
Abstract
Small cell lung cancer (SCLC) continues to cause poor clinical outcomes due to limited advances in sustained treatments for rapid cancer cell proliferation and progression. The transcriptional factor Forkhead Box M1 (FOXM1) regulates cell proliferation, tumor initiation, and progression in multiple cancer types. However, its biological function and clinical significance in SCLC remain unestablished. Analysis of the Cancer Cell Line Encyclopedia and SCLC datasets in the present study disclosed significant upregulation of FOXM1 mRNA in SCLC cell lines and tissues. Gene set enrichment analysis (GSEA) revealed that FOXM1 is positively correlated with pathways regulating cell proliferation and DNA damage repair, as evident from sensitization of FOXM1-depleted SCLC cells to chemotherapy. Furthermore, Foxm1 knockout inhibited SCLC formation in the Rb1fl/flTrp53fl/flMycLSL/LSL (RPM) mouse model associated with increased levels of neuroendocrine markers, Ascl1 and Cgrp, and decrease in Yap1. Consistently, FOXM1 depletion in NCI-H1688 SCLC cells reduced migration and enhanced apoptosis and sensitivity to cisplatin and etoposide. SCLC with high FOXM1 expression (N = 30, 57.7%) was significantly correlated with advanced clinical stage, extrathoracic metastases, and decrease in overall survival (OS), compared with the low-FOXM1 group (7.90 vs. 12.46 months). Moreover, the high-FOXM1 group showed shorter progression-free survival after standard chemotherapy, compared with the low-FOXM1 group (3.90 vs. 8.69 months). Our collective findings support the utility of FOXM1 as a prognostic biomarker and potential molecular target for SCLC.
Similar content being viewed by others

Introduction
Small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) are two major lung cancer histotypes that constitute the leading causes of death among all malignancies worldwide [1]. SCLC is an aggressive cancer type characterized by a short doubling time, high genomic instability, early metastatic spread, and rapid chemotherapy resistance [2, 3]. While the majority of SCLC (60–80%) cases are initially responsive to frontline chemotherapy with etoposide plus platinum drugs (cisplatin and carboplatin), high rates of cancer recurrence and resistance within a few months are observed in most patients at all stages after chemotherapy [4,5,6]. The lack of targeted therapy and effective second-line drugs for chemoresistant SCLC underlies poor prognosis. Median overall survival (OS) time of patients with advanced NSCLC harboring epidermal growth factor receptor (EGFR) or ALK gene mutations reached over 3–5 years in clinical trials [7, 8]. In contrast, <5% extensive-stage SCLC patients survived for more than 5 years [9]. Fundamental progress in SCLC treatment has been far lower compared to NSCLC therapy over the past few decades.
Genetic studies have shown that loss-of-function mutations in tumor suppressor genes TP53 and retinoblastoma 1 (RB1) are the most frequently occurring alterations in SCLC [2]. The p53 tumor suppressor responds to DNA damage and hypoxia to maintain genomic stability while RB1 protein mainly regulates cell cycle progression and cellular differentiation. Loss-of-function mutations in RB1 and TP53 genes deregulate the cell cycle, leading to abnormal cell proliferation, accumulation of mutations, and oncogenesis [10]. For instance, conditional deletion of TP53 and RB1 in lung neuroendocrine cells is reported to induce deregulation of pulmonary neuroendocrine stem cell self-renewal and cell cycle regulatory genes, resulting in SCLC development in Rb1fl/flTrp53fl/fl (RP) and Rb1fl/flTrp53fl/flMycLSL/LSL (RPM) mouse models [11, 12]. However, the molecular basis of carcinogenesis of SCLC originating from variant neuroendocrine cells remains unclear.
Forkhead Box M1 (FOXM1) is a proliferation-specific transcription factor expressed in all proliferating cell types, including tumor cell lines [13]. We and others previously demonstrated that FOXM1 plays a critical role in cell proliferation by directly regulating the transcriptional network essential for G1/S and G2/M progression, in particular, positive regulation of cell cycle genes, including cyclin B1, cdc25B, Aurora B, polo-like kinase 1 (Plk-1), Survivin, and Cenp A, B and F isoforms [14, 15]. Increased FOXM1 expression in multiple human cancer types has been reported, including breast cancer, NSCLC, and gastric cancer. Moreover, overexpression of FOXM1 is associated with adverse clinical outcomes, including survival and chemoresistance [16,17,18]. Accumulating reports suggest that inactivation of TP53 and RB1 induce elevated FOXM1 expression and activity, and consequently, tumor proliferation and metastases [19, 20]. However, limited studies to date have focused on the role of FOXM1 in SCLC with loss of TP53 and RB1 function. Here, we have focused on the biological function of FOXM1 in SCLC and potential associations among FOXM1 expression, drug resistance, and clinical outcomes.
Results
FOXM1 expression is elevated in small cell lung cancer cells in vitro and in vivo
While FOXM1 regulates cell proliferation and is correlated to many cancer types, its mechanisms of action in SCLC, a highly aggressive type of lung cancer characterized by rapid proliferation, are not completely understood at present. Initial analysis of the RNA-seq dataset in Cancer Cell Line Encyclopedia (CCLE) revealed FOXM1 mRNA level (median log2 value) of 5.98 (interquartile range (IQR): 5.27–6.34) in SCLC cell lines, which was significantly higher than that in NSCLC (5.43; IQR: 4.87–5.85; P < 0.001) and other cancer cell lines (5.17; IQR: 4.62–5.65; P < 0.001) (Fig. 1A). Moreover, SCLC samples from a Japanese population (GSE43346 dataset) showed significantly higher expression of FOXM1 mRNA (median GeneChip score 133.70 [IQR: 88.00–199.00]) than normal tissues, including normal lung (median GeneChip score 6.30 [IQR: 1.80–23.75]) (P < 0.001) (Fig. 1B). Consistently, in the SCLC cohort dataset, GSE60052, containing 79 SCLC and 7 normal lung samples, the FOXM1 mRNA level (median log2 value 7.69 [IQR: 7.10–8.11]) was significantly higher than that in normal lung (median log2 value 5.71 [IQR: 4.71–6.06]; P < 0.001) (Fig. 1C). These results suggest that FOXM1 is correlated with rapid proliferation in highly aggressive SCLC.

