
Abstract
Disruption of the cellular pathway modulating endogenous 24-h rhythms, referred to as “the circadian clock”, has been recently proven to be associated with cancer risk, development, and progression. This pathway operates through a complex network of transcription-translation feedback loops generated by a set of interplaying proteins. The expression of core circadian clock genes is frequently dysregulated in human tumors; however, the specific effects and underlying mechanisms seem to vary depending on the cancer types and are not fully understood. In addition, specific oncogenes may differentially induce the dysregulation of the circadian clock in tumors. Pharmacological modulation of clock components has been shown to result in specific lethality in certain types of cancer cells, and thus holds great promise as a novel anti-cancer therapeutic approach. Here we present an overview of the rationale and current evidence for targeting the clock in cancer treatment.
Similar content being viewed by others

Introduction
The circadian clock consists of a hierarchical multi-oscillator network of intracellular and intercellular mechanisms throughout the body that contributes to anticipating metabolic activity and maintaining system homeostasis in response to environmental cues and intrinsic stimuli. Any biological process that has roughly a 24-h pattern can be said to have a circadian rhythm. Although most, perhaps all, organisms display circadian rhythms in their respective biological processes, circadian rhythms in mammals help optimize the transition between day and night for different behavioral and physiological activities [1]. The circadian clock is a highly conserved self-sustained mechanism that regulates and synchronizes a wide variety of functions including sleep-wake cycles, rhythmical fluctuations of blood pressure, heart rate, body temperature and hormone secretion, and feeding behavior [2]. The clock molecular mechanism is operated at the single-cell level through a network of transcription-translation feedback loops (TTFLs) generated by a set of interplaying clock proteins, which regulate according to a circadian rhythm the expression of specific target genes and their products. Up to 20% of genes expressed in any particular cell or tissue have been found to undergo circadian oscillations at the mRNA level, indicating the extensive role of circadian gene regulation [3]. As the genes showing circadian rhythmicity in different tissues are non-overlapping, more than 50% of genes in the body may be clock-regulated.
The disruption of the circadian rhythms in humans, resulting from sleep deprivation, jet-lag, shiftwork involving nightshifts, or unnatural light exposure, has been shown to have a deep impact on many physiological functions and has been related to several disorders including insomnia, depression, metabolic and cardiovascular diseases, and cancer. This can be explained by the clock-dependent modulation of several key cellular pathways regulating proliferation and apoptosis, DNA damage repair, cellular senescence, metabolic homeostasis, xenobiotic detoxification, oxidative stress, inflammatory and immune response [4]. Over the past few years, genetic variations of core clock genes have been associated with cancer risk in several epidemiological studies and germline polymorphisms in these genes have been proposed as biomarkers of cancer risk, although genetic associations are not always consistent across different studies [5]. This may partly be explained by the fact that the effect of gene variants on protein expression levels or function is often unknown. Mutations or loss of function of different clock proteins have, however, been confirmed in animal models to correlate with different pathological phenotypes and cancer risk.
Chronochemotherapy, as a way to apply chemotherapy against malignant tumors on a circadian schedule in order to optimize treatment efficacy and reduce toxicities by exploiting the physiology of cellular circadian rhythms, has been explored in several studies. Clinical trials of chronochemotherapy continue to be an active area of research, but have not consistently resulted in improved treatment efficacy to date [6]. More recently, growing evidence suggests that small molecule drugs targeting the core components of the circadian machinery have promising anti-tumor activities. As such, circadian clock components are emerging as novel intriguing therapeutic targets for cancer treatment. Here we present an overview of the current evidence supporting targeting the clock as an actionable anti-cancer strategy.
The mammalian circadian clock
Signaling to the circadian network
“Zeitgebers” or “time-givers” are any natural cues in the environment that can entrain an organism’s biological clock. Zeitgebers include: light and dark cycle, food and drink, exercise, rest, social interactions, and temperature. The most influential and potent zeitgeber of the mammalian circadian system is light and secondary to that is the feeding schedule. Consequently, in addition to genetic perturbations, many animal studies to date focus on altering 12 h light/dark cycles or feeding schedules in order to investigate the effects of disruption of the circadian system [1].
The suprachiasmatic nucleus (SCN), referred to as the central or master clock, is a cluster of about 20,000 neurons that is located in the hypothalamus of the brain. The SCN transduces light-driven signals from melanopsin, a G-protein-coupled receptor in the retina, into electrical and hormonal signals to other regions of the brain and body in order to entrain circadian rhythms of many different biological processes, such as the sleep-wake cycle [7]. Clocks located outside of the SCN are known as peripheral clocks. As noted above, clocks are present in every cell and are often coordinated at the organ level. Peripheral clocks that can act independently of the SCN may be present in every tissue and have been identified in most organs, including the metabolic, cardiovascular, reproductive, endocrine, immune, and gastrointestinal systems. Input from non-photic zeitgebers acts upon these peripheral clocks which, consequently, feedback to the SCN to align the master clock with peripheral clocks [8]. The interplay between the central clock and peripherical clocks, therefore, has implications on not only biological processes but also the pathophysiology of various diseases.
The transcription-translation feedback loop
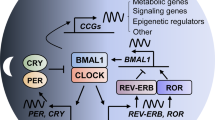
At the cellular level, a series of TTFLs make up the molecular circadian machinery that controls the expression of clock-controlled genes (CCGs) and lead to rhythmic cycling of gene expression and, consequently, behavioral and physiological processes (Fig. 1). The core clock transcriptional activators, brain and muscle ARNT-Like 1 (BMAL1) and circadian locomotor output cycles kaput (CLOCK), form a heterodimer and act upon conserved enhancer “E-box” elements of the promoters of CCGs to initiate their transcription. The set of genes with E-box elements include the negative regulators of the core clock, period 1/2 (PER1/2), and cryptochrome 1/2 (CRY1/2) [8]. Interestingly, CLOCK itself does not exhibit rhythmic expression but its circadian control and activity are dependent upon BMAL1 in that its nuclear accumulation is BMAL1-dependent [9]. Very similarly, PER1/2 is dependent upon CRY1/2 for their nuclear accumulation [10]. Following their translation and accumulation in the nucleus, the PER and CRY heterodimer suppresses the transcriptional activity of BMAL1::CLOCK, likely by recruiting Casein Kinase 1 δ (CK1δ), resulting in the phosphorylation of the transcriptional complex and dissociation from the E-box [11]. This serves as a negative feedback loop in that PER::CRY suppresses its own transcription. The decrease in PER1/2 and CRY1/2 transcripts combined with the degradation of PER1/2 and CRY1/2 proteins relieve the suppression of BMAL1::CLOCK transcriptional activity, thereby reinstating CCG output and generating a new cycle of oscillation [8]. Besides this core negative feedback loop, there are additional loops that are generated by positive and negative regulators of BMAL1 transcription—retinoic acid-related orphan receptor α/β/γ (RORα/β/γ) and REV-ERBα/β. The positive regulator RORα/β/γ is encoded by RORA, RORB, and RORC, respectively. The negative regulator REV-ERBα/β is encoded by the nuclear receptor subfamily 1 Group D Member 1/2 (NR1D1/2) genes. RORs and REV-ERBs compete for interaction with the ROR response element (RORE) motif on the BMAL1 promoter. RORs promote BMAL1 transcription while REV-ERBs interact with nuclear repressor corepressor (NCoR) to suppress BMAL1 transcription. As with PER1/2 and CRY1/2, BMAL1::CLOCK controls the expression of RORs and REV-ERBs by acting upon the E-boxes of their gene promoters [12]. In addition to their positive and negative regulators, BMAL1::CLOCK’s transcriptional targets include many proteins including other transcription factors such as differentiated embryonic chondrocyte gene 1/2 (DEC1/2), D-box binding protein, hepatic leukemia factor, thyrotroph embryonic factor, and E4-binding protein 4 [8].

