
Abstract
Lung squamous cell carcinoma (LUSC) represents a major subtype of non-small cell lung cancer with limited treatment options. Previous studies have elucidated the complex genetic landscape of LUSC and revealed multiple altered genes and pathways. However, in stark contrast to lung adenocarcinoma, few targetable driver mutations have been established so far and targeted therapies for LUSC remain unsuccessful. Immunotherapy has revolutionized LUSC treatment and is currently approved as the new standard of care. To gain a better understanding of the LUSC biology, improved modeling systems are urgently needed. Preclinical models, particularly those mimicking human disease with an intact tumor immune microenvironment, are an invaluable tool to study cancer development and evaluate new therapeutic targets. Here, we discuss recent advances in LUSC preclinical models, with a focus on genetically engineered mouse models (GEMMs) and organoids, in the context of evolving precision medicine and immunotherapy.
Similar content being viewed by others


Introduction
Lung cancer remains the most commonly diagnosed malignancy and the leading cause of cancer death worldwide [1]. The majority of lung cancer is categorized as non-small cell carcinoma (NSCLC), of which are mainly lung adenocarcinoma (LUAD) and lung squamous carcinoma (LUSC). LUSC is characterized by its unique squamous appearance; it typically occurs at the proximal part of the lung and originates primarily from basal cells of the bronchi [2]. Genomic analysis of LUSC patient tumors revealed numerous highly altered genes and pathways, but actionable driver mutations are rare [3,4,5]. Several targeted therapies tested in LUSC patients have demonstrated very limited clinical benefits [6]. This is in contrast to LUAD, which has targetable driver mutations, such as EGFR, ALK and ROS1 [7]. In the past decade, treatments enhancing the immune system to target cancer have dramatically shifted the paradigm of cancer therapies [8]. Compared to conventional chemotherapy, immunotherapies such as anti-programmed cell death 1 (PD-1)/programmed cell death ligand 1 (PD-L1), lead to a durable response and manageable adverse effects [9]. Of note, several immunotherapy drugs have been approved for LUSC patients based on their substantial clinical benefits [6]. However, there is still a large proportion of LUSC patients who fail to respond to current immunotherapy. Identifying biomarkers for immunotherapy and exploring more effective therapeutics represent an urgent unmet need for LUSC patients.
Preclinical models have been essential in studying lung cancer development and in testing therapeutics [10, 11]. Due to the lack of established driver mutations, the development of LUSC preclinical models that recapitulate human LUSC genetics and pathology remains challenging. Based on the frequently mutated genes in LUSC, multiple genetically engineered mouse models (GEMMs) of LUSC have been successfully characterized [12]. These de novo LUSC models provide instrumental tools to study cell origin, pathogenesis and tumor microenvironment of LUSC. Moreover, recent progress in organoids technology allows culturing and engineering primary tumor cells and normal stem cells in vitro [13, 14]. Several studies have utilized the organoids system to successfully generate LUSC models, which has enabled driver mutation evaluation and therapeutic exploration [15,16,17]. Here, we discuss the latest progress in LUSC preclinical model development, focusing on GEMMs and organoids, in the era of targeted therapies and immunotherapies for lung cancer management.
Genomic hallmarks of lung squamous cell carcinoma
LUSC is strongly associated with smoking and has a relatively high mutational burden, with an average of 261–360 exonic mutations per tumor, which corresponds to a mean mutation rate of 8.1–8.71 mutations per megabase [3, 4]. Comprehensive molecular profiling of LUSC revealed numerous genomic alterations including TP53, CDKN2A, PTEN, PIK3CA, KEAP1, KMT2D and NFE2L2 [3, 4, 18, 19]. Of note, TP53 is the most commonly mutated gene (73–81%) and chromosome 3q26 amplification (including SOX2, PIK3CA and other genes) represents a distinct hallmark for LUSC. In addition, 8p11 (FGFR1, WHSC1L1), 7p11 (EGFR), 11q13 (CCND1) and 4q12 (KDR, KIT, PDGFRA), are also the regions of frequent amplification, while 2q37, 4q35 (CASP3), 9p21 (CDKN2A) and 10q23 (PTEN) are the regions of common deletion [3, 4].
These somatic alterations in LUSC are involved in numerous major signaling pathways essential for cancer formation and progression, which include oxidative stress response (NFE2L2, KEAP1 and CUL3), squamous differentiation (SOX2, TP63, NOTCH1), cell proliferation/apoptosis (TP53, CDKN2A, PTEN, PIK3CA) and chromatin remodeling (KMT2D, KDM6A) [3, 4]. How these pathways contribute to LUSC formation and progression is being actively investigated. Of note, the genomic landscape of LUSC is quite distinct from LUAD, while it is more similar to other squamous carcinomas such as head and neck squamous cell carcinoma [5, 20, 21]. In contrast to LUAD, driver mutations in LUSC are not clear. The most common drivers in LUAD, EGFR and KRAS, are rarely mutated in LUSC. Future research is warranted to elucidate the leading, or a combination of, driver mutations in LUSC tumor formation and progression.