FOXM1 mRNA levels are significantly elevated in SCLC cell lines from Cancer Cell Line Encyclopedia (CCLE) (A) and SCLC patient tumors from GSE43346 (B) and GSE60052 (C) datasets. Box plots represent the median and interquartile range of FOXM1 mRNA levels in SCLC and non-SCLC groups.
FOXM1 is correlated with cell proliferation and DNA repair gene expression in SCLC tumors
We compared the mRNA expression profiles in SCLC and normal lung tissue samples of the GSE60052 dataset. The heat map depicted the mRNA levels of FOXM1 and genes involved in cell proliferation, anti-apoptosis, and neuroendocrine markers that were significantly correlated with SCLC (P value <0.05) (Fig. 2A). Neuroendocrine markers were positively correlated with SCLC while Notch expression was decreased. High FOXM1 expression in SCLC was positively correlated with genes involved in response to DNA damage, including double-strand break (DSB) repair, base-excision repair (BER), and mismatch repair (MMR) (Fig. 2A). To identify the master regulators of SCLC-associated genes, a total of 2196 differentially expressed genes (DEG) between control and SCLC patients from GSE60052 (fold change >1.5 and P value <0.05) were applied for transcription factor enrichment analysis. Both web-based Enrichr and ChEA3 transcription factor enrichment analysis (TFEA) prioritized FOXM1 as a top ranking putative transcription factor based on SCLC-related differentially expressed genes (Fig. 2B, C). To further explore the genes involved in crucial biological processes of SCLC, we performed PANTHER of statistical Gene Ontology (GO) enrichment analysis for biological processes (Fig. 2D). The top two major functional groups, specifically, cellular process (GO:0009987) and biological regulation (GO:0065007), contain the top 15 enriched GO biological processes mostly involved in regulating mitotic cell cycle progression and cell division (Fig. 2E). GO enrichment analysis showed that SCLC tumors are positively correlated with genes that regulate DNA replication and damage responses and negatively with those involved in the regulation of epithelial cell apoptosis and adherens junction (Fig. 2F). Consistently, increased transcription of FOXM1, BIRC5, and NEUROD1 was detected in SCLC relative to normal tissue after normalization, while YAP1 and MYC mRNA levels were downregulated (Fig. 2A, G).

A Heat map of expression patterns of FOXM1 and selected genes correlated with human SCLC tumors from patients (GSE60052) (p value <0.05). B, C Enrichr (B) and ChEA3 (C) transcription factor enrichment analysis for identification of putative transcription factors based on SCLC-related differentially expressed genes (DEGs) from the GSE60052 dataset (fold change >1.5 and p value <0.05). D Pie chart showing the distribution of functional groups of the 2196 DEGs classified using PANTHER of Gene Ontology (GO) biological processes. E Statistical GO enrichment analysis of the two major functional groups (D) (GO:0009987 and GO:0065007) containing the top enriched GO biological processes. F GO enrichment analysis showing that SCLC is significantly positively correlated with DNA replication, DNA damage repair, apoptosis, and cell adherens junctions. Nominal p value (NOM) < 0.05. Abbreviations: NES normalized enrichment score, FDR false discovery rate. G mRNA expression data extracted from GSE60052 used to determine levels of FOXM1 and neuroendocrine-related genes between SCLC and normal tissues after normalization. Data are presented as mean ± s.d. *p < 0.05; **p < 0.01 and ***p < 0.001.
FOXM1 is essential for SCLC tumor growth
The Rb1fl/flTrp53fl/flMycLSL/LSL (RPM) mouse model recapitulates the formation of human SCLC showing loss of function of both RB1 and TP53 genes and frequent amplification of the MYC gene [12]. To ascertain whether Foxm1 is critical for SCLC initiation and development, wild-type (WT), RPM, and RPM:Foxm1fl/fl mouse lungs were intratracheally infected with Cre-expressing adenovirus (Ad-Cre). Computational reconstruction of 3D mouse lungs based on lung microCT images revealed that the RPM mice had developed nodules relative to RPM:Foxm1-/- and WT mice at 7 weeks post infection (Fig. 3A and Supplementary Figs. S1–S2). Foxm1 depletion in RPM:Foxm1-/- mouse lungs was determined via immunoblotting (Fig. 3B). Aggressive lung tumors were detected in the central regions of RPM mouse lungs. In contrast, no tumor was evident in RPM:Foxm1-/- lungs (Fig. 3C and Supplementary Fig. S2). Gross nodule/tumor volume was significantly decreased in RPM:Foxm1-/- lungs, compared with RPM and WT lungs, as determined by microCT imaging quantification (Fig. 3D and Supplementary Fig. S2).

A Seven weeks after intratracheal (I.T.) injection of one dose of 1 × 106 Ad-Cre into RPM and RPM:Foxm1fl/fl mice. Lung tumors are presented in the 3D-reconstituted lung from microCT images. Deletion of Foxm1 prevented RPM-mediated small cell lung tumorigenesis. B Immunoblotting results indicate that Foxm1 was depleted in the RPM:Foxm1-/- mouse lung tissue. C Lung tumors are indicated with arrows in transversal mouse lung microCT slides. D Gross nodule volume in mouse lung determined from microCT images of RPM mice (n = 10), RPM:Foxm1-/- mice (n = 7) and wild-type (WT) mice (n = 2) was quantitated and represented in a Dot Plot. *p < 0.05; **p < 0.01.