The molecular mammalian clock is made up of interlocking TTFLs. The BMAL1::CLOCK heterodimer binds to E-box containing promoters to induce expression of CCGs and their negative regulators, CRY1/2, PER1/2, and REV-ERBα/β, and positive regulators, RORα/β/γ. PER1/2 and CRY1/2 heterodimerize in the cytoplasm and are translocated to the nucleus where they inhibit the transcriptional activity of BMAL1::CLOCK. In doing so, they regulate their expression as well as the expression of other CCGs. Kinases regulate the phosphorylation of these integral clock proteins. PER1/2 is phosphorylated by either CK1δ/ε or CK2 and this is followed by the recruitment of the SCFβ-TrCP complex to ubiquitinate and target p-PER1/2 for proteasomal degradation. CRY1/2 is phosphorylated by AMPK and is then ubiquitinated by the SCFFBXL3 complex. These events result in CRY1/2 being targeted for proteasomal degradation. Degradation of PER1/2 and CRY1/2 help to relieve repression upon BMAL1::CLOCK transcriptional activity. RORα/β/γ and REV-ERBα/β on the other hand act upon the RORE promoter element of the BMAL1 promoter to control transcription of BMAL1. RORα/β/γ promote BMAL1 transcription while REV-ERBα/β interact with NCoR peptides to inhibit BMAL1 transcription. These TTFLs as a whole generate roughly 24-h rhythmicity in clock-controlled gene expression. Created with Biorender.com.
Post-translational modifications and protein turnover add an additional layer of regulation to clock components in order to generate a period length of roughly 24-h in gene expression cycling. Following synthesis in the cytoplasm, BMAL1, CLOCK, CRY1/2, and PER1/2 nuclear translocation and heterodimerization are regulated by their phosphorylation status [12]. For example, phosphorylation of CLOCK at Serine-38 and -42 is important for its transcriptional activity, while phosphorylation at Serine-845 by AKT inhibits its nuclear localization, and phosphorylation at Serine-427 by glycogen synthase kinase-3 beta (GSK-3β) results in its degradation [13, 14]. Adenosine monophosphate-activated protein kinase (AMPK) phosphorylates and destabilizes CRY1/2. This event is then followed by the subsequent ubiquitination of CRY1/2 by the Skp1/cullin/F-box protein (SCF) E3 ubiquitin ligase F-box/LRR-repeat protein 3 (FBXL3) complex (SCFFbxl3), resulting in the proteasomal degradation of CRY1/2 [8]. However, it is important to note that, like CLOCK, phosphorylation at some residues of CRY1/2 results in degradation while phosphorylation at other residues affects its subcellular localization [15]. PER1/2 is phosphorylated by CK1δ/ε, which leads to the recruitment of a complex containing SCF and β-Transducin repeat-containing protein, another F-box protein, (SCFβ-TrCP) and, consequently, to the ubiquitination and degradation of PER1/2 [8]. CK2 is another circadian-related kinase that phosphorylates both BMAL1 (on Serine-90), which results in its nuclear accumulation, and PER2, which leads to either its accumulation in the nucleus or its degradation after additional phosphorylation by CK1δ/ε [16, 17]. Such events suggest that there is a defined ratio in the level of phosphorylation and/or the site of phosphorylation of clock proteins that determines their subcellular localization, stabilization, or degradation.
The dynamics of transcriptional activation and/or repression, protein subcellular localization, and protein degradation as a whole generates oscillations in circadian CCG expression and, consequently, rhythms in behavioral and physiological processes.
Clock and cancer
A growing body of evidence reveals that the circadian clock is tightly linked to cancer on multiple levels. Early evidence came from epidemiological studies showing that the disruption of circadian rhythms by factors such as shift work was associated with increased risk of breast cancer [18, 19], later supported by additional epidemiological data confirming the association between shift work and cancer risk [20,21,22]. This evidence motivated the international agency for research on cancer to list “shift work leading to a disruption in circadian rhythm” as a probable human carcinogen [23]. Although this connection remains controversial, this connection inspired researchers to investigate the direct correlation between the clock and cancer using prospective physiological and genetic experiments in animal models. For example, chronic jet lag mimicked by shifted lighting or SCN ablation in mice can induce spontaneous hepatocellular carcinoma (HCC) [24, 25]. In mice that harbor mutated p53 or Ras, alteration or knock-out of core clock genes such as Bmal1 and Per can accelerate the initiation and progression of multiple cancers [26, 27]. Epidemiological and animal model studies are reviewed in detail by Pariollaud et al. [28]. More recently, a pan-cancer analysis of the cancer genome atlas showed that alteration of clock genes at transcriptional and genetic levels is pervasive in cancer, and transcriptional dysregulation is strongly associated with survival. Furthermore, the transcription of clock genes and many drug target genes are closely associated with each other [29]. This work underscored the clinical significance of clock genes in cancer treatment.
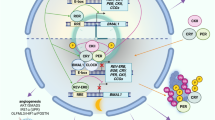
Although a broad association between a disrupted clock and tumorigenesis is well established, the detailed molecular relationship remains elusive. First, it is clear that many cancer cells harbor disrupted clock machinery where the circadian rhythm of the cells is dampened or completely diminished, but no data thus far have argued clearly whether the disruption of the clock is the cause or effect of malignant cell transformation. Furthermore, in certain types of cancer such as glioblastoma (GBM), the circadian rhythm in tumor cells remains intact and is in fact required for the maintenance of the disease. Second, the isoform-specificity, activity, and function of the clock components highly depend on their tissue background, which implies that different roles may be played by the clock in cancers originating from different organs. This explains how certain clock genes can act as tumor suppressors in some tumor types, but oncogenes in others. Third, because of the essential role of the circadian clock in regulating cellular physiology, the downstream processes regulated by the clock machinery in the cells are also broad, weaving complex regulating networks and making it difficult to build clear and definite relationships. The interplay between clock components and the generally recognized hallmarks of cancer (summarized in Fig. 2) have been recently reviewed by Sulli et al. [30]. More detailed studies are being carried out to scrutinize the roles of clock molecules in cancer. Here we reviewed some current studies that elucidate the roles of core molecules of the clock machinery in cancer.

Components of the circadian clock either regulate the transcription of tumor suppressors or oncogenes or directly interact with proteins that are part of cell signaling and regulatory pathways that contribute to cancer hallmarks. Cell-cycle regulators such as cyclin-dependent kinases (CDKs), CDK inhibitors, and cyclin proteins exhibit rhythmicity in their expression, and some are directly regulated by BMAL1::CLOCK or ROR/REV-ERB transcriptional activity, thereby contributing to proliferation and evasion of growth suppression [30]. In pancreatic cancer cells, DEC1/2 and CRY1 transcription were dependent on the SMAD family member 4 (SMAD4) and activation of the TGFβ pathway led to circadian dysregulation and increased cell invasiveness, suggesting crosstalk between clock components and metastasis [92]. BMAL1::CLOCK was found to upregulate levels of a chemokine, olfactomedin-like 3 (OLFML3), resulting in recruitment of immune-suppressive microglia in GBM, and separate studies show that disruption of clock components or jet-lag in rodents led to the upregulation of cytokines, resulting in chronic inflammation and immunosuppression [30, 41]. An abnormal circadian clock also contributes to T cell exhaustion and immune evasion via global upregulation of inhibitory molecules such as programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [91]. BMAL1 regulation of REV-ERBα led to upregulation of vascular endothelial growth factor A (VEGFA) transcription by acting upon its RORE promoter element and increased proliferation of human CRC cell lines. High BMAL1 expression in patients was found to be associated with clinical non-response to combined chemotherapy and bevacizumab, an anti-angiogenesis treatment [89]. DNA replication occurs at night in many organisms, suggesting tight regulation of these processes by the circadian clock; therefore, modulation of clock components or circadian disruption can lead to sustained proliferative signaling and DNA replication [30]. Although both proteins negatively regulate BMAL1 and CLOCK, DEC1 positively regulates while DEC2 negatively regulates proteins involved in apoptosis, suggesting their roles as a circadian-regulated tumor suppressor or oncogene, respectively [93]. Not only do components of the DNA damage response pathway and DNA repair mechanisms display rhythms in their mRNA and protein levels but they also directly interact with clock proteins, suggesting daily rhythms in the detection and reparation of damaged DNA. BMAL1::CLOCK may regulate the transcription of telomerase reverse transcriptase (TERT) at the E-boxes of its promoter as it displays rhythm in expression, thereby contributing to circadian control of telomerase. Not only do BMAL1 and CLOCK directly regulate key transcription factors, proteins, and signaling pathways that contribute to increased glycolysis in cancer cells and tumorigenesis but they also regulate BMAL1::CLOCK heterodimerization and transcriptional activity. Lastly, BMAL1, CLOCK, and CRY1/2 modulate the function of the NF-kB pathway, one of the key regulators of inflammation, either by directly or indirectly modulating the p65 subunit [30]. Created with Biorender.com.