Current treatment in advanced LUSC
Targeted therapies in LUSC
Targeted therapies against receptor tyrosine kinases have transformed the treatment in subsets of LUAD patients. For example, EGFR is one of the most prevalent oncogenes among LUAD patients and targeting EGFR with tyrosine kinase inhibitors (TKIs) substantially benefits patients with EGFR mutations [22,23,24,25,26,27]. Unfortunately, attempts for targeted treatment in LUSC patients remain mostly unsuccessful [6]. FGFR1 is frequently amplified in LUSC, which makes it a potential actionable target. Several FGFR inhibitors (BGJ398, AZD4547 and JNJ-42756493) are under investigation in advanced stage LUSC patients. However, data from early phase trials did not show significant clinical benefits [28,29,30]. Another potential target is PI3K, and early phase studies explored several potential drug candidates including Taselisib, Buparlisib and LY302341 in PI3K-deregulated LUSC patients [31,32,33]. However, these trials did not meet the primary objective of overall response rate (ORR) and progression-free survival (PFS) [31, 33]. In 2014, the Lung Master Protocol (Lung-MAP), a biomarker-driven protocol supported by the National Cancer Institute (NCI), was initiated to advance the development of targeted therapies for genetically stratified LUSC [34]. Similarly, the National Lung Matrix Trial is another systematic trial aimed to identify genotype-based therapies for NSCLC patients including LUSC [35]. Current data from both studies indicated the clinical benefits for targeted therapy remain low in LUSC patients [35,36,37]. The umbrella design of these trials enables assessing biomarkers and treatments under a unified protocol, facilitating precision medicine and targeted therapies in LUSC.
Immunotherapies in LUSC
Immunotherapies such as immune checkpoint blockades (ICBs) have revolutionized the treatment of many types of cancer, including LUSC. ICBs promote antitumor response by targeting against immune suppressive pathways modulated by cytotoxic T lymphocyte–associated protein 4 (CTLA-4) or PD-1/PD-L1 signaling [8]. So far, several ICBs have been approved for the first- and second-line treatment for LUSC patients (Table 1). In the first-line treatment, pembrolizumab (an anti-PD-1 antibody) and atezolizumab (an anti-PD-L1 antibody), was approved as monotherapy in LUSC patients with tumor PD-L1 expression ≥50% [38,39,40]. In addition, the combination of nivolumab (an anti-PD-1 antibody) plus ipilimumab (an anti-CTLA-4 antibody), was approved for these patients [41]. For LUSC patients with median expression of tumor PD-L1 (1–49%), pembrolizumab monotherapy [42] and the combination of nivolumab plus ipilimumab also received approval [41]. Additionally, pembrolizumab plus chemotherapy, as well as nivolumab plus ipilimumab combined with two cycles of chemotherapy was also approved in the first line setting for LUSC patients regardless of tumor PD-L1 expression [43, 44]. As the second-line treatment, nivolumab [45], pembrolizumab [46] and atezolizumab [47, 48] have been approved as monotherapy by the FDA based on the substantial clinical benefits.
In summary, ICBs have become the new standard of care and no targeted therapies have been approved for LUSC. Future studies are urgently needed to identify actionable driver targets in genetically stratified patients, and to further explore effective combinational immunotherapies.
Preclinical models for LUSC
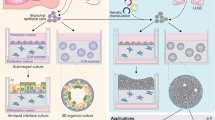
Preclinical models of lung cancer are powerful tools to study disease development and evaluate novel therapeutics. The development of LUSC preclinical models has been challenging, partially due to its complex genetics. As seen with their wide application in modeling other cancer types, human cancer cell lines and patient-derived xenografts (PDXs) have also been used to characterize LUSC signature and disease progression [12]. Additionally, several carcinogens including 3-methylcholanthrene (MCA), N-nitroso-methyl-bis-chloroethylurea (NMBCU) or N-nitroso-tris-chlo-oethylurea (NTCU), have been used to induce LUSC formation in mouse lungs [49,50,51,52]. The PDXs and carcinogen-induced models have been extensively discussed in several recent reviews [10, 12, 53, 54]. GEMMs in cancer research are de novo tumor mouse models generated through conditionally activating oncogenes or deleting tumor suppressor genes in the targeted tissue regions. For LUAD, a series of oncogene-driven GEMMs have been successfully generated and have contributed significantly to our understanding of LUAD tumorigenesis and discovery of new treatment [10, 55]. In the past decade, there is also a growing interest in utilizing GEMMs for modeling LUSC. Tumors arising from GEMMs develop in the tissue native environment that is immune-proficient. Thus, GEMMs are particularly useful for immuno-oncology research, such as testing immunotherapies. In addition, with the recent development of 3D culture and genetic engineering technologies, organoids have emerged as a promising platform for studying LUSC. Here, we discuss recent progress in LUSC preclinical models, with a focus on GEMMs and organoids models (Fig. 1).

Schematic summary of different preclinical models for LUSC.
Genetically engineered mouse models (GEMMs)
Sox2 overexpression models
The human LUSC genomics revealed that squamous differentiation signaling is one of the major deregulated pathways in patients [3, 4]. As a key player in squamous differentiation, SOX2 is amplified in 20–40% and overexpressed in more than 60% LUSC [3, 56, 57]. Sox2 overexpression alone, however, is not able to drive LUSC formation in mouse models [58]. Overexpression of Sox2 with Lkb1 loss promotes LUSC formation in mice [59, 60]. 6–10 months after viral induction, lung tumors with predominantly squamous histology were observed in mice [59]. The Lkb1−/− Sox2OE (Sox2 overexpression) tumors can be further facilitated by loss of Nkx2-1, a transcription factor in LUSC with low expression [60]. Moreover, Sox2 overexpression with Nkx2-1 inactivation also leads to LUSC formation [61]. Interestingly, Sox2 overexpression in combination with Cdkn2a, Cdkn2b and Pten loss, drives pure LUSC formation in multiple cell origins including basal cells, club cells and AT2 cells of the mouse lungs [62]. These studies indicate that Sox2 plays a major role in LUSC tumorigenesis.