Foxm1 regulates tumor growth and neuroendocrine differentiation in SCLC
Tumors were not detected in RPM:Foxm1-/- and WT mouse lung sections subjected to H&E staining, indicating that depletion of Foxm1 suppresses RPM-induced tumor formation (Fig. 4A). Moreover, we observed a significant decrease in the number of Ki67 and pH3-positive cells in RPM:Foxm1-/- lungs compared with RPM tumors, suggesting that Foxm1 is essential for rapid tumor cell proliferation and aggressiveness of SCLC. Levels of the neuroendocrine marker, calcitonin gene-related peptide (CGRP), lung epithelial cell marker, Ttf1, and Sox2, were determined in RPM tumors (Fig. 4A) [12, 21]. The ratio changes of these proteins were significantly decreased in RPM:Foxm1-/- lungs based on IHC intensity semi-quantification (Fig. 4A and Supplementary Fig. S3). In addition to a regular expression in airway epithelium, the majority of Cgrp was detected in the early-stage nodules or smaller tumors of RPM lungs (Supplementary Fig. S4B–E). The mRNA expression levels of Foxm1, c-Myc, Survivin (Birc5), Ascl1, Cgrp, and Sox2 were significantly decreased in Foxm1-depleted RPM mouse lung whereas Yap1 and Tft1 were significantly increased compared with RPM (Fig. 4B). These RNA expression data supported previous research that FOXM1 directly regulated c-Myc [22], Survivin [14, 16], and SOX2 [23, 24] in cell lines. Furthermore, we analyzed mouse Cgrp promoter and found a potential Foxm1 binding site at -1316~-1302 region (Supplementary Fig. S5A). The 1.4 kb length mouse Cgrp promoter (−1385 to +87) was PCR cloned into the pGL3 basic vector for dual luciferase assay. Co-transfection experiment showed that human GFP-FOXM1 significantly stimulated 1.4 kb mouse Crgp promoter activity in both HEK-293T and NCI-H1688 cells (Supplementary Fig. S5B, C), suggesting that Foxm1 regulates mouse Cgrp gene promoter. Our collective data suggest that Foxm1 regulates cell proliferation, survival, and neuroendocrine differentiation of SCLC.

Paraffin sections of lungs from WT, RPM and RPM:Foxm1-/- mice stained with hematoxylin and eosin (H&E) (A) or used for immunohistochemistry. Slides were counterstained with Nuclear Fast Red (red nuclei); magnification: 200X and 400X (inset). IHC staining intensity levels were semi-quantified and normalized. The ratio changes of IHC intensities were shown in the bar graphs. B Total RNAs isolated from WT, RPM, and RPM:Foxm1-/- mouse lung samples were subjected to quantitative reverse transcription PCR (qRT-PCR) to determine mRNA levels of SCLC-correlated genes. The error bars represent ± s.d. in triplicate experiments. *p < 0.05; **p < 0.01 and ***p < 0.001.
Inhibition of FOXM1 sensitizes cisplatin and etoposide-induced cytotoxicity in SCLC cells
The development of drug resistance to frontline chemotherapy is a major limitation in SCLC treatment. FOXM1 has been characterized as a transcriptional regulator of DNA repair genes, facilitating resistance to doxorubicin and paclitaxel (Taxol) cytotoxicity [25, 26]. To determine whether high FOXM1 expression in SCLC affects the efficacy of etoposide/cisplatin, NCI-H1688 pIND-shFOXM1 stable cells were treated with doxycycline (DOX). Successful FOXM1 depletion via shFOXM1 RNA interference was confirmed via Western blot (Fig. 5A). Depletion of FOXM1 in NCI-H1688 cells (pIND-shFOXM1; +DOX) led to a significant increase in apoptosis ratio (Fig. 5B, C) and sensitization to cisplatin, determined as a decrease in IC50 from 3314 nM to 2199 nM (P = 0.032), compared with non-DOX treated (-DOX) or control shRNA-expressing cells (pIND-shCtrl; +DOX; Fig. 5D). Similarly, depletion of FOXM1 sensitized cells to etoposide, as evident by the decrease in IC50 from 1479 nM to 696 nM (P = 0.006) (Fig. 5E). Consistently, siRNA-mediated FOXM1 depletion in NCI-H1688 cells caused significantly decreased IC50 of both cisplatin (Supplementary Fig. S6A and C) and etoposide (Supplementary Fig. S6B and D) as determined by clonogenic growth assays. Additionally, three FOXM1 inhibitors, Honokiol, Thiostrepton, and RCM-1, caused decreased expression of FOXM1 protein and its downstream target genes BIRC5, Cyclin B1, and Aurora B mRNA levels in NCI-H1688 cells (Supplementary Fig. S7A–D), leading to synergistic cytotoxicity of cisplatin or etoposide in the combined treatment (Supplementary Fig. S7E–J). The finding that suppression of FOXM1 sensitizes cells to cisplatin and etoposide in SCLC NCI-H1688 cells supports the theory that FOXM1 regulates the mechanisms underlying resistance to standard etoposide and cisplatin regimens. Moreover, siRNA-mediated suppression of FOXM1 led to significantly reduced NCI-H1688 cell migration on culture plates in the wound healing assay (Fig. 5F–G) and anchorage-independent growth on soft agar (Fig. 5H–I). Consistently, the GO analysis for the RNA-seq results from the FOXM1-depleted NCI-H1688 groups (GSE174462) also revealed that the differential expression genes (DEGs) were enriched in cell adhesion, cell division, and migration (Supplementary Fig. S8A–C).

A Western blot of FOXM1 protein from NCI-H1688-pIND stable cell lines treated with doxycycline (DOX) for 48 h or left untreated. Diminished FOXM1 expression was detected in shFOXM1-expressing cells (shFOXM1 + DOX). B Apoptotic H1688 cells (red) were detected with the TUNEL assay after FOXM1 depletion with inducible shFOXM1. Cell nuclei were stained with DAPI (blue). C Quantification of apoptosis in cells 48 h after treatment with DOX-inducible shRNA or left untreated. Significant increase in the TUNEL-positive cell ratio was detected in FOXM1-depleted NCI-H1688 cells. D, E IC50 curves of FOXM1-depleted NCI-H1688 cells treated with cisplatin (D) and etoposide (E) for 48 h. Percentage cell survival from MTT assays plotted against the logarithm of treatment concentrations. IC50 values for cisplatin or etoposide in FOXM1-depleted cells (shFOXM1 + DOX) were significantly decreased, compared with control groups (shCtrl + DOX or shFOXM1). F NCI-H1688 cells were transfected with siRNA and grown to confluence before generation of scratch wounds. Phase-contrast micrographs of scratch wounds at the time of wounding (0 h, between two dashed yellow lines) and 18 h post wounding. G Percentage wound closure presented as a bar graph. H siRNA-treated NCI-H1688 cells were trypsinized and replated in soft agar. Microphotographs showing colonies after 12 days of soft agar culture. I Numbers of colonies per microscopic field. Error bars represent ± s.d. of experiments conducted in triplicate. *p < 0.05; **p < 0.01; ***p < 0.001.