The core clock in cancer
BMAL1 and CLOCK are the central modulators of circadian output in cells and their pathological functions in cancer are the most thoroughly studied so far. BMAL1::CLOCK was initially suggested to be tumor suppressive. Low expression of BMAL1 is reported to be associated with tumor progression and poor diagnosis in melanoma, pancreatic adenocarcinoma, and breast cancer [31,32,33]. Consistently, overexpression of BMAL1 in HCC, osteosarcoma, ovarian and hematologic cancer cells reduced tumor cell growth, and BMAL1 knockout in hamsters results in HCC [24, 34,35,36,37]. In addition, it is becoming clear that BMAL1::CLOCK works differently across organs by interacting with tissue-specific factors. The molecular mechanism of this tissue-specific control has been exemplified by the liver, where the hepatic nuclear receptor 4a (HNF4A) was shown to repress the BMAL1::CLOCK transcriptional activity, adding a layer of modulation to the circadian network [38]. HNF4A and BMAL1 seem to collaboratively regulate the proliferation of HCC cell lines, pointing out the functional role of tissue-specific circadian clocks in carcinogenesis [34]. In acute myeloid leukemia (AML) and GBM, BMAL1 and CLOCK are essential for the survival of the cancer stem cell (CSC) population. In AML, knockdown of BMAL1 and CLOCK led to stem cell differentiation and disease regression in a murine model, while keeping physical hematopoiesis intact [39]. This selective role of BMAL1::CLOCK was also observed in GBM, where more details were revealed [40]. Glioblastoma stem cells (GSCs), normal neural stem cells (NSCs), and non-malignant (NM) neuronal cultures excised from epilepsy patients all display rhythmic expression of BMAL1 but disruption of BMAL1 by shRNA only specifically blocked cell proliferation of GSCs, showing that GSCs have a specific and exquisite reliance on BMAL1 activity for proliferation. GSC, but not NSC, self-renewal was also impaired upon BMAL1 or CLOCK disruption. This observation was explained by chromatin immunoprecipitation sequencing (ChIP-seq) analysis, which showed that in GSCs, BMAL1 had largely skewed binding activities compared to NSCs, including newly gained binding sites and motifs that are enriched in genes involved with glucose and lipid metabolism. Functional assays confirmed that BMAL1 and CLOCK indeed maintained metabolic homeostasis in GSCs. These data highlight epigenetic regulation and metabolism as primary processes regulated by the clock proteins in cancer. Disruption of BMAL1 or CLOCK or BMAL1::CLOCK transcriptional activity by shRNA or small molecules drastically reduced the tumor burden and improved survival in the mice model of GBM, indicating that BMAL1::CLOCK-fueled tumor progression is a viable target for anti-cancer therapy [40]. In an independent study gain-of-function screen of epigenetic regulators, CLOCK was shown to enhance GSC self-renewal. CLOCK::BMAL1 induced the recruitment of immune-suppressive microglia to the GBM microenvironment and triggered pro-tumor immune activity, hence helping tumor maintenance and progression. Depletion of the Clock improved the survival in a GBM murine model [41]. These studies underscore the role of the clock machinery in cancer and suggest there is great potential for clinical benefits as a result of directly targeting the clock to treat cancer.
Repressors of the core clock in cancer
The negative regulator arms of the clock are also clearly implicated in cancer etiology. Levels of the PER family genes were found repressed in clinical samples from many cancer types, including PER1 in glioma, stomach, non-small-cell lung cancer (NSCLC), breast and prostate cancers, PER2 in lymphoma, leukemia, lung, stomach, and breast cancers, as well as PER3 in colorectal cancer (CRC) [42]. Interestingly, an increase in PER3 expression was observed in patients with AML and acute lymphoid leukemia (ALL) in remission, but not in those who relapsed after treatment, suggesting that upregulation of PER3 is associated with better clinical outcome and may have a therapeutic potential in acute leukemia [43]. Reduced PER1 and PER2 expression were associated with shorter survival in glioma and gastric cancer. Accordingly, overexpression of PER1 in cell lines can suppress tumor growth and induce apoptosis. Based on reporter assay in cultured cell lines, PER1 was shown to bind to the androgen receptor (AR) and inhibited AR-mediated gene activation thus repressing prostate cancer cell growth [44]. PER2 suppresses estrogen receptor-α (ERα) transcriptional activation and is required for proteasomal-mediated degradation of ERα in the MCF-7 breast cancer cell line. Furthermore, the addition of 17-b estradiol (E2) induces PER2 expression, and PER2 overexpression results in inhibition of growth and colony formation and induced apoptosis, implying the clock’s potential role as part of a feedback mechanism in ER + breast cancer [45]. In addition, PER2 may act as a negative regulator of epithelial-mesenchymal transition (EMT) in normal mammalian epithelial cells by interacting with OCT1 [46].
The nuclear receptor REV-ERBs are relatively less studied in cancer. High expression of REV-ERBβ was reported to be associated with poor overall survival in HCC [47]. In a cell line study, REV-ERBα was shown to inhibit the proliferation of gastric cancer cells by regulating glucose metabolism and the pentose phosphate pathway [48]. Remarkably, pharmacological activation of REV-ERBs was reported to induce apoptosis by regulating autophagy and de novo lipogenesis and to be lethal to multiple types of tumor cells, including brain cancer, CRC, breast cancer, leukemia, and melanoma [49].
The CRY family genes also play apparently complex roles in many cancer types, perhaps explained by the differential role and tissue expression of the two isoforms, CRY1, and CRY2. Single nucleotide polymorphisms (SNPs) in CRY1 were associated with a higher risk of breast cancer. Increased CRY1 in CRC cells correlated with tumor advancement and poor prognosis [50]. Recently, it was found that CRY1 expression is regulated by androgen hormones in that androgen stimulation via the addition of dihydrotestosterone leads to AR binding to the CRY1 locus in prostate cancer cells. CRY1 was found to regulate DNA repair, homologous recombination, and G2/M transition in the cell cycle, thereby promoting prostate cancer cell survival and metastasis [51]. By contrast, high expression levels of CRY2 correlated with better survival in breast cancer, and, consistently, a low level of CRY2 was associated with shorter survival in HCC [29, 52]. Expression of CRY2 was also found to be lowered in papillary and follicular thyroid carcinoma [53]. These data imply a tumor-suppressive role of CRY2. On the other hand, ablation of both Cry1/2 seems to suppress tumor development and improve prognosis in mice with mutated p53, implying that CRY2 can also be tumorigenic [54]. Knocking out both Cry genes in hamsters resulted in the development of HCC [24]. The role of CRY2 in tumor suppression may involve its direct regulation of the oncogene MYC which is discussed in the following section.
The expression of core clock genes and negative regulators of the clock has been also found to be dysregulated in esophageal and cervical cancer cell lines compared to their normal counterparts, with low levels of CLOCK, CRY1, and RORA expression in cancer cells due in part to promoter hypermethylation. Notably, overexpression of CLOCK and PER2 attenuated cell proliferation while activation of RORα and REV-ERBα via small molecule agonists (SR9011 and SR1078, respectively) led to increasing in apoptosis in cervical and esophageal cancer cells, with a lesser effect on non-cancerous control cells [55].
MYC and the clock
MYC is one of the most well-known oncogenes and its abnormal activation is pervasive in cancer, being deregulated (normally overexpressed) in many cancers. Like BMAL1 and CLOCK, MYC has a basic helix-loop-helix (bHLH) structure with a high affinity to E-box motifs. Therefore, it is not surprising that the functions of MYC and clock genes are correlated. Cellular studies showed that MYC can disrupt circadian oscillations in at least two ways, by either activating REV-ERBs to suppress BMAL1 and CLOCK transcription or repressing BMAL1 and CLOCK directly in complex with Myc-interacting zinc finger protein 1 (MIZ1) [56, 57]. These results provide a potential mechanism for how the clock is disrupted during tumorigenesis. Conversely, MYC is also regulated by the clock network. MYC expression oscillates in a 24-h period and the dynamics of the MYC protein are directly regulated by clock proteins. CRY2 can promote the ubiquitylation of c-MYC by acting as a co-factor of FBXL3, leading to MYC degradation. Loss of Cry2 stabilizes c-MYC and enhances cell transformation and proliferation in mouse fibroblasts [58]. CRY modulation is therefore potentially protective against malignant transformation and is thus a potential molecular target for cancer therapy.
The increasing molecular connections between cancer and the clock machinery give exciting inspiration for designing strategies to target clock components for the treatment of cancer.