Lkb1 deletion models
LKB1/STK11 serine/threonine kinase is highly mutated in lung cancer [63], and Lkb1 deletion has been widely used in mouse models of LUSC. It was originally reported in 2007 that simultaneous activation of KrasG12D with deletion of Lkb1, results in mixed adenocarcinoma and squamous carcinoma histology in the mouse lung [64]. Subsequent research demonstrated that Lkb1 is involved in the trans-differentiation from LUAD into LUSC in the KrasG12D driven tumors [65,66,67]. KRAS has relatively low mutation frequency in LUSC and occurs at 0.9%, 3.9%, and 3.6% in the TCGA, GENIE and COSMIC database respectively. Therefore, the KrasG12D Lkb1−/− model only represents a small subset of human LUSC. Other LUSC GEMMs harboring Lkb1 deletion have also been reported. One is established through Lkb1 deletion and Sox2 overexpression as described above [59, 60]. Another model involves biallelic inactivation of Lkb1 and Pten [68], which develops squamous tumors in the lung 40–50 weeks after Ad5-CMV-Cre induction. Similar to human LUSC, these Lkb1−/− Pten−/− mouse tumors have elevated expression of SOX2, P63 and KRT5, and are enriched for a squamous differentiation signature [68]. Interestingly, a recent study reported that inactivation of Lkb1 alone is sufficient to give rise to lung tumors with squamous histology [69]. Instead of using the Cre that targets all cells in the lung, the authors used CCSPi-Cre; Lkb1fl/fl mouse to specifically delete Lkb1 in clara/club cells. Lkb1 inactivation through this strategy leads to mixed histology lung tumors of squamous carcinoma and adenocarcinoma after one year. The authors also found that the formation of CCSPi-Cre; Lkb1fl/fl driven tumors was further accelerated by Jnk1/2 loss [69]. In summary, these studies highlight the essential role of Lkb1 in LUSC development and trans-differentiation from LUAD into LUSC in mouse lungs. Despite the pivotal function of Lkb1 in mouse LUSC, it is worth noting that less than 3% of LUSC patients harbor LKB1 mutations based on several cancer databases (1.8% in TCGA, 2.3% in GENIE and 2.2% in COSMIC). Given that LKB1 genetic alterations involve point mutation, exonic loss and deletion of whole gene allele [70], it is possible that LKB1 mutation frequency is underestimated due to the limitations of current sequencing methods [71]. Indeed, using multiplex ligation-dependent probe amplification (MLPA) analysis, LKB1 mutation was identified in ~13% (13/101) of LUSC patients [67]. Furthermore, loss of Lkb1 in mouse lung could recapitulate the biochemical changes and dysregulated signaling observed in human LUSC. For example, mTOR, one of the major downstream pathways of LKB1 [72], is frequently deregulated in human LUSC by the PTEN-PI3K signaling axis [3, 73, 74]. Future research needs to explore the role of LKB1 and associated signaling in human LUSC.
LUSC GEMMs driven by other mutations
In addition to Sox2 and Lkb1 alterations, other mutations have also shown promise in generating LUSC mouse models. Constitutive activation of a dominant negative mutant form of IKKα leads to squamous cell carcinoma in the mice lungs [75]. These IKKαKA/KA mice, however, usually die at 6–10 months after induction, which is possibly caused by tumor lesions in the skin and other tissues. Rescuing IKKα expression in the skin of these mice leads to LUSC in all mice [75]. FBXW7 is mutated in around 5–6% of LUSC. Simultaneous activation of KrasG12D and deletion of Fbxw7 results in adenocarcinoma and squamous cell carcinoma [76]. Interestingly, using virus that express Cre recombinase under specific promoters, the authors further uncovered that the CC10+ club cells, rather than the CK5+ basal cells, are likely to be the cell origin of LUSC in this model [76]. Furthermore, Camolotto et al. revealed that transcriptional factors FoxA1 and FoxA2 contribute to squamous identity in lung cancer [77]. They demonstrated that FoxA1/2 are downregulated in the squamous compartment of mouse tumor, as well as in human adeno-squamous carcinoma. Conditionally deleting Foxa1 or Foxa2 in KrasG12D Nkx2-1−/− tumors leads to trans-differentiation of lung tumors toward squamous identity [77].
GEMMs for LUSC immuno-oncology research
As discussed above, lung tumors from GEMMs are developed in the native immunocompetent microenvironment manifested by extensive infiltrating immune populations. Multiple studies have utilized GEMMs to characterize the LUSC immune microenvironment. For example, The Lkb1−/− Pten−/− LUSC tumors display the signature of immune-suppression including high PD-L1 expression in tumor and a large number of tumor-associated neutrophils (TANs) infiltration [68]. Similarly, using the Lkb1−/− Sox2OE model, Mollaoglu et al. reported that mouse LUSC tumors are enriched with TANs [60]. Mechanistically, SOX2 enhances TANs recruitment through repressing NKX2-1 activity, which elevates the expression of chemokine CXCL5. Moreover, the Sox2OE Cdkn2a−/− Cdkn2b−/− Pten−/− LUSC tumors have high percentages of neutrophil infiltration and express high levels of PD-L1 [62]. All together, these findings from LUSC GEMMs harboring different genetic alterations suggest that TANs might play an important role in shaping the immune suppressive environment in LUSC.
