CA mutation profiling in patients with breast cancer, using a highly sensitive detection system. Cancer Sci. 2018;109:2558–66.
Cizkova M, Dujaric ME, Lehmann-Che J, Scott V, Tembo O, Asselain B, et al. Outcome impact of PIK3CA mutations in HER2-positive breast cancer patients treated with trastuzumab. Br J Cancer. 2013;108:1807–9.
Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68:9221–30.
Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, Muller WJ. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc Natl Acad Sci USA. 2000;97:3444–9.
Kinross KM, Montgomery KG, Kleinschmidt M, Waring P, Ivetac I, Tikoo A, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122:553–7.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Pandey PR, Saidou J, Watabe K. Role of myoepithelial cells in breast tumor progression. Front Biosci (Landmark Ed). 2010;15:226–36.
Li YR, Yang WX. Myosins as fundamental components during tumorigenesis: diverse and indispensable. Oncotarget. 2016;7:46785–812.
Koren S, Bentires-Alj M. Mouse models of PIK3CA mutations: one mutation initiates heterogeneous mammary tumors. FEBS J. 2013;280:2758–65.
Adams JR, Xu K, Liu JC, Agamez NM, Loch AJ, Wong RG, et al. Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Res. 2011;71:2706–17.
Julien SG, Dube N, Read M, Penney J, Paquet M, Han Y, et al. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat Genet. 2007;39:338–46.
Meyer DS, Brinkhaus H, Muller U, Muller M, Cardiff RD, Bentires-Alj M. Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Res. 2011;71:4344–51.
Davies BR, Davies MP, Gibbs FE, Barraclough R, Rudland PS. Induction of the metastatic phenotype by transfection of a benign rat mammary epithelial cell line with the gene for p9Ka, a rat calcium-binding protein, but not with the oncogene EJ-ras-1. Oncogene. 1993;8:999–1008.
Kalinsky K, Jacks LM, Heguy A, Patil S, Drobnjak M, Bhanot UK, et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res. 2009;15:5049–59.
Acknowledgements
This work was supported by a Canada Research Councils Chair in Molecular Oncology (950-2310-33), and a Foundation award from the Canadian Institutes of Health Research (CIHR-FDN-148373) (all to W.J.M.).
Author information
Authors and Affiliations
Contributions
Conceptualization: AMS, TR, WJM; investigation: AMS, TB, VS, DM; analysis: AMS, TB, RDC; resources: WAP; writing: AMS, TB, WJM; review and editing: AMS, TB, WAP, WJM; visualization: AMS, TB; funding acquisition: WJM; supervision: WJM.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Simond, A.M., Bui, T., Zuo, D. et al. Physiological expression of PI3K H1047R mutation reveals its anti-metastatic potential in ErbB2-driven breast cancer. Oncogene 41, 3445–3451 (2022). https://doi.org/10.1038/s41388-022-02323-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-022-02323-9
- Springer Nature Limited