Baseline characteristics of SCLC patients
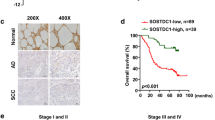
Elevated FOXM1 levels in cancer are associated with dysregulation of cellular proliferative capability and tumor aggressiveness [27, 28]. In 52 eligible SCLC patients with adequate tumor tissues at National Taiwan University Hospital Hsinchu Branch (NTUH-HC), we investigated whether high expression of FOXM1 is correlated with clinical outcomes. Nuclear localization of FOXM1 was specifically detected in SCLC tumor cells but not adjacent normal lung tissue (Fig. 6A). Overall, 30 patients (57.7%) were classified as high FOXM1 expression (grades 2+ and 3+) and 22 patients (42.3%) as low FOXM1 expression (grades 0 and 1+) for analysis (Fig. 6A, B; Table 1). TTF-1 staining was detected in the FOXM1 grade 0 SCLC samples, indicating that low or non-FOXM1 staining was not caused by failure of IHC or poor quality of the specimen. Interestingly, TTF-1 was not always detected in the SCLC sample categorized as FOXM1 expression grade 3+ (Fig. 6A).

A H&E staining (upper panel) and immunohistochemistry (IHC) for TTF-1 and FOXM1 with Nuclear Fast Red counterstain (middle and lower panel). FOXM1 IHC results were evaluated and graded based on unclear staining intensity by pathologists. Selected images represent IHC staining scores, classified as 0, 1+, 2+, and 3+. FOXM1 protein was detected in the nuclei of SCLC, but not adjacent normal tissue. Magnification: 200X; Inset 40X. B Flow chart represents SCLC patient enrollment, classification and grouping.
The 52 patients had a median age of 67.3 years (range, 40–90 years), and included 45 men (86.5%) and 49 smokers (94.2%). Two patients were diagnosed with stage I/II disease (3.8%) and 17 (32.7%) and 33 patients (63.5%) as stage III and IV, respectively, including 17 (32.7%) with bone metastasis, 11 (21.2%) with liver metastasis, and 6 (11.5%) with brain metastasis. The clinical cancer stages at diagnosis and extrathoracic metastases, particularly bone metastasis, were significantly different between the high and low-FOXM1 patient groups. However, we observed no marked differences in clinical characteristics between groups in terms of age, gender, or smoking status (Table 1).
High FOXM1 expression is associated with poor clinical outcomes of SCLC patients
Median OS of the 52 enrolled SCLC patients (stage I-IV) was 10.20 (95% confidence interval [CI]: 7.02–13.38) months. Notably, median OS of the high-FOXM1 SCLC group was significantly shorter than that of the low-FOXM1 group (7.90 [95% CI: 4.51–11.30] vs. 12.46 [95% CI: 9.07–15.85] months, hazard ratio [HR]: 2.07; 95% CI: 1.08–3.97; P = 0.029) (Fig. 7A). Within our patient group, 40 received standard chemotherapy (etoposide plus platinum drugs) and were regularly followed up at our hospital. Among the 21 patients receiving chemotherapy in the high-FOXM1 group, median PFS was 3.90 (95% CI: 3.22–4.59) months, while that of the other 19 patients in the low-FOXM1 group was 8.69 (95% CI: 7.10–10.27) months (HR: 2.31; 95% CI: 1.12–4.80; P = 0.024) (Fig. 7B). The finding that high FOXM1 expression in SCLC is significantly correlated to poor outcomes highlights a potential function as a critical regulator of genes contributing to cell proliferation, anti-apoptosis, migration, chemoresistance, and metastasis in SCLC.

A SCLC patients with high FOXM1 levels (dashed red line) were associated with significantly higher risk than patients with low FOXM1 levels (solid blue line) in OS. B SCLC patients with low FOXM1 expression (solid blue line) presented a trend of significantly longer PFS, compared with the high-FOXM1 group (dashed red line).
Discussion
SCLC is one of the most aggressive cancer types characterized by rapid proliferation and high recurrence rates after chemotherapy. Here, we showed that SCLC expresses higher Forkhead box M1 (FOXM1) levels than other human cancer cell lines and normal tissues. Our initial experiments disclosed that high-FOXM1 SCLC patients display 4.56-month shorter in median OS and 4.8-month shorter in median PFS than the low-FOXM1 group receiving etoposide plus platinum-based chemotherapy. GO analysis of the SCLC dataset showed that the FOXM1 level is associated with cell growth, anti-apoptosis, migration, metastasis, and DNA damage repair, supporting a correlation with poor prognosis. Diminished FOXM1 expression led to reduced SCLC cell migration and anchorage-independent growth (Fig. 5F–I), as reported previously [29], and increase in SCLC cell sensitivity to the DNA-damaging chemotherapeutic agents, cisplatin and etoposide, consistent with data from GO analysis showing that FOXM1 is correlated with assembly of SCLC cell adherens junction and DNA damage repair (Figs. 5 and 2F). Moreover, genetic deletion of Foxm1 in RPM mouse lung inhibited SCLC tumor initiation and proliferation and reduced the expression of neuroendocrine markers detected in SCLC patients. These results concur with the Enrichr and ChEA3 TFEA prediction that FOXM1 is a master regulator of gene transcription networks for SCLC carcinogenesis and further support the finding that FOXM1 regulates the transcription network of cell cycle, migration, tumorigenesis, epithelial-to-mesenchymal transition (EMT), and cancer stemness genes in other cancer types (Fig. 2) [14, 29,30,31]. In view of the collective data, we conclude that FOXM1 is a critical regulator of SCLC development and progression, and a prognostic marker for poor outcomes.