In conclusion, LUSC GEMMs (summarized in Table 2) provide a precious platform to study de novo tumor formation and progression in the native tissue environment. Due to the long breeding time and tumor latency, only a few genes have been manipulated to model LUSC GEMMs. Recently, several approaches have been established to facilitate GEMMs development in cancer research. One involves engineering the embryonic stem cells (ESC) from preexisting GEMMs. The ESC derived from established GEMMs can be used to introduce additional genetic modifications [78,79,80]. For example, this GEMM-ESC strategy has been used to study the role of MET in BRCA1 deficient metaplastic breast cancer [81] and the function of PTEN and MYC in pancreatic cancer [82]. Another major advancement of GEMMs in cancer research is to generate in situ somatic mutations using CRISPR/Cas9 technology in the targeted tissue regions [83,84,85]. Recently, the CRISPR/Cas9 system has been extensively used to model different types of cancer including LUAD [84, 86] and small cell carcinoma [87]. Compared with the traditional GEMMs which require laborious breeding, the CRISPR/Cas9-based GEMMs are rapid and less costly for evaluating driver mutations and their roles during tumor progression [88, 89]. Moreover, multiplexed approaches can be incorporated to study a number of genetic alterations at the same time [88, 90,91,92,93,94]. Rogers et al, for example, deciphered the tumor suppressive effects and genetic interactions of 31 commonly mutated genes in LUAD by combining CRISPR/Cas9, tumor barcoding and deep sequencing [93]. Until now, neither GEMM-ESC nor CRISPR/Cas9-based in situ genetic engineering have been reported for LUSC modeling. Considering the complex genetic alterations in LUSC, it would be intriguing to incorporate these rapid and multiplexed strategies in validating potential oncogenic drivers for LUSC. Moreover, only a few studies have utilized GEMMs to evaluate experimental therapeutics for LUSC, while no immunotherapies have been tested. More research is needed to examine mono- and combinational immunotherapies in these immune-competent models.
Organoids models
The recent development of 3D culture technologies has enabled long-term culture of adult stem cells and primary cancer cells in more physiological conditions in vitro. These stem cells or cancer cells can self-organize into organotypic structures, known as organoids [13, 95, 96]. Organoids are increasingly appreciated as an important tool for basic and translational cancer research and success has been made in culturing many mouse and human epithelia organoids including colon, liver, pancreas, prostate, stomach and lung (reviewed in [13, 97]). Many living biobanks of human healthy and cancer organoids have been generated and served as a useful platform for personalized cancer treatment testing and drug screening [98,99,100,101,102]. Furthermore, genetic engineering in organoids allows de novo cancer modeling to examine the role of driver mutations in cancer initiation and progression [96]. Here we discuss the recent development in organoids for LUSC modeling and characterization (Fig. 2).

Schematic summary of organoids for LUSC study. Human lung normal and tumor organoids can be used for genetic cancer modeling, personalized medicine and testing immunotherapy. Mouse basal cell-derived organoids are ideal for testing LUSC driver mutations and generating novel syngeneic models.
Human lung cancer organoids
Several groups have successfully generated human lung cancer organoids [103,104,105] (Table 3). These organoids recapitulate the genetic and pathologic characteristics of the original tumors. When transplanted into mice, some cancer organoids maintain their tumorigenic capability. Sachs et al. first reported the long-term expansion of human airway organoids from both healthy and cancer tissues [103]. They generated different subtypes of human NSCLC organoids, including LUSC organoids. Subsequent orthotopic transplantation of these cancer organoids into immunocompromised mice enabled lung cancer formation in vivo. In addition, Kim and colleagues established the largest and most inclusive biobank of lung organoids to date, comprising of 80 human lung cancer organoids and 5 normal bronchial organoids [104]. The banked lung cancer organoids include five subtypes of lung cancer: LUAD, LUSC, adenosquamous carcinoma, large cell carcinoma, and small cell carcinoma. Similarly, Shi et al. successfully established human NSCLC organoids from both primary lung tumor and PDX tumor tissues [105]. Using organoids derived from PDXs, the authors further revealed that the combination of FGFR and MEK inhibition suppressed the growth of a FGFR1 amplified LUSC organoid line in vitro and in vivo.
Mouse lung cancer organoids
Mouse organoids are ideal to study de novo transformation and identify novel oncogenic drivers, as normal adult stem cell organoids can be cultured and passaged for long term in vitro and are easy to manipulate using genetic approaches. For instance, driver mutations such as Kras, Apc, Trp53 and Smad4, were introduced into mouse colon organoids followed by transplantation into mice to study colon cancer progression [106,107,108]. To date, genetic engineering of normal mouse organoids has been performed to study driver mutations and disease progression in several cancer types including pancreatic, gastric, brain and ovarian [96].