Targeting the circadian clock in cancer
BMAL1 and CLOCK are transcription factors that are incredibly challenging to target in the human body due to general difficulties in targeting protein-DNA or protein-protein interactions that arise with transcription factors. Consequently, various small pharmacological molecules that either inhibit or promote the activity of the negative regulators of the clock have been developed in order to enhance or suppress BMAL1::CLOCK transcriptional activity. To date, numerous clock compounds have been applied as potential therapeutic agents in a variety of diseases, including cancer (Table 1). As noted above, BMAL1 and CLOCK can either play an oncogenic or tumor-suppressive role depending on the cancer type and the downstream targets of BMAL1::CLOCK transcriptional activity that are actively driving tumorigenesis. It is therefore imperative to account for the specific functions of these compounds before utilizing them to target cancer cells.
REV-ERB agonists
REV-ERB agonists have perhaps the richest history amongst the existing collection of clock-targeting small molecules. GSK4112 or SR6452 was the first synthetic REV-ERB agonist to be identified and targets both REVERB isoforms, mimicking the action of heme, the physiological ligand for REV-ERBs. It was able to reset circadian rhythms and regulate metabolic pathways but displayed low systemic exposure in vivo [59]. GSK4112 derivatives SR9009 and SR9011 display improved potency, efficacy, and pharmacokinetic properties compared to GSK4112 and have been thoroughly studied in numerous disease models [60]. They have been found to be potent against a number of different cancer types containing a variety of different oncogenic drivers [40, 41, 49, 61]. Notably, SR9009 and SR9011 have both been reported to not display overt toxicity in animal models [60]. It is important to note, however, that high concentrations were needed to observe anti-tumor effects in in vivo experiments, suggesting that more potent derivatives of these agonists may need to be developed for therapeutic potential in the clinic [49]. It has been reported that SR9009 may display REV-ERB independent effects given that SR9009 still retained its activity in mouse hepatocytes and embryonic stem cells under Nr1d1/2 (REV-ERBα/β) knockout conditions [62]. In previous studies, however, loss of REV-ERBs led to attenuation of SR9009 treatment effects in various disease models [49, 60]. Given such findings, SR9009 may not be completely ruled out for pharmacological modulation of REV-ERBα/β activity, but investigators must properly determine whether SR9009 exclusively displays effects on REV-ERBα/β in the disease, tissue, and model of interest in their studies.
Given that SR9009 has an improved but still sub-optimal pharmacokinetic profile and has potential off-target effects, emerging novel REV-ERB agonist scaffolds may pose to be better options for future cancer studies and clinical applications, and further in vivo investigation of these scaffolds are pertinent moving forward especially in cancer types and other diseases where BMAL1::CLOCK activity are drivers of disease pathophysiology.
REV-ERB antagonists
SR8278 was the first identified REV-ERB antagonist with a structure similar to GSK4112 but it competes for heme rather than substitutes for it and thus represses REV-ERBα/β target transcription rather than activates it. SR8278 enhances luciferase reporter activity of target genes that are normally repressed by REV-ERBα/β: BMAL1, glucose 6-phosphatase (G6Pase), and phosphoenolpyruvate carboxykinase (PEPCK). SR8278 inhibited the repressive transcriptional activity of REV-ERBα/β in a dose-dependent manner. Interestingly, SR8278 treatment of HCC HepG2 cells led to a significant increase in the mRNA levels of G6Pase and PEPCK, genes that play tumor-suppressive roles in HCC and renal cell (RCC) carcinomas [63].
ARN5187 is a REV-ERB antagonist that exclusively interacts directly with the ligand-binding domain (LBD) of REV-ERBβ. Treatment of BT-474 breast cancer cells leads to an increase in BMAL1, PER1, and PEPCK expression levels in a dose-dependent manner. ARN5187 also inhibited autophagy by blocking lysosomal function and preventing autophagolysosome maturation and increased cleaved poly[ADP-ribose] polymerase levels. REV-ERBβ levels were higher than REV-ERBα in breast, liver, prostate, melanoma, and colon cancers, suggesting that specific inhibition of REV-ERBβ may be a promising therapeutic strategy for these cancer types [64].
As REV-ERBs were found to play a role in inflammatory responses, additional studies will need to be conducted on the action and pharmacokinetic properties of REV-ERB antagonists in inflammatory disease as well as cytotoxicity and chronic inflammation in cancer [65].
ROR agonists
SR1078 was identified to be a direct agonist of RORɑ/γ in a biochemical screen. Co-transfection assays in which human embryonic kidney (HEK) 293 cells were transfected with RORα or γ and a luciferase reporter driven by the mouse promoter of G6Pase, a target of RORs, showed that addition of SR1078 was able to significantly increase reporter activity. This increase was lost when the RORE domain was mutated in the G6Pase promoter, indicating compound specificity for RORɑ/γ. The agonistic effects of SR1078 were confirmed by treating human liver cancer HepG2 cells with the compound, which resulted in a twofold and threefold increase in G6Pase and fibroblast growth factor-21 (FGF21) mRNA levels, respectively. Intraperitoneal injection of SR1078 into mice showed that the compound displays acceptable pharmacokinetic properties and increased expression of ROR target genes in the liver [66]. Interestingly, HepG2 cells treated with SR1078 showed an increase in p53 protein levels, expression of p53 target genes, and apoptosis as indicated by the increase in the fraction of cells in the sub-G1 phase [67].
Nobiletin (NOB) is another direct agonist of RORɑ/γ identified through radioligand binding assays in which NOB displayed robust competitive binding to RORɑ/γ LBD. NOB increased luciferase reporter activity driven by the mouse Bmal1 promoter at a dose-dependent manner in HCC Hepa1-6 cells. This effect of NOB was lost when either the RORE element of the Bmal1 promoter was mutated or RORA/C was knocked down. Pharmacokinetic studies of NOB revealed significant plasma, liver, and brain exposure and NOB treatment significantly increased expression of several ROR target genes in mice [68]. Across a variety of cancer cell types, NOB has been found to induce apoptosis and cell cycle arrest, suppress EMT, inhibit many oncogenic drivers, upregulate tumor suppressors, and increase chemotherapy sensitivity [69].
The specificity and favorable pharmacokinetic properties of these ROR agonists suggest that they can be applied in future in vivo studies for disease treatment in which BMAL1 transcription and transcriptional activity might be advantageous.
CRY stabilizers
KL001 was shown to cause period lengthening and amplitude reduction of luciferase activity in mBmal1-dLuc reporter cells when it was identified in a cell-based phenotypic screen of 60,000 structurally unique compounds [70]. KL001 was able to lengthen the period and amplitude of a signal in NIH-3T3 fibroblast cells transfected with mBmal1-dLuc or mPer2-dLuc reporters as well as mice SCN and lung explants containing a mPer2Luc knock-in reporter. KL001 was found to directly interact with CRY1/2 and stabilized their protein levels by inhibiting FBXL3 mediated ubiquitination of CRY1/2 by occupying the flavin adenine dinucleotide binding pocket. KL001 treatment impacted hepatocyte glucose synthesis and is therefore promising for the treatment of metabolic disorders [70]. There has been an undertaking to identify derivatives of KL001 to improve upon its potency for different disease applications and to improve on its pharmacokinetic characteristics [71, 72]. Application of KL001 as well as SHP-656, a derivative of KL001 with increased bioavailability, was found to specifically target GSCs in vitro compared to normal neuronal cells by downregulating stemness genes and inducing apoptosis. SHP-656 treatment of mice inoculated with GSCs was able to extend their survival [40].
Recent work was able to identify CRY stabilizers that are specific for either one of the two CRY isoforms, and these pharmacological molecules are structurally different from any other identified clock compound identified thus far [73, 74]. The selectivity for CRY stabilizers for different CRY isoforms will be a powerful tool to investigate the similarities and differences between the CRY isoforms, their functional roles, and how the regulation of the CRYs plays a role in a variety of diseases, including cancer.
CRY inhibitors
KS15 was identified from a two-step cell-based screening method of 1000 different compounds that utilized BMAL1::CLOCK heterodimer driven luciferase reporter or an artificial E-box luciferase reporter in cultured cell lines. The addition of KS15 obstructed CRY1/2-mediated binding and transcription repression allowing for the activation of BMAL1::CLOCK E-box-mediated transcription and resulted in the increase of Per1/2 and Nr1d1 (REV-ERBα) mRNA levels, all of which is lost under Cry1/2 knockout conditions [75]. Treatment of MCF-7 breast cancer cells with KS15 induced changes in the expression of cycle-cell regulators and pro-apoptotic genes resulting in cell cycle arrest and induction of apoptosis and repressed proliferation in in vitro wound healing assays. KS15 led to an increased sensitivity to doxorubicin and tamoxifen treatment, indicating that KS15 increases the chemosensitivity of some breast cancer cells [76]. Optimization of KS15 for in vivo application may result in a new therapeutic strategy for the treatment of different cancers that are linked to CRY activity.