The neurogenic transcription factor, ASCL1, mediates distinct molecular signatures of neuroendocrine tumor formation and heterogeneity of SCLC [32]. In contrast to ASCL1-high and NeuroD1-high SCLC subtypes, YAP1 is expressed predominantly in a non-neuroendocrine (NE) SCLC subtype associated with intact RB [33, 34]. Another recent study suggests that Yap1 is dynamically temporally elevated to facilitate the evolution of an invasive subtype of SCLC from a NE to non-NE state in a Myc-driven manner [35]. In our experiments, depletion of Foxm1 in RPM mouse lung led to significant suppression of Ascl1, Cgrp, and c-Myc while Yap1 mRNA was markedly decreased in RPM lung, compared with WT lung, consistent with the expression pattern in the SCLC patient dataset (Figs. 4B and 2A) [36]. Furthermore, suppression of Yap1 in RPM lung was rescued after deletion of Foxm1 (RPM:Foxm1-/-), which exhibited lower numbers and sizes of SCLC tumors (Fig. 4B). The data suggest that Foxm1 regulates the initiation of SCLC and neuroendocrine-associated differentiation of the SCLC subtype in RPM mouse lungs.
Previous studies by our group on an inducible oncogenic KrasG12D mouse lung cancer model with SPC-Cre/Foxm1fl/fl mice demonstrated that Foxm1-deficient lung epithelial cells fail to develop into lung adenocarcinoma due to blockade of oncogenic signaling pathways [37]. In contrast to most lung cancer types, which are driven by specific oncogenes, SCLC tumors generally display functional inactivation of both TP53 and RB1 tumor suppressor genes. The mechanisms by which loss of tumor suppressors drive cancer initiation are still unclear. FOXM1 expression and activity are known to be inhibited by the tumor suppressor genes TP53 and RB1 [28]. Loss of TP53 and RB1 function in SCLC may thus result in remarkable elevation of FOXM1, compared with the other cancer types (Fig. 1A). Furthermore, amplification of the Myc family has been reported in ~20% SCLC cases in association with increased tumor cell survival [2, 38]. FOXM1 and c-Myc proteins mutually transactivate each other’s promoters, giving rise to a positive feedback loop stimulating gene expression and cell proliferation [22]. Inactivation of tumor suppressor TP53 and RB1 genes or additional c-Myc gene amplification could thus affect gene expression of FOXM1, contributing to rapid cancer cell proliferation and high levels of aggressive phenotypes in SCLC.
The SCLC tumors rapidly progressed to advanced stage about 7 weeks post Ad-Cre intratracheal (I.T.) injection to RPM mouse lungs, which displayed high levels of Foxm1 as well as c-Myc mRNA that could be transcribed from both endogenous c-MYC gene and the CAG promoter-driven transgenic MYC-T58A (Fig. 4B). In contrast, conditional deletion of Foxm1 (RPM: Foxm1-/-) inhibited SCLC development, which was associated with a decrease of c-MYC expression. Consistently, depletion of FOXM1 in NCI-H1688, an advanced stage of SCLC cells that were isolated from liver metastasis, also caused a decrease in c-MYC expression (Supplementary Fig. S8). Both in vivo mouse model and in vitro cell culture experiments reinforced the concept that FOXM1 positively regulates c-Myc in SCLC (Fig. 4B).
Of the GSE60052 dataset that we analyzed, interestingly, we found only 4 of 99 SCLC patients (4%) were diagnosed with stage IV disease [36], and the mean MYC level was significantly lower in tumors than normal tissues in the SCLC dataset (Fig. 2G). Since the MYC expression is more correlated to the advanced stage of SCLC [12], therefore, the mean expression level of MYC in tumor depends on the composition of cancer stages in the SCLC dataset. Even though most SCLC patients in this dataset were in the earlier stages of cancer, a high FOXM1 level was still detected in SCLC when compared with the tumor-matched normal lung tissues because FOXM1 is a mitotic-specific protein that is expressed as early as the initial stage of tumorigenesis and continues toward the advanced tumor stages. Besides, genetic studies indicated only 20% MYC paralog genomic amplification occurs in SCLC [39]. Among the Myc family, MYCL and MYCN amplification is more commonly found than MYC (c-Myc) in SCLC patients [2]. Some research indicated that amplification of MYC family members is mutually exclusive in SCLC in some datasets [12, 38]. Indeed, we found that MYC mRNA level was low in the GSE60052 dataset, which was associated with high expression levels of MYCL and MYCN (Supplementary Fig. S9). Thus, the relative reduction of MYC average mRNA level in the GSE60052 dataset may be due to Myc family heterogeneity, and a majority of samples were in earlier stages. The discrepancy of c-Myc levels between the human SCLC GSE60052 dataset and RPM mouse model could be caused by the heterogeneity of the Myc family and the composition of cancer stages in the dataset, as well as the transgenic expression of c-Myc in the genetically engineered RPM mouse model.
The CGRP-positive nodules were detected in the small tumors, but not in the big tumors, of the RPM mouse lungs that were harvested 7 weeks post Ade-Cre IT injection (Fig. 4A). Previous studies demonstrated that expression of the hyperactive form of c-Myc (MycT58A) promoted SCLC tumor progression in RPM mice and caused low expression level of neuroendocrine markers, such as CGRP, in the advanced stage RPM tumors [12]. To further validate the association between RPM tumor progression and CGRP level, we have harvested the RPM mice at 3, 4, 5, 6 weeks time-points after adenoviral Cre intratracheal (IT) injection. We found that lung tumor areas were remarkably increased in the advanced stage (at 5 and 6-week time-points, Supplementary Fig. S4D, E) compared with the early-stage (at 3 and 4-week time-points, Fig. 4SB, C). Consistent with the previous report, Cgrp was detected in the nodules or smaller (early stage) RPM lung tumors by immunohistochemistry (IHC) (Supplementary Fig. S4). Notably, not all the smaller tumors were Cgrp positive, indicating the heterogeneity of SCLC in the RPM mouse model.
Compared with the significant advances in translational research on targeted therapeutic drugs and immunotherapies for NSCLC, the development of targeted agents for SCLC has been stagnant [9, 13]. Notably, several anti-tumor agents for SCLC are under clinical development, including therapeutic inhibitors for poly [ADP-ribose] polymerase (PARP1) [40], enhancer of zeste homolog 2 (EZH2) [41], histone deacetylase (HDAC) [42], and proteasome [43]. FOXM1 is a critical downstream protein involved in PARP, EZH2, and HDAC regulatory mechanisms [44,45,46]. Bortezomib, the first proteasome inhibitor drug approved for cancer, was also used for SCLC, both as monotherapy and combination therapy with cisplatin/etoposide, in an earlier clinical trial [43]. Recently, bortezomib was reported to inhibit FOXM1 expression in cell culture and enhance the effect of standard chemotherapy in a xenograft mouse model of SCLC [47, 48]. These results unanimously highlight the potential utility of FOXM1 as a therapeutic target for SCLC. In conclusion, with its regulatory roles in tumorigenesis, progression, heterogeneity, and chemoresistance of SCLC, FOXM1 may serve as an effective prognostic biomarker in standard frontline chemotherapy.