Mouse lung organoids derived from basal cells, secretory cells and alveolar type-II cells (AT2 cells), have been reported [109]. In 2009, Rock et al. first reported mouse lung basal cell-derived organoids (basal epithelia cells or lung basal cell spheres/tracheospheres described in some literature) [110]. When cultured in vitro, these basal cells can self-renew and differentiate into luminal club cells and ciliated cells [109, 110]. As basal cells are proposed to be the main cell of origin for LUSC, these basal cell-derived organoids are optimal to study LUSC in vitro and in vivo (Table 4). Jeong et al. discovered that inactivation of Keap1 or Trp53 promotes airway basal stem cell self-renewal in vitro and in vivo [15]. By transducing Ade-Cre virus in vitro, the authors generated the Trp53−/− Keap1−/− basal cells. Interestingly, these genetically modified basal cells formed LUSC subcutaneously in mice. Furthermore, BCL11A is a potential oncogenic driver amplified in a subset of LUSC patients. Utilizing the mouse organoids system, Lazarus et al. found that overexpressing Bcl11a in mouse basal cell-derived organoids leads to a hyper-proliferative and abnormal structure, while Bcl11a knock-out organoids were unable to form the 3D structure in vitro [111]. However, whether the Bcl11a overexpressing organoids can form LUSC in vivo was not studied. Chromosome 3q26 copy number gain (CNG) is a genetic hallmark of LUSC, but its functional significance in LUSC formation is not well understood. Liu et al. discovered that overexpression of Sox2, Prkci and Ect2 in the context of Trp53 loss, significantly promotes mouse lung basal stem cells growth in vitro [17]. Intriguingly, the transformed basal cells formed LUSC when orthotopically injected into mouse lungs. Using CRISPR/Cas9 genomic engineering technology, our laboratory recently characterized the potential of tumor formation from multiple mouse lung basal organoids with the deletion of LUSC specific tumor suppressor genes [16]. Our findings revealed that Sox2OE Trp53−/− Pten−/− Cdkn2a−/− organoids, but not the Sox2OE Trp53−/− Cdkn2a−/− organoids, are able to efficiently form LUSC in vivo which closely mimics the human disease.
Organoids for LUSC immuno-oncology research
Human lung organoids are a promising tool for personalized medicine and drug screening [103,104,105]. However, one of the main drawbacks is that most of the current human lung organoids lack tumor infiltrating immune cells and stroma, thus they are not suitable for immuno-oncology research. Recent studies from our group and others attempted to incorporate tumor-associated immune cells to mimic the immune tumor microenvironment in human lung organoid culture systems [112]. One approach is using the patient-derived organotypic tumor spheroids (PDOTS) from human primary tumors [113, 114]. PDOTS contain the autologous lymphoid and myeloid cell populations and they respond to immunotherapy and other therapies in vitro. Another way involves the utilization of an air-liquid interface method to culture patient-derived organoids with native embedded immune cells (T, B, NK cells and macrophages). Thus far, this method has enabled the establishment of organoids with immune microenvironment originated from different types of cancer, including lung cancer [112, 115]. The preservation of tumor infiltrating immune cells enables evaluation of the response to ICBs such as anti-PD-1 and/or anti-PD-L1 treatment. Currently, the in vitro maintenance of infiltrating immune cells is challenging, therefore future research is needed to improve the lifespan of immune cells and to illustrate how effective these organoids are at predicting an immunotherapy response.
On the other hand, the main advantage of mouse lung organoids is its application in in vivo immuno-oncology research, when the recipient animals are in the same genomic background with the organoids. As described previously, Sox2OE Trp53−/− Pten−/− Cdkn2a−/− organoids were successfully established from C57BL/6J mice in our laboratory. Upon injection back into C57BL/6J mice, LUSC tumors were formed and LUSC syngeneic cell lines were subsequently generated. We further examined the effect of WEE1 inhibition in enhancing immunotherapy in immune-competent C57BL/6J mice. Our study uncovered that WEE1 inhibition induces DNA damage and enhances the immune response of anti-PD-1 therapy in LUSC [16].
In summary, organoids have become an important component of preclinical LUSC models in recent years (Fig. 2), although lung organoids are still at an early stage of development. It is worth noting that the current culturing method has limitations to generate pure human lung cancer organoids. For example, the growth of normal airway organoids could outcompete the tumor organoids in vitro [116]. In addition, developing a better organoids system for long-term culturing lung cancer organoids with the tumor infiltrating immune cells, would greatly facilitate LUSC immuno-oncology research. Genetic engineering in normal human lung organoids is currently ongoing in our laboratory and we foresee this will be a powerful strategy to study driver mutations, disease progression and to evaluate treatment of LUSC in vitro. Likewise, mouse basal cell-derived organoids are a powerful tool for genetic LUSC modeling; they are optimal for efficiently testing genetic drivers and performing genome-wide screens. Moreover, mouse organoids can be used to develop novel syngeneic allograft models with defined genetic alterations. Given the long and variable latency of LUSC GEMMs (6–10 months) [12], these basal cell-derived organoids hold great promise to model LUSC and facilitate immuno-oncology studies in an efficient manner.
Conclusion and future direction
The past decade has witnessed substantial progress in LUSC genetics and therapeutics, as comprehensive genomic sequencing has begun dissecting the genetic mutational landscape and immunotherapy has transformed treatment in patients with advanced stage disease. In parallel, characterization of preclinical models mimicking human LUSC has proven to be an invaluable tool in understanding tumor biology and developing better treatment strategies. In particular, GEMMs harboring key genetic mutations that recapitulate human tumor physiology and pathology have shed light on the essential role of functional driver mutations in tumor intrinsic signaling as well as on how genetic characteristics affect the tumor microenvironment. With the help of emerging approaches such as GEMM-ESC and CRISPR/Cas9, GEMMs are positioned to continue playing pivotal roles in preclinical LUSC modeling. Combining CRISPR/Cas9 with next generation sequencing will be a powerful approach to decipher LUSC driver mutations and their genetic interactions. Furthermore, the evolving organoids technology is an encouraging and complementary ex vivo and transplantable model system to study LUSC biology and test therapeutics.