CK1 inhibitors
Longdaysin was identified from a screen of about 120,000 compounds using U2OS osteosarcoma cells containing a mBmal1-dLuc reporter and causes a strong period-lengthening effect [77]. Longdaysin displayed consistent dose-dependent response in mice fibroblasts and lung and SCN explants containing a mPer2Luc knock-in reporter and in zebrafish harboring a per3-luc reporter. It was revealed that CK1ɑ/δ and ERK2 are both targets of longdaysin. Treatment of U2OS mBmal1-dLuc reporter showed that longdaysin strongly upregulated the amount of PER1 protein by influencing the interaction of CK1ɑ/δ with PER1 [77]. Longdaysin attenuated Wnt/β-catenin signaling by blocking LDL receptor-related protein 6 and dishevelled-2, reduced levels of active and total β-catenin, and decreased the expression of Wnt target genes in Hs578T and MDA-MB-231 breast cancer cells. In vitro and in vivo breast cancer studies demonstrated that longdaysin suppressed colony and sphere formation, cell migration and invasion, expression of stemness markers, and tumor growth and proliferation [78]. A derivative of longdaysin, NCC007, has been shown to have stronger period effects, improved IC50s against CK1ɑ/δ, and lengthened periods of in vivo mouse behavioral rhythms, suggesting that further studies of this compound can prove to be more potent against cancer cells [79].
IC261 was first identified through a small molecule screen for antagonistic activity against CK1ɑ/δ/ε in the presence of low and high concentrations of ATP, the nucleotide substrate of CK1. IC261 selectivity inhibits all CK1 isoforms in comparison to protein kinases that are unrelated to CK1 [80]. Studies using MEF and C57MG mammary epithelial cells demonstrated that IC261 treatment leads to cell cycle arrest at the post-mitotic G1 phase in a p53-dependent manner while cells that lacked p53 overrode checkpoint control and had high concentrations of DNA, resulting in the development of micronuclei. IC261 caused centrosome amplification, resulting in multipolar mitosis, and induced apoptosis regardless of p53 status [81].
SR-2890 and SR-3029 are two CK1δ/ε specific inhibitors that were found to have potent anti-proliferative activity in an MTT assay against the A375 melanoma cell line. They also displayed suitable in vitro and in vivo pharmacokinetic properties and activity in experiments that assessed microsome stability, cytochrome P450 inhibition, and IC50 against CK1δ/ε. SR-3029 also exhibited brain penetration, which suggests its potential for the treatment of brain cancers [82]. In separate studies, SR-3029 showed anti-tumor effects across breast cancer subtypes [83].
Additional studies will need to be performed to elucidate the value of CK1 inhibitors as chronotherapeutic agents for disease modulation but pharmacological targeting of CK1 has proven to be a promising anti-cancer therapy option to pursue in a number of different cancer types.
CK2 inhibitors
CK2 is another clock-related protein kinase that has important functions in cell growth and apoptosis and, consequently, tumorigenesis. GO289 was identified to be a period lengthening compound through a chemical screen using U2OS mBmal1-dLuc reporter cells. Target identification and in vitro kinase assays revealed that GO289 potently inhibited CK2 and only had minimal effects on other kinases [84]. GO289 was found to inhibit phosphorylation of PER2. GO289 was identified to have anti-cancer effects on human renal cell carcinoma (RCC) lines, Caki-2, A498, and 769-P, and mice harboring MLL-AF9 based AML tumors. Researchers also found that GO289 had less potent effects on other RCC cell lines and, unlike AML stem cells which have intact circadian rhythms, AML cancer cells themselves have a disrupted circadian clock. These findings suggest that CK2 regulates growth depending on cell type and the application of GO289 must be thoroughly investigated in the cancer model of interest [39, 84]. There have been other identified and well-studied CK2 inhibitors including TBB, DMAT, and CX-4945 (Silmitasertib), which have displayed clinical effects on the circadian clock and are currently being investigated in a number of cancer clinical trials, however, it has to be noted that they do not display optimal selectivity against CK2 [84, 85].
GSK-3β inhibitors
As noted above, phosphorylation of CLOCK Serine-427 by GSK-3β results in its degradation. There is emerging evidence highlighting aberrant activation of GSK-3β in certain cancer types, resulting in sustained survival, stemness, proliferation, invasion, and insensitivity or resistance to chemoradiation. More specifically, GSK-3β was found to have increased expression and deregulated activity due to changes in phosphorylation, contributing to tumorigenesis and cancer progression. Genetic or pharmacological inhibition of GSK-3β led to attenuation of survival and proliferation and induced apoptosis in a number of different cancer cells. GSK-3β is thought to primarily function through β-catenin and c-MYC to confer anti-tumor effects [86]. Interestingly, compound library screens using the U2OS mBmal1-dLuc reporter cell line and LOPAC chemical library identified some GSK-3β inhibitors to have period shortening effects [87]. Further investigation is necessary to elucidate whether these GSK-3β inhibitors mediate anti-tumor effects through attenuation of circadian cycling in addition to targeting key oncogenic pathways.
Expert opinion and future perspectives
The growing research on the interplay between the circadian machinery and cancer has been unveiling the promising therapeutic potential of targeting the clock pathway in oncology. However, the biological understanding of the role of clock genes in cancer dynamics has yet to be fully clarified. In fact, based on the available evidence on pre-clinical studies in different cancer types, a unifying theory modeling the circadian biology of cancer cannot be formulated, implying a unique impact of clock pathway alterations according to each specific tumor type. This specificity reflects a differential effect of individual clock genes on the initiation and progression of specific cancers, the complex interaction between the circadian clock and cancer in which cancer-related alterations may, in turn, disrupt the clock function, and distinct molecular interactions between clock genes and specific oncogenes in each tissue. In this challenging scenario, when approaching the genetic validation of the role of clock genes and the identification of the optimal target for future drug development, a tailored disease-specific approach will be paramount to yield actionable insights. Nevertheless, the strong rationale behind this intriguing therapeutic approach is opening new horizons in cancer research which warrant further exploration. Moving forward, a deeper mechanistic understanding and dedicated studies exploiting in vivo cancer models are needed as a proof of concept to lay the basis for the use of clock proteins-targeting compounds as cancer therapeutics.
Notably, targeting the clock machinery may hold great potential to be effective as a combination treatment with available anti-cancer drugs. The link between clock genes disruption and several druggable cancer-related pathways has been established (Fig. 2), including angiogenesis, DNA repair, and RAS/MAPK signaling; furthermore, a few studies highlighted the predictive and prognostic impact of SNPs in clock genes on treatment outcomes in different cancer types (including CRC and gastric cancer) [88,89,90]. Hence, pharmacological modulation of proteins in the circadian machinery may offer a novel opportunity to improve other targeted treatment efficacy operating through its effects on major cancer-related pathways. To this extent, the connection between the circadian clock and the immune system may offer a valuable opportunity to enhance immunotherapy efficacy and possibly overcome resistance mechanisms. In fact, core components of the molecular clock—most notably BMAL1, CLOCK, and REV-ERBα—also control fundamental aspects of the immune response modulating immune cell development, function, and trafficking, thus impacting cancer immune surveillance [41, 65, 91]. Moreover, the core BMAL1::CLOCK transcription apparatus has been shown to play an essential role in the maintenance of stemness in at least two different cancer types (AML and GBM), therefore the use of small molecules targeting the clock to reduce BMAL1::CLOCK activity and inhibit CSCs may be an effective strategy to enhance cellular sensitivity to non-targeted conventional therapy. Further study into these mechanisms and possible synergistic effects of clock-targeting agents may provide fundamental evidence that will allow the design of rational combination strategies and promote innovative therapeutic advances in the next future.