Materials and methods
Dataset acquisition
Raw RNA sequencing (RNA-seq) data were obtained from the Cancer Cell Line Encyclopedia (CCLE) comprising 54 SCLC, 136 NSCLC, and 829 other cancer cell lines [49]. The gene expression profiles of the SCLC cancer dataset (GSE43346) with 23 SCLC and 42 normal tissues (only one normal lung tissue) [50] and GSE60052 consisting of 79 SCLC and seven normal samples [36] were used for analysis.
Gene Set Enrichment Analysis (GSEA) and transcription factor enrichment analysis (TFEA)
The gene expression dataset, GSE60052, was downloaded. Differentially expressed genes (DEG) (fold change >1.5, P value <0.05) between SCLC and control tissue samples were used for web-based Enrichr and ChEA3 transcription factor enrichment analyses [51, 52]. Gene Ontology (GO) was performed using GSEA software (http://software.broadinstitute.org/gsea/index.jsp) [36, 53]. FOXM1 mRNA levels in SCLC patients and associated regulatory pathways were evaluated.
Mouse model of SCLC
Rb1fl/flTrp53fl/flMycLSL/LSL (RPM, JAX #029971) mice were crossed with Foxm1fl/fl mice and the offspring self-crossed to generate Rb1fl/flTrp53fl/flMycLSL/LSLFoxm1fl/fl mice (RPM:Foxm1fl/fl) [12, 54]. Mice were maintained in specific pathogen-free vivarium-filtered cages at the animal facility of the National Health Research Institute (NHRI) under a 12 h light/dark cycle. Five-week-old RPM and RPM:Foxm1fl/fl mice were intratracheally (I.T.) injected with 1 × 106 plaque-forming units (PFU) of Ad-Cre [12, 55]. Both genders were used. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of NHRI and National Tsing Hua University (NTHU).
Generation of rabbit antisera specific for the FOXM1 protein C-terminal region
The His-tagged human FoxM1b 696-748 amino acid construct was cloned into p15Cool2 expression vector and expressed in Escherichia coli BL21 cells. Affinity-purified His-tagged FoxM1b 696-748 aa protein was obtained after nickel chromatography (ThermoFisher) and used as an antigen to immunize rabbits (LTK BioLaboratories, New Taipei City, Taiwan). Subsequent antibody production consisted of initial immunization, followed by six boosts with the antigen.
Immunohistochemical staining and grading of tumor specimens
Paraffin-embedded mouse lung lobes were sectioned and subjected to immunohistochemistry (IHC) with antibodies against p-Histone H3 (1:1000; #SC-8656-R; Santa Cruz Biotechnology, Santa Cruz, CA), Ki67 (1:500; #Clone SP6; Abcam), Ttf1 (1:1000; #WRAB-TTF1, Seven Hills Bioreagents, Cincinnati, OH), TTF1(1:300; #NCL-L-TTF-1; Leica), Sox2 (1:2000; #SC-365823; Santa Cruz), Cgrp (1:5000; #C8198; Sigma-Aldrich) or FOXM1 C-terminus (1:3000). Microscopic images were obtained using a Carl Zeiss Axio Scope.A1 microscope (Oberkochen, Germany). Human SCLC biopsy samples were requested from the pathological department of NTUH-HC. Paraffin-sectioned samples were used for hematoxylin and eosin (H&E, Leica Biosystems Surgipath, Richmond, IL) staining and IHC with antibodies specific for human FOXM1 (1:1000, #SC-32855, Santa Cruz) [37]. Following exclusion of specimens due to inadequate tumor material by two pathologists (H-L S and C-W K), patients with adequate and qualified specimens were enrolled and graded independently based on staining intensity, classified as negative staining (grade 0), weak positive staining (grade 1+), moderate positive staining (grade 2+) and strong positive staining (grade 3+) [56]. Discordant grading interpretations were re-evaluated by the pathologists until a consensus was reached.
Quantitative real-time RT-PCR (qRT-PCR)
Total RNA was prepared from homogenized whole lung tissue using TRIzol reagent (#15596018, ThermoFisher) according to the manufacturer’s instructions. Total RNA (500 ng) was treated with RQ1 RNase-free DNase (#M6101, Promega, Madison, WI) to remove contaminating genomic DNA, followed by cDNA amplification using the High-Capacity cDNA Reverse Transcription Kit (#4368814, ThermoFisher). cDNA samples (500 ng) were amplified with Fast SYBR Green Master Mix (#4385612, ThermoFisher) combined with primers for the gene of interest using a QuantStudio 3 Real-Time PCR system (ThermoFisher). Reactions were analyzed in triplicate. Expression levels were normalized to that of endogenous cyclophilin mRNA and presented as means ± standard deviation (s.d.) [14]. The sense (S) and antisense (AS) primer sequences and annealing temperatures (Ta) used for real-time RT-PCR of mRNA were described in Supplementary Methods.
Patients and study design
Between November 2014 and August 2019, 101 patients were diagnosed with SCLC at NTUH-HC. After retrospective screening, 59 patients were recruited with informed consent. We investigated 52 eligible SCLC patients with adequate tissues. This study was approved by the Institutional Review Broad (IRB) committee of NTUH-HC with IRB No. 104-011-F. Baseline characteristics, clinical data, and treatment outcomes were obtained until November 2019. Cancer staging data at diagnosis were obtained using the International Association for the Study of Lung Cancer (IASLC) 7th edition lung cancer staging system [57].