Utilizing these preclinical models through genetic engineering and 3D culturing technologies, future research is needed to characterize the role of underappreciated driver genetic alterations in tumorigenesis and to develop combination therapy to target dysregulated genes/pathways identified in LUSC patients. It is worthwhile highlighting that from GEMMs to organoids, and transplantable mouse models, each system has its strengths and shortcomings (Table 5). In LUSC immuno-oncology research, selecting an appropriate model is the first and crucial step in investigating the interactions between cancer cells and immune cells, as well as evaluating immunotherapies. Combining these different toolsets in a complementary manner will greatly advance our research in this field. Ultimately, improved understanding of tumor immune microenvironment and identifying biomarkers of response to immunotherapy will have significant translational impact in tailored treatment for LUSC patients.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Sanchez-Danes A, Blanpain C. Deciphering the cells of origin of squamous cell carcinomas. Nat Rev Cancer. 2018;18:549–61.
Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25.
Kim Y, Hammerman PS, Kim J, Yoon JA, Lee Y, Sun JM, et al. Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. J Clin Oncol. 2014;32:121–8.
Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48:607–16.
Paik PK, Pillai RN, Lathan CS, Velasco SA, Papadimitrakopoulou V. New treatment options in advanced squamous cell lung cancer. Am Soc Clin Oncol Educ Book. 2019;39:e198–206.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–54.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–5.
Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14.
Gazdar AF, Hirsch FR, Minna JD. From mice to men and back: an assessment of preclinical model systems for the study of lung cancers. J Thorac Oncol. 2016;11:287–99.
Kwon MC, Berns A. Mouse models for lung cancer. Mol Oncol. 2013;7:165–77.
Singh AP, Adrianzen Herrera D, Zhang Y, Perez-Soler R, Cheng H. Mouse models in squamous cell lung cancer: impact for drug discovery. Expert Opin Drug Disco. 2018;13:347–58.
Clevers H. Modeling development and disease with organoids. Cell. 2016;165:1586–97.
Neal JT, Kuo CJ. Organoids as models for neoplastic transformation. Annu Rev Pathol. 2016;11:199–220.
Jeong Y, Hoang NT, Lovejoy A, Stehr H, Newman AM, Gentles AJ, et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Disco. 2017;7:86–101.
Hai J, Zhang H, Zhou J, Wu Z, Chen T, Papadopoulos E. et al. Generation of genetically engineered mouse lung organoid models for squamous cell lung cancers allows for the study of combinatorial immunotherapy. Clin Cancer Res. 2020;26:3431–3442.
Liu Y, Yin N, Wang X, Khoor A, Sambandam V, Ghosh AB. et al. Chromosome 3q26 gain is an early event driving coordinated overexpression of the PRKCI, SOX2, and ECT2 oncogenes in lung squamous cell carcinoma. Cell Rep. 2020;30:771–82.e776.
Li C, Gao Z, Li F, Li X, Sun Y, Wang M, et al. Whole exome sequencing identifies frequent somatic mutations in cell-cell adhesion genes in chinese patients with lung squamous cell carcinoma. Sci Rep. 2015;5:14237.
Paik PK, Shen R, Won H, Rekhtman N, Wang L, Sima CS, et al. Next-generation sequencing of stage IV squamous cell lung cancers reveals an association of PI3K aberrations and evidence of clonal heterogeneity in patients with brain metastases. Cancer Disco. 2015;5:610–21.
Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–44.
Dotto GP, Rustgi AK. Squamous cell cancers: a unified perspective on biology and genetics. Cancer Cell. 2016;29:622–37.
Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–8.
Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol. 2011;29:2866–74.
Inoue A, Kobayashi K, Maemondo M, Sugawara S, Oizumi S, Isobe H, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol. 2013;24:54–59.
Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16:141–51.
Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann Oncol. 2015;26:1877–83.
Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. N Engl J Med. 2018;378:113–25.
Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2015;33:3401–8.
Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord JP, Hidalgo M, et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study. J Clin Oncol. 2017;35:157–65.
Aggarwal C, Redman MW, Lara PN Jr, Borghaei H, Hoffman P, Bradley JD, et al. SWOG S1400D (NCT02965378), a phase II study of the fibroblast growth factor receptor inhibitor AZD4547 in previously treated patients with fibroblast growth factor pathway-activated stage IV squamous cell lung cancer (Lung-MAP Substudy). J Thorac Oncol. 2019;14:1847–52.
Vansteenkiste JF, Canon JL, De Braud F, Grossi F, De Pas T, Gray JE, et al. Safety and efficacy of buparlisib (BKM120) in patients with PI3K pathway-activated non-small cell lung cancer: results from the phase II BASALT-1 study. J Thorac Oncol. 2015;10:1319–27.
Bendell JC, Varghese AM, Hyman DM, Bauer TM, Pant S, Callies S, et al. A first-in-human phase 1 study of LY3023414, an oral PI3K/mTOR dual inhibitor, in patients with advanced cancer. Clin Cancer Res. 2018;24:3253–62.