Several promising compounds targeting core circadian components and clock-related proteins have shown signs of anti-cancer activity and hold the potential to be explored as novel anti-cancer agents (Table 1). However, the development of these drugs may face several challenges. A crucial step before moving forward will be the genetic and pharmacological validation of their target specificity and activity in different cancer types. The heterogeneous and sometimes conflicting role of clock genes according to different cancer types will have to be addressed to dissect the molecular mechanism of action and therapeutic efficacy of different compounds in each tumor of interest. An additional issue worthy of attention is that of potential off-target effects which may differentially contribute in varying degrees to the biological effect of these molecules. On a positive note, evidence shows a selective effect of clock targeting agents (particularly REV-ERB agonists and CRY stabilizers) on cancer cells versus normal cells, supporting the tailored activity of these drugs in cancer [40, 49]. Another major challenge in the future development of these compounds will be the selection and optimization of the lead molecules for further study, as several new-generation compounds are emerging in an effort to improve pharmacological potency and achieve more favorable pharmacokinetic/pharmacodynamic profiles and better bioavailability. The identification of cancer molecular characteristics which could impact clock-targeting agents’ efficacy will also be of paramount importance to select the most appropriate models for in vivo studies and eventually individualize treatment when moving towards clinical applications. Finally, the safety profile and potential adverse effects of these drugs will have to be carefully assessed in light of the pleiotropic effects of clock genes in human pathophysiology.
Conclusion
The integration of circadian biology into cancer research is leading to the development of a novel therapeutic paradigm with a unique potential to leverage the interplay between the circadian machinery and major cancer-related pathways. While extremely promising, further study is needed to fully establish the efficacy of circadian rhythm modulation as a therapeutic approach in cancer and to inform the modeling of circadian rhythm regulation of cancer biology. If successful, targeting of clock components may significantly improve anti-cancer treatment efficacy by exploiting differential strategies and distinct targets across multiple tumor types.
References
Roenneberg T, Merrow M. The circadian clock and human health. Curr Biol. 2016;26:R432–43.
Schibler U, Sassone-Corsi P. A web of circadian pacemakers. Cell. 2002;111:919–22.
Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18:164–79.
Sulli G, Manoogian ENC, Taub PR, Panda S. Training the circadian clock, clocking the drugs, and drugging the clock to prevent, manage, and treat chronic diseases. Trends Pharmacol Sci. 2018;39:812–27.
Benna C, Helfrich-Forster C, Rajendran S, Monticelli H, Pilati P, Nitti D, et al. Genetic variation of clock genes and cancer risk: a field synopsis and meta-analysis. Oncotarget. 2017;8:23978–95.
Sancar A, Van Gelder RN. Clocks, cancer, and chronochemotherapy. Science. 2021;371:eabb0738.
Panda S, Sato TK, Castrucci AM, Rollag MD, DeGrip WJ, Hogenesch JB, et al. Melanopsin (Opn4) requirement for normal light-induced circadian phase shifting. Science. 2002;298:2213–6.
Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. 2012;35:445–62.
Kondratov RV, Chernov MV, Kondratova AA, Gorbacheva VY, Gudkov AV, Antoch MP. BMAL1-dependent circadian oscillation of nuclear CLOCK: posttranslational events induced by dimerization of transcriptional activators of the mammalian clock system. Genes Dev. 2003;17:1921–32.
Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–67.
Cao X, Yang Y, Selby CP, Liu Z, Sancar A. Molecular mechanism of the repressive phase of the mammalian circadian clock. Proc Natl Acad Sci USA. 2021;118:e2021174118.
Solt LA, Kojetin DJ, Burris TP. The REV-ERBs and RORs: molecular links between circadian rhythms and lipid homeostasis. Fut Med Chem. 2011;3:623–38.
Yoshitane H, Takao T, Satomi Y, Du NH, Okano T, Fukada Y. Roles of CLOCK phosphorylation in suppression of E-box-dependent transcription. Mol Cell Biol. 2009;29:3675–86.
Luciano AK, Zhou W, Santana JM, Kyriakides C, Velazquez H, Sessa WC. CLOCK phosphorylation by AKT regulates its nuclear accumulation and circadian gene expression in peripheral tissues. J Biol Chem. 2018;293:9126–36.
Liu N, Zhang EE. Phosphorylation regulating the ratio of intracellular CRY1 protein determines the circadian period. Front Neurol. 2016;7:159.
Tamaru T, Hirayama J, Isojima Y, Nagai K, Norioka S, Takamatsu K, et al. CK2alpha phosphorylates BMAL1 to regulate the mammalian clock. Nat Struct Mol Biol. 2009;16:446–8.
Tsuchiya Y, Akashi M, Matsuda M, Goto K, Miyata Y, Node K, et al. Involvement of the protein kinase CK2 in the regulation of mammalian circadian rhythms. Sci Signal. 2009;2:ra26.
Schernhammer ES, Kroenke CH, Laden F, Hankinson SE. Night work and risk of breast cancer. Epidemiology. 2006;17:108–11.
Wegrzyn LR, Tamimi RM, Rosner BA, Brown SB, Stevens RG, Eliassen AH, et al. Rotating night-shift work and the risk of breast cancer in the nurses’ health studies. Am J Epidemiol. 2017;186:532–40.
Walasa WM, Carey RN, Si S, Fritschi L, Heyworth JS, Fernandez RC, et al. Association between shiftwork and the risk of colorectal cancer in females: a population-based case–control study. Occup Environ Med. 2018;75:344–50.
Viswanathan AN, Hankinson SE, Schernhammer ES. Night shift work and the risk of endometrial cancer. Cancer Res. 2007;67:10618–22.
Flynn-Evans EE, Mucci L, Stevens RG, Lockley SW. Shiftwork and prostate-specific antigen in the national health and nutrition examination survey. J Natl Cancer Inst. 2013;105:1292–7.
Straif K, Baan R, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, et al. Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 2007;8:1065–6.
Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, et al. Circadian homeostasis of liver metabolism suppresses hepatocarcinogenesis. Cancer Cell. 2016;30:909–24.
Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS ONE. 2010;5:e10995.
Fu L, Pelicano H, Liu J, Huang P, Lee CC. The circadian gene period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50.
Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, et al. Circadian rhythm disruption promotes lung tumorigenesis. Cell Metab. 2016;24:324–31.
Pariollaud M, Lamia KA. Cancer in the fourth dimension: what is the impact of circadian disruption? Cancer Discov. 2020;10:1455–64.
Ye Y, Xiang Y, Ozguc FM, Kim Y, Liu CJ, Park PK, et al. The genomic landscape and pharmacogenomic interactions of clock genes in cancer chronotherapy. Cell Syst. 2018;6:314–28. e2
Sulli G, Lam MTY, Panda S. Interplay between circadian clock and cancer: new frontiers for cancer treatment. Trends Cancer. 2019;5:475–94.
de Assis LVM, Kinker GS, Moraes MN, Markus RP, Fernandes PA, Castrucci AML. Expression of the circadian clock gene BMAL1 positively correlates with antitumor immunity and patient survival in metastatic melanoma. Front Oncol. 2018;8:185.
Li W, Liu L, Liu D, Jin S, Yang Y, Tang W, et al. Decreased circadian component Bmal1 predicts tumor progression and poor prognosis in human pancreatic ductal adenocarcinoma. Biochem Biophys Res Commun. 2016;472:156–62.
Ramos CA, Ouyang C, Qi Y, Chung Y, Cheng CT, LaBarge MA, et al. A non-canonical function of BMAL1 metabolically limits obesity-promoted triple-negative breast cancer. iScience. 2020;23:100839.
Fekry B, Ribas-Latre A, Baumgartner C, Deans JR, Kwok C, Patel P, et al. Incompatibility of the circadian protein BMAL1 and HNF4α in hepatocellular carcinoma. Nat Commun. 2018;9:4349.
Taniguchi H, Fernández AF, Setién F, Ropero S, Ballestar E, Villanueva A, et al. Epigenetic inactivation of the circadian clock gene BMAL1 in hematologic malignancies. Cancer Res. 2009;69:8447–54.
Zhang S, Zhang J, Deng Z, Liu H, Mao W, Jiang F, et al. Circadian clock components RORα and Bmal1 mediate the anti-proliferative effect of MLN4924 in osteosarcoma cells. Oncotarget. 2016;7:66087–99.
Yeh CM, Shay J, Zeng TC, Chou JL, Huang TH, Lai HC, et al. Epigenetic silencing of ARNTL, a circadian gene and potential tumor suppressor in ovarian cancer. Int J Oncol. 2014;45:2101–7.