Statistical analysis
For dataset analysis, one-way ANOVA and multiple comparison tests were applied for continuous variables. To compare the association of clinical features with FOXM1 expression, we used chi-square or Fisher’s exact test (at n < 5) to analyze categorical variables. Progression-free survival (PFS) and OS between subgroups of interest were calculated using the Kaplan–Meier method and Cox proportional hazards regression model for presenting hazard ratio (HR) together with 95% confidence interval (CI). The 50% inhibitory concentration (IC50) was calculated from cell survival curves using GraphPad Prism 6 software (San Diego, CA). Two-sided P values <0.05 were considered statistically significant. Box plot and other analyses were performed using SPSS version 18.0 software (SPSS Inc., Chicago, IL) and both PFS and OS curves were plotted using Stata Version 14 software (StataCorp, College Station, TX).
Detailed procedures describing Micro-computed tomography imaging (microCT) and analysis, Immunohistochemistry (IHC) semi-quantification, Mouse Cgrp promoter cloning and dual luciferase assay, RT-qPCR primers, Cell culture and generation of pInducer-shFOXM1 stable cell lines, Western blot analysis, TUNEL assay, Cell viability assay, Cell clonogenic assay, Cell migration assay, Soft agar colony formation assays, and Bulk RNA-seq and analysis of FOXM1-depleted NCI-H1688 are included in the Supplemental Materials and Methods.
Change history
12 October 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41388-021-02044-5
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53.
Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–22.
Jackman DM, Johnson BE. Small-cell lung cancer. Lancet. 2005;366:1385–96.
Kalemkerian GP, Akerley W, Bogner P, Borghaei H, Chow LQ, Downey RJ, et al. Small cell lung cancer. J Natl Compr Canc Netw. 2013;11:78–98.
Karim SM, Zekri J. Chemotherapy for small cell lung cancer: a comprehensive review. Oncol Rev. 2012;6:e4.
Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl J Med. 2020;382:41–50.
Mok T, Camidge DR, Gadgeel SM, Rosell R, Dziadziuszko R, Kim DW, et al. Updated overall survival and final progression-free survival data for patients with treatment-naive advanced ALK-positive non-small-cell lung cancer in the ALEX study. Ann Oncol. 2020;31:1056–64.
Nicholson AG, Chansky K, Crowley J, Beyruti R, Kubota K, Turrisi A, et al. The international association for the study of lung cancer lung cancer staging project: proposals for the revision of the clinical and pathologic staging of small cell lung cancer in the forthcoming eighth edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11:300–11.
Semenova EA, Nagel R, Berns A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev. 2015;29:1447–62.
Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–9.
Mollaoglu G, Guthrie MR, Bohm S, Bragelmann J, Can I, Ballieu PM, et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell. 2017;31:270–85.
Koo CY, Muir KW, Lam EW. FOXM1: from cancer initiation to progression and treatment. Biochim Biophys Acta. 2012;1819:28–37.
Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25:10875–94.
Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–36.
Nestal de Moraes G, Delbue D, Silva KL, Robaina MC, Khongkow P, Gomes AR, et al. FOXM1 targets XIAP and Survivin to modulate breast cancer survival and chemoresistance. Cell Signal. 2015;27:2496–505.
Okada K, Fujiwara Y, Takahashi T, Nakamura Y, Takiguchi S, Nakajima K, et al. Overexpression of forkhead box M1 transcription factor (FOXM1) is a potential prognostic marker and enhances chemoresistance for docetaxel in gastric cancer. Ann Surg Oncol. 2013;20:1035–43.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–45.
Pandit B, Halasi M, Gartel AL. p53 negatively regulates expression of FoxM1. Cell Cycle. 2009;8:3425–7.
Barsotti AM, Prives C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene. 2009;28:4295–305.
Karachaliou N, Rosell R, Viteri S. The role of SOX2 in small cell lung cancer, lung adenocarcinoma and squamous cell carcinoma of the lung. Transl Lung Cancer Res. 2013;2:172–9.
Wierstra I, Alves J. FOXM1c transactivates the human c-myc promoter directly via the two TATA boxes P1 and P2. Febs J. 2006;273:4645–67.
Wang Z, Park HJ, Carr JR, Chen YJ, Zheng Y, Li J, et al. FoxM1 in tumorigenicity of the neuroblastoma cells and renewal of the neural progenitors. Cancer Res. 2011;71:4292–302.
Ustiyan V, Wert SE, Ikegami M, Wang IC, Kalin TV, Whitsett JA, et al. Foxm1 transcription factor is critical for proliferation and differentiation of Clara cells during development of conducting airways. Dev Biol. 2012;370:198–212.
Bhat UG, Halasi M, Gartel AL. FoxM1 is a general target for proteasome inhibitors. PLoS One. 2009;4:e6593.
Carr JR, Park HJ, Wang Z, Kiefer MM, Raychaudhuri P. FoxM1 mediates resistance to herceptin and paclitaxel. Cancer Res. 2010;70:5054–63.
Wang IC, Meliton L, Tretiakova M, Costa RH, Kalinichenko VV, Kalin TV. Transgenic expression of the forkhead box M1 transcription factor induces formation of lung tumors. Oncogene. 2008;27:4137–49.
Raychaudhuri P, Park HJ. FoxM1: a master regulator of tumor metastasis. Cancer Res. 2011;71:4329–33.
Wang IC, Chen YJ, Hughes DE, Ackerson T, Major ML, Kalinichenko VV, et al. FoxM1 regulates transcription of JNK1 to promote the G1/S transition and tumor cell invasiveness. J Biol Chem. 2008;283:20770–8.
Gemenetzidis E, Elena-Costea D, Parkinson EK, Waseem A, Wan H, Teh MT. Induction of human epithelial stem/progenitor expansion by FOXM1. Cancer Res. 2010;70:9515–26.
Laoukili J, Stahl M, Medema RH. FoxM1: at the crossroads of ageing and cancer. Biochim Biophys Acta. 2007;1775:92–102.
Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, et al. ASCL1 and NEUROD1 reveal heterogeneity in pulmonary neuroendocrine tumors and regulate distinct genetic programs. Cell Rep. 2016;16:1259–72.
McColl K, Wildey G, Sakre N, Lipka MB, Behtaj M, Kresak A, et al. Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget. 2017;8:73745–56.
Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer. 2019;19:289–97.
Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, et al. MYC drives temporal evolution of small cell lung cancer subtypes by reprogramming neuroendocrine fate. Cancer Cell. 2020;38:60–78. e12
Jiang L, Huang J, Higgs BW, Hu Z, Xiao Z, Yao X, et al. Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PLoS Genet. 2016;12:e1005895.
Wang IC, Ustiyan V, Zhang Y, Cai Y, Kalin TV, Kalinichenko VV. Foxm1 transcription factor is required for the initiation of lung tumorigenesis by oncogenic Kras(G12D.). Oncogene. 2014;33:5391–6.
Dammert MA, Bragelmann J, Olsen RR, Bohm S, Monhasery N, Whitney CP, et al. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat Commun. 2019;10:3485.
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–10.
Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Disco. 2012;2:798–811.
Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, et al. Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 Axis. Cancer Cell. 2017;31:286–99.
Pan CH, Chang YF, Lee MS, Wen BC, Ko JC, Liang SK, et al. Vorinostat enhances the cisplatin-mediated anticancer effects in small cell lung cancer cells. BMC Cancer. 2016;16:857.
Davies AM, Lara PN Jr., Mack PC, Gandara DR. Incorporating bortezomib into the treatment of lung cancer. Clin Cancer Res. 2007;13:s4647–4651.
Pal S, Kozono D, Yang X, Fendler W, Fitts W, Ni J, et al. Dual HDAC and PI3K inhibition abrogates NFkappaB- and FOXM1-mediated DNA damage response to radiosensitize pediatric high-grade gliomas. Cancer Res. 2018;78:4007–21.
Kim SH, Joshi K, Ezhilarasan R, Myers TR, Siu J, Gu C, et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep. 2015;4:226–38.
Fang P, Madden JA, Neums L, Moulder RK, Forrest ML, Chien J. Olaparib-induced adaptive response is disrupted by FOXM1 targeting that enhances sensitivity to PARP Inhibition. Mol Cancer Res. 2018;16:961–73.
Gartel AL. A new target for proteasome inhibitors: FoxM1. Expert Opin Investig Drugs. 2010;19:235–42.
Taromi S, Lewens F, Arsenic R, Sedding D, Sanger J, Kunze A, et al. Proteasome inhibitor bortezomib enhances the effect of standard chemotherapy in small cell lung cancer. Oncotarget. 2017;8:97061–78.
Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the cancer cell line encyclopedia. Nature. 2019;569:503–8.
Sato T, Kaneda A, Tsuji S, Isagawa T, Yamamoto S, Fujita T, et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci Rep. 2013;3:1911.
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–97.
Keenan AB, Torre D, Lachmann A, Leong AK, Wojciechowicz ML, Utti V, et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019;47:W212–W224.
Varemo L, Nielsen J, Nookaew I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013;41:4378–91.
Krupczak-Hollis K, Wang X, Kalinichenko VV, Gusarova GA, Wang IC, Dennewitz MB, et al. The mouse Forkhead Box m1 transcription factor is essential for hepatoblast mitosis and development of intrahepatic bile ducts and vessels during liver morphogenesis. Dev Biol. 2004;276:74–88.
Stec DE, Davisson RL, Haskell RE, Davidson BL, Sigmund CD. Efficient liver-specific deletion of a floxed human angiotensinogen transgene by adenoviral delivery of Cre recombinase in vivo. J Biol Chem. 1999;274:21285–90.
van Diest PJ, van Dam P, Henzen-Logmans SC, Berns E, van der Burg ME, Green J, et al. A scoring system for immunohistochemical staining: consensus report of the task force for basic research of the EORTC-GCCG. European Organization for Research and Treatment of Cancer-Gynaecological Cancer Cooperative Group. J Clin Pathol. 1997;50:801–4.
Goldstraw P, Crowley J, Chansky K, Giroux DJ, Groome PA, Rami-Porta R, et al. The IASLC Lung Cancer Staging Project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J Thorac Oncol. 2007;2:706–14.
Acknowledgements
We thank all study participants and staff at NTUH-HC for their contribution to this project, Feng-Yuan Tsai (NHRI), and NHRI Laboratory Animal Center (LA-109-PP-01), and I-BEN Service (TSSI-BN-31) for technical assistance, Stephen Elledge (Harvard Medical School) for kindly providing the pInducer10-mir-RUP-PheS plasmid, Pradip Raychaudhuri (UIC) and Vladimir Kalinichenko (CCHMC) for kindly providing the Foxm1fl/fl mice. This work was supported by the National Taiwan University Hospital Hsinchu Branch research grant NTUH-HC 108-s267 (S-K L), the Ministry of Science and Technology of Taiwan grant MOST105-2628‐B‐007‐003‐MY3 and MOST109-2314-B-007-005-MY3 (I-C W), National Tsing Hua University grant 108Q2502E1, 109Q2714E1, 110Q2501E1 (I-C W), and by the Brain Research Center under the Higher Education Sprout Project, co-funded by the Ministry of Education and the Ministry of Science and Technology in Taiwan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41388_2021_1895_MOESM8_ESM.pdf
Fig. S6_The clonogenic cell survival assay for the FOXM1-depleted NCI-H1688 cells after cisplatin-based chemotherapeutic reagents treatment
Rights and permissions
About this article

Cite this article
Liang, SK., Hsu, CC., Song, HL. et al. FOXM1 is required for small cell lung cancer tumorigenesis and associated with poor clinical prognosis. Oncogene 40, 4847–4858 (2021). https://doi.org/10.1038/s41388-021-01895-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-021-01895-2
- Springer Nature Limited
This article is cited by
-
FOXM1: a new therapeutic target of extramammary Paget disease
Scientific Reports (2024)
-
The splicing factor WBP11 mediates MCM7 intron retention to promote the malignant progression of ovarian cancer
Oncogene (2024)
-
HNRNPA2B1-mediated m6A modification of FOXM1 promotes drug resistance and inhibits ferroptosis in endometrial cancer via regulation of LCN2
Functional & Integrative Genomics (2024)
-
USP39 interacts with SIRT7 to promote cervical squamous cell carcinoma by modulating autophagy and oxidative stress via FOXM1
Journal of Translational Medicine (2023)
-
FOXM1 promotes TGF-β2-induced injury of human lens epithelial cells by up regulating VEGFA expression
Graefe's Archive for Clinical and Experimental Ophthalmology (2023)