Langer CJ, Redman MW, Wade JL 3rd, Aggarwal C, Bradley JD, Crawford J, et al. SWOG S1400B (NCT02785913), a phase II study of GDC-0032 (Taselisib) for previously treated PI3K-positive patients with stage IV squamous cell lung cancer (Lung-MAP sub-study). J Thorac Oncol. 2019;14:1839–46.
Herbst RS, Gandara DR, Hirsch FR, Redman MW, Leblanc M, Mack PC, et al. Lung master protocol (Lung-MAP)-A biomarker-driven protocol for accelerating development of therapies for squamous cell lung cancer: SWOG S1400. Clin Cancer Res. 2015;21:1514–24.
Middleton G, Fletcher P, Popat S, Savage J, Summers Y, Greystoke A, et al. The National Lung Matrix Trial of personalized therapy in lung cancer. Nature. 2020;583:807–12.
Targeted drugs fall short in squamous lung cancer. Cancer Discov. 2021;11:OF3–OF3.
Redman MW, Papadimitrakopoulou VA, Minichiello K, Hirsch FR, Mack PC, Schwartz LH, et al. Biomarker-driven therapies for previously treated squamous non-small-cell lung cancer (Lung-MAP SWOG S1400): a biomarker-driven master protocol. Lancet Oncol. 2020;21:1589–601.
Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–33.
Herbst RS, Giaccone G, de Marinis F, Reinmuth N, Vergnenegre A, Barrios CH, et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. N Engl J Med. 2020;383:1328–39.
Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–50.
Hellmann MD, Ciuleanu T-E, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus Ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–104.
Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393:1819–30.
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab plus chemotherapy for squamous non–small-cell lung cancer. N Engl J Med. 2018;379:2040–51.
Paz-Ares L, Ciuleanu TE, Cobo M, Schenker M, Zurawski B, Menezes J, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:198–211.
Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–35.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28.
Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–65.
Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837–46.
Nettesheim P, Hammons AS. Induction of squamous cell carcinoma in the respiratory tract of mice. J Natl Cancer Inst. 1971;47:697–701.
Rehm S, Lijinsky W, Singh G, Katyal SL. Mouse bronchiolar cell carcinogenesis. Histologic characterization and expression of Clara cell antigen in lesions induced by N-nitrosobis-(2-chloroethyl) ureas. Am J Pathol. 1991;139:413–22.
Wang Y, Zhang Z, Yan Y, Lemon WJ, LaRegina M, Morrison C, et al. A chemically induced model for squamous cell carcinoma of the lung in mice: histopathology and strain susceptibility. Cancer Res. 2004;64:1647–54.
Azpilikueta A, Agorreta J, Labiano S, Perez-Gracia JL, Sanchez-Paulete AR, Aznar MA, et al. Successful immunotherapy against a transplantable mouse squamous lung carcinoma with anti-PD-1 and anti-CD137 monoclonal antibodies. J Thorac Oncol. 2016;11:524–36.
Gengenbacher N, Singhal M, Augustin HG. Preclinical mouse solid tumour models: status quo, challenges and perspectives. Nat Rev Cancer. 2017;17:751–65.
Morgan KM, Riedlinger GM, Rosenfeld J, Ganesan S, Pine SR. Patient-derived xenograft models of non-small cell lung cancer and their potential utility in personalized medicine. Front Oncol. 2017;7:2.
Kellar A, Egan C, Morris D. Preclinical murine models for lung cancer: clinical trial applications. Biomed Res Int. 2015;2015:621324.
Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238–42.
Hussenet T, Dali S, Exinger J, Monga B, Jost B, Dembelé D, et al. SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS ONE. 2010;5:e8960.
Lu Y, Futtner C, Rock JR, Xu X, Whitworth W, Hogan BL, et al. Evidence that SOX2 overexpression is oncogenic in the lung. PLoS ONE. 2010;5:e11022.
Mukhopadhyay A, Berrett KC, Kc U, Clair PM, Pop SM, Carr SR, et al. Sox2 cooperates with Lkb1 loss in a mouse model of squamous cell lung cancer. Cell Rep. 2014;8:40–9.
Mollaoglu G, Jones A, Wait SJ, Mukhopadhyay A, Jeong S, Arya R. et al. The lineage-defining transcription factors SOX2 and NKX2-1 determine lung cancer cell fate and shape the tumor immune microenvironment. Immunity. 2018;49:764–79.e769.
Tata PR, Chow RD, Saladi SV, Tata A, Konkimalla A, Bara A. et al. Developmental history provides a roadmap for the emergence of tumor plasticity. Dev Cell. 2018;44:679–93.e675.
Ferone G, Song JY, Sutherland KD, Bhaskaran R, Monkhorst K, Lambooij JP, et al. SOX2 Is the determining oncogenic switch in promoting lung squamous cell carcinoma from different cells of origin. Cancer Cell. 2016;30:519–32.
Gao Y, Ge G, Ji H. LKB1 in lung cancerigenesis: a serine/threonine kinase as tumor suppressor. Protein Cell. 2011;2:99–107.
Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–10.
Zhang H, Fillmore Brainson C, Koyama S, Redig AJ, Chen T, Li S, et al. Lkb1 inactivation drives lung cancer lineage switching governed by Polycomb Repressive Complex 2. Nat Commun. 2017;8:14922.