Qu M, Duffy T, Hirota T, Kay SA. Nuclear receptor HNF4A transrepresses CLOCK:BMAL1 and modulates tissue-specific circadian networks. Proc Natl Acad Sci USA. 2018;115:E12305–e12.
Puram RV, Kowalczyk MS, de Boer CG, Schneider RK, Miller PG, McConkey M, et al. Core circadian clock genes regulate leukemia stem cells in AML. Cell 2016;165:303–16.
Dong Z, Zhang G, Qu M, Gimple RC, Wu Q, Qiu Z, et al. Targeting glioblastoma stem cells through disruption of the circadian clock. Cancer Discov. 2019;9:1556–73.
Chen P, Hsu WH, Chang A, Tan Z, Lan Z, Zhou A, et al. Circadian regulator CLOCK recruits immune-suppressive microglia into the GBM tumor microenvironment. Cancer Discov. 2020;10:371–81.
Kinouchi K, Sassone-Corsi P. Metabolic rivalry: circadian homeostasis and tumorigenesis. Nat Rev Cancer. 2020;20:645–61.
Yang MY, Lin PM, Hsiao HH, Hsu JF, Lin HY, Hsu CM, et al. Up-regulation of PER3 Expression Is correlated with better clinical outcome in acute leukemia. Anticancer Res. 2015;35:6615–22.
Cao Q, Gery S, Dashti A, Yin D, Zhou Y, Gu J, et al. A role for the clock gene per1 in prostate cancer. Cancer Res. 2009;69:7619–25.
Gery S, Virk RK, Chumakov K, Yu A, Koeffler HP. The clock gene Per2 links the circadian system to the estrogen receptor. Oncogene. 2007;26:7916–20.
Hwang-Verslues WW, Chang P-H, Jeng Y-M, Kuo W-H, Chiang P-H, Chang Y-C, et al. Loss of corepressor PER2 under hypoxia up-regulates OCT1-mediated EMT gene expression and enhances tumor malignancy. Proc Natl Acad Sci USA. 2013;110:2331–6.
Tong H, Liu X, Li T, Qiu W, Peng C, Shen B, et al. NR1D2 accelerates hepatocellular carcinoma progression by driving the epithelial-to-mesenchymal transition. OncoTargets Ther. 2020;13:3931–42.
Tao L, Yu H, Liang R, Jia R, Wang J, Jiang K, et al. Rev-erbα inhibits proliferation by reducing glycolytic flux and pentose phosphate pathway in human gastric cancer cells. Oncogenesis. 2019;8:57.
Sulli G, Rommel A, Wang X, Kolar MJ, Puca F, Saghatelian A, et al. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature. 2018;553:351–5.
Yu H, Meng X, Wu J, Pan C, Ying X, Zhou Y, et al. Cryptochrome 1 overexpression correlates with tumor progression and poor prognosis in patients with colorectal cancer. PLoS ONE. 2013;8:e61679.
Shafi AA, McNair CM, McCann JJ, Alshalalfa M, Shostak A, Severson TM, et al. The circadian cryptochrome, CRY1, is a pro-tumorigenic factor that rhythmically modulates DNA repair. Nat Commun. 2021;12:401.
Cadenas C, van de Sandt L, Edlund K, Lohr M, Hellwig B, Marchan R, et al. Loss of circadian clock gene expression is associated with tumor progression in breast cancer. Cell Cycle. 2014;13:3282–91.
Mannic T, Meyer P, Triponez F, Pusztaszeri M, Le Martelot G, Mariani O, et al. Circadian clock characteristics are altered in human thyroid malignant nodules. J Clin Endocrinol Metab. 2013;98:4446–56.
Ozturk N, Lee JH, Gaddameedhi S, Sancar A. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci USA. 2009;106:2841–6.
van der Watt PJ, Roden LC, Davis KT, Parker MI, Leaner VD. Circadian oscillations persist in cervical and esophageal cancer cells displaying decreased expression of tumor-suppressing circadian clock genes. Mol Cancer Res. 2020;18:1340–53.
Shostak A, Ruppert B, Ha N, Bruns P, Toprak UH, Eils R, et al. MYC/MIZ1-dependent gene repression inversely coordinates the circadian clock with cell cycle and proliferation. Nat Commun. 2016;7:11807.
Altman BJ, Hsieh AL, Sengupta A, Krishnanaiah SY, Stine ZE, Walton ZE, et al. MYC disrupts the circadian clock and metabolism in cancer cells. Cell Metab. 2015;22:1009–19.
Huber AL, Papp SJ, Chan AB, Henriksson E, Jordan SD, Kriebs A, et al. CRY2 and FBXL3 cooperatively degrade c-MYC. Mol Cell. 2016;64:774–89.
Noel R, Song X, Shin Y, Banerjee S, Kojetin D, Lin L, et al. Synthesis and SAR of tetrahydroisoquinolines as Rev-erbα agonists. Bioorg Med Chem Lett. 2012;22:3739–42.
Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485:62–8.
Shen W, Zhang W, Ye W, Wang H, Zhang Q, Shen J, et al. SR9009 induces a REV-ERB dependent anti-small-cell lung cancer effect through inhibition of autophagy. Theranostics. 2020;10:4466–80.
Dierickx P, Emmett MJ, Jiang C, Uehara K, Liu M, Adlanmerini M, et al. SR9009 has REV-ERB-independent effects on cell proliferation and metabolism. Proc Natl Acad Sci USA. 2019;116:12147–52.
Kojetin D, Wang Y, Kamenecka TM, Burris TP. Identification of SR8278, a synthetic antagonist of the nuclear heme receptor REV-ERB. ACS Chem Biol. 2011;6:131–4.
De Mei C, Ercolani L, Parodi C, Veronesi M, Lo Vecchio C, Bottegoni G. et al. Dual inhibition of REV-ERBβ and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene. 2015;34:2597–608.
Amir M, Chaudhari S, Wang R, Campbell S, Mosure SA, Chopp LB, et al. REV-ERBα regulates T(H)17 cell development and autoimmunity. Cell Rep. 2018;25:3733–49. e8
Wang Y, Kumar N, Nuhant P, Cameron MD, Istrate MA, Roush WR, et al. Identification of SR1078, a synthetic agonist for the orphan nuclear receptors RORα and RORγ. ACS Chem Biol. 2010;5:1029–34.
Wang Y, Solt LA, Kojetin DJ, Burris TP. Regulation of p53 stability and apoptosis by a ROR agonist. PLoS ONE. 2012;7:e34921.
He B, Nohara K, Park N, Park YS, Guillory B, Zhao Z, et al. The small molecule nobiletin targets the molecular oscillator to enhance circadian rhythms and protect against metabolic syndrome. Cell Metab. 2016;23:610–21.
Ashrafizadeh M, Zarrabi A, Saberifar S, Hashemi F, Hushmandi K, Hashemi F, et al. Nobiletin in cancer therapy: how this plant derived-natural compound targets various oncogene and onco-suppressor pathways. Biomedicines. 2020;8:110.
Hirota T, Lee JW, St John PC, Sawa M, Iwaisako K, Noguchi T, et al. Identification of small molecule activators of cryptochrome. Science. 2012;337:1094–7.
Oshima T, Yamanaka I, Kumar A, Yamaguchi J, Nishiwaki-Ohkawa T, Muto K, et al. C-H activation generates period-shortening molecules that target cryptochrome in the mammalian circadian clock. Angew Chem. 2015;54:7193–7.
Lee JW, Hirota T, Kumar A, Kim NJ, Irle S, Kay SA. Development of small-molecule cryptochrome stabilizer derivatives as modulators of the circadian clock. ChemMedChem. 2015;10:1489–97.
Miller S, Son YL, Aikawa Y, Makino E, Nagai Y, Srivastava A, et al. Isoform-selective regulation of mammalian cryptochromes. Nat Chem Biol. 2020;16:676–85.
Miller S, Aikawa Y, Sugiyama A, Nagai Y, Hara A, Oshima T, et al. An isoform-selective modulator of cryptochrome 1 regulates circadian rhythms in mammals. Cell Chem Biol. 2020;27:1192–8. e5
Chun SK, Jang J, Chung S, Yun H, Kim NJ, Jung JW, et al. Identification and validation of cryptochrome inhibitors that modulate the molecular circadian clock. ACS Chem Biol. 2014;9:703–10.
Chun SK, Chung S, Kim HD, Lee JH, Jang J, Kim J, et al. A synthetic cryptochrome inhibitor induces anti-proliferative effects and increases chemosensitivity in human breast cancer cells. Biochem Biophys Res Commun. 2015;467:441–6.