Li F, Han X, Li F, Wang R, Wang H, Gao Y, et al. LKB1 inactivation elicits a redox imbalance to modulate non-small cell lung cancer plasticity and therapeutic response. Cancer Cell. 2015;27:698–711.
Han X, Li F, Fang Z, Gao Y, Li F, Fang R, et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat Commun. 2014;5:3261.
Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25:590–604.
Liu J, Wang T, Creighton CJ, Wu SP, Ray M, Janardhan KS, et al. JNK(1/2) represses Lkb(1)-deficiency-induced lung squamous cell carcinoma progression. Nat Commun. 2019;10:2148.
Chow E, Meldrum CJ, Crooks R, Macrae F, Spigelman AD, Scott RJ. An updated mutation spectrum in an Australian series of PJS patients provides further evidence for only one gene locus. Clin Genet. 2006;70:409–14.
Fang R, Zheng C, Sun Y, Han X, Gao B, Li C, et al. Integrative genomic analysis reveals a high frequency of LKB1 genetic alteration in Chinese lung adenocarcinomas. J Thorac Oncol. 2014;9:254–8.
Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75.
Ekman S, Wynes MW, Hirsch FR. The mTOR pathway in lung cancer and implications for therapy and biomarker analysis. J Thorac Oncol. 2012;7:947–53.
Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, et al. The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell. 2018;33:905–921.e905.
Xiao Z, Jiang Q, Willette-Brown J, Xi S, Zhu F, Burkett S, et al. The pivotal role of IKKalpha in the development of spontaneous lung squamous cell carcinomas. Cancer Cell. 2013;23:527–40.
Ruiz EJ, Diefenbacher ME, Nelson JK, Sancho R, Pucci F, Chakraborty A, et al. LUBAC determines chemotherapy resistance in squamous cell lung cancer. J Exp Med. 2019;216:450–65.
Camolotto SA, Pattabiraman S, Mosbruger TL, Jones A, Belova VK, Orstad G. et al. FoxA1 and FoxA2 drive gastric differentiation and suppress squamous identity in NKX2-1-negative lung cancer. Elife. 2018;7:e38579.
Huijbers IJ, Bin Ali R, Pritchard C, Cozijnsen M, Kwon MC, Proost N, et al. Rapid target gene validation in complex cancer mouse models using re-derived embryonic stem cells. EMBO Mol Med. 2014;6:212–25.
Premsrirut PK, Dow LE, Kim SY, Camiolo M, Malone CD, Miething C, et al. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell. 2011;145:145–58.
Huijbers IJ, Del Bravo J, Bin Ali R, Pritchard C, Braumuller TM, van Miltenburg MH, et al. Using the GEMM-ESC strategy to study gene function in mouse models. Nat Protoc. 2015;10:1755–85.
Henneman L, van Miltenburg MH, Michalak EM, Braumuller TM, Jaspers JE, Drenth AP, et al. Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1-deficient metaplastic breast cancer. Proc Natl Acad Sci USA. 2015;112:8409–14.
Saborowski M, Saborowski A, Morris JPT, Bosbach B, Dow LE, Pelletier J, et al. A modular and flexible ESC-based mouse model of pancreatic cancer. Genes Dev. 2014;28:85–97.
Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–55.
Sánchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature. 2014;516:428–31.
Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514:380–4.
Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23:1362–8.
Ng SR, Rideout WM 3rd, Akama-Garren EH, Bhutkar A, Mercer KL, Schenkel JM, et al. CRISPR-mediated modeling and functional validation of candidate tumor suppressor genes in small cell lung cancer. Proc Natl Acad Sci USA. 2020;117:513–21.
Winters IP, Murray CW, Winslow MM. Towards quantitative and multiplexed in vivo functional cancer genomics. Nat Rev Genet. 2018;19:741–55.
Yin H, Xue W, Anderson DG. CRISPR-Cas: a tool for cancer research and therapeutics. Nat Rev Clin Oncol. 2019;16:281–95.
Maresch R, Mueller S, Veltkamp C, Ollinger R, Friedrich M, Heid I, et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat Commun. 2016;7:10770.
Chow RD, Guzman CD, Wang G, Schmidt F, Youngblood MW, Ye L, et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat Neurosci. 2017;20:1329–41.
Rogers ZN, McFarland CD, Winters IP, Naranjo S, Chuang CH, Petrov D, et al. A quantitative and multiplexed approach to uncover the fitness landscape of tumor suppression in vivo. Nat Methods. 2017;14:737–42.
Rogers ZN, McFarland CD, Winters IP, Seoane JA, Brady JJ, Yoon S, et al. Mapping the in vivo fitness landscape of lung adenocarcinoma tumor suppression in mice. Nat Genet. 2018;50:483–6.
Wang G, Chow RD, Ye L, Guzman CD, Dai X, Dong MB, et al. Mapping a functional cancer genome atlas of tumor suppressors in mouse liver using AAV-CRISPR-mediated direct in vivo screening. Sci Adv. 2018;4:eaao5508.
Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5.
Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer. 2018;18:407–18.
Ballard DH, Boyer CJ, Alexander JS. Organoids - preclinical models of human disease. N. Engl J Med. 2019;380:1981–2.
van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–45.
Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell. 2016;18:827–38.
Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell. 2018;173:515–28.e517.
Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172:373–386.e310.
Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Disco. 2018;8:1112–29.
Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, Heo I, Bottinger L, Klay