Hirota T, Lee JW, Lewis WG, Zhang EE, Breton G, Liu X, et al. High-throughput chemical screen identifies a novel potent modulator of cellular circadian rhythms and reveals CKIα as a clock regulatory kinase. PLoS Biol. 2010;8:e1000559.
Xiong Y, Zhou L, Su Z, Song J, Sun Q, Liu SS, et al. Longdaysin inhibits Wnt/β-catenin signaling and exhibits antitumor activity against breast cancer. OncoTargets Ther. 2019;12:993–1005.
Lee JW, Hirota T, Ono D, Honma S, Honma KI, Park K, et al. Chemical control of mammalian circadian behavior through dual inhibition of casein kinase Iα and δ. J Med Chem. 2019;62:1989–98.
Mashhoon N, DeMaggio AJ, Tereshko V, Bergmeier SC, Egli M, Hoekstra MF, et al. Crystal structure of a conformation-selective casein kinase-1 inhibitor. J Biol Chem. 2000;275:20052–60.
Behrend L, Milne DM, Stöter M, Deppert W, Campbell LE, Meek DW, et al. IC261, a specific inhibitor of the protein kinases casein kinase 1-delta and -epsilon, triggers the mitotic checkpoint and induces p53-dependent postmitotic effects. Oncogene. 2000;19:5303–13.
Bibian M, Rahaim RJ, Choi JY, Noguchi Y, Schürer S, Chen W, et al. Development of highly selective casein kinase 1δ/1ε (CK1δ/ε) inhibitors with potent antiproliferative properties. Bioorg Med Chem Lett. 2013;23:4374–80.
Rosenberg LH, Lafitte M, Quereda V, Grant W, Chen W, Bibian M, et al. Therapeutic targeting of casein kinase 1δ in breast cancer. Sci Transl Med. 2015;7:318ra202.
Oshima T, Niwa Y, Kuwata K, Srivastava A, Hyoda T, Tsuchiya Y, et al. Cell-based screen identifies a new potent and highly selective CK2 inhibitor for modulation of circadian rhythms and cancer cell growth. Sci Adv. 2019;5:eaau9060.
Lian H, Su M, Zhu Y, Zhou Y, Soomro SH, Fu H. Protein kinase CK2, a potential therapeutic target in carcinoma management. Asian Pac J Cancer Prev. 2019;20:23–32.
Duda P, Akula SM, Abrams SL, Steelman LS, Martelli AM, Cocco L, et al. Targeting GSK3 and associated signaling pathways involved in cancer. Cells. 2020;9:1110.
Hirota T, Lewis WG, Liu AC, Lee JW, Schultz PG, Kay SA. A chemical biology approach reveals period shortening of the mammalian circadian clock by specific inhibition of GSK-3beta. Proc Natl Acad Sci USA. 2008;105:20746–51.
Battaglin F, Cao S, Millstein J, Puccini A, Togunaka R, Naseem M, et al. Polymorphism in the circadian clock pathway to predict outcome in patients (pts) with metastatic colorectal cancer (mCRC): data from TRIBE and FIRE-3 phase III trials. J Clin Oncol. 2018;36(15_suppl):3576.
Burgermeister E, Battaglin F, Eladly F, Wu W, Herweck F, Schulte N, et al. Aryl hydrocarbon receptor nuclear translocator-like (ARNTL/BMAL1) is associated with bevacizumab resistance in colorectal cancer via regulation of vascular endothelial growth factor A. EBioMedicine. 2019;45:139–54.
Qu F, Qiao Q, Wang N, Ji G, Zhao H, He L, et al. Genetic polymorphisms in circadian negative feedback regulation genes predict overall survival and response to chemotherapy in gastric cancer patients. Sci Rep. 2016;6:22424.
Wu Y, Tao B, Zhang T, Fan Y, Mao R. Pan-cancer analysis reveals disrupted circadian clock associates with T cell exhaustion. Front Immunol. 2019;10:2451.
Li Y, Basti A, Yalçin M, Relógio A. Circadian dysregulation of the TGFβ/SMAD4 pathway modulates metastatic properties and cell fate decisions in pancreatic cancer cells. iScience. 2020;23:101551.
Sato F, Bhawal UK, Yoshimura T, Muragaki Y. DEC1 and DEC2 crosstalk between circadian rhythm and tumor progression. J Cancer. 2016;7:153–9.
Westermaier Y, Ruiz-Carmona S, Theret I, Perron-Sierra F, Poissonnet G, Dacquet C, et al. Binding mode prediction and MD/MMPBSA-based free energy ranking for agonists of REV-ERBα/NCoR. J Comput-Aided Mol Des. 2017;31:755–75.
Zou K, Li Z, Zhang Y, Zhang H-Y, Li B, Zhu W-L, et al. Advances in the study of berberine and its derivatives: a focus on anti-inflammatory and anti-tumor effects in the digestive system. Acta Pharm Sin. 2017;38:157–67.
Yuan F, Li D, Guo M, Fang T, Sun J, Qi F, et al. IC261 suppresses progression of hepatocellular carcinoma in a casein kinase 1 δ/ε independent manner. Biochem Biophys Res Commun. 2020;523:809–15.
Zhang Y, Zhang Y, Li M, Meng F, Yu Z, Chen Y, et al. Combination of SB431542, CHIR99021 and PD0325901 has a synergic effect on abrogating valproic acid‑induced epithelial‑mesenchymal transition and stemness in HeLa, 5637 and SCC‑15 cells. Oncol Rep. 2019;41:3545–54.
Kitabayashi T, Dong Y, Furuta T, Sabit H, Jiapaer S, Zhang J, et al. Identification of GSK3β inhibitor kenpaullone as a temozolomide enhancer against glioblastoma. Sci Rep. 2019;9:10049.
Tao NN, Zhang ZZ, Ren JH, Zhang J, Zhou YJ, Wai Wong VK, et al. Overexpression of ubiquitin-conjugating enzyme E2 L3 in hepatocellular carcinoma potentiates apoptosis evasion by inhibiting the GSK3β/p65 pathway. Cancer Lett. 2020;481:1–14.
Duffy DJ, Krstic A, Schwarzl T, Higgins DG, Kolch W. GSK3 inhibitors regulate MYCN mRNA levels and reduce neuroblastoma cell viability through multiple mechanisms, including p53 and Wnt signaling. Mol Cancer Therap. 2014;13:454–67.
Sha Z, Zhou J, Wu Y, Zhang T, Li C, Meng Q, et al. BYSL promotes glioblastoma cell migration, invasion, and mesenchymal transition through the GSK-3β/β-catenin signaling pathway. Front Oncol. 2020;10:565225.
Lo WY, Chang NW. An indirubin derivative, indirubin-3′-monoxime suppresses oral cancer tumorigenesis through the downregulation of survivin. PLoS ONE. 2013;8:e70198.
Acknowledgements
We thank Tsuyoshi Hirota and Jamie Cope for the critical reading of the manuscript.
Funding
Partly supported by the USC/Norris Translational Team Accelerator Project Initiative, the National Cancer Institute (grant numbers P30CA014089, UG1CA180830, R01CA23866201), the National Institute of Neurological Disorders and Stroke (grant number 1F31NS120654-01), the Gloria Borges WunderGlo Foundation, the Gene Gregg Pancreas Research Fund, and the Victoria Wilson Research Fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SAK serves on the board of Synchronicity Pharma and received research support from the company. Other authors declare no potential competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Battaglin, F., Chan, P., Pan, Y. et al. Clocking cancer: the circadian clock as a target in cancer therapy. Oncogene 40, 3187–3200 (2021). https://doi.org/10.1038/s41388-021-01778-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-021-01778-6
- Springer Nature Limited
This article is cited by
-
Evaluation of small-molecule modulators of the circadian clock: promising therapeutic approach to cancer
Molecular Biology Reports (2024)
-
The circadian clock as a potential biomarker and therapeutic target in pancreatic cancer
Molecular and Cellular Biochemistry (2024)
-
Targeted screening and identification of chlorhexidine as a pro-myogenic circadian clock activator
Stem Cell Research & Therapy (2023)
-
Integrated multi-omics analysis reveals the molecular interplay between circadian clocks and cancer pathogenesis
Scientific Reports (2023)
-
ROR activation by Nobiletin enhances antitumor efficacy via suppression of IκB/NF-κB signaling in triple-negative breast cancer
Cell Death & Disease (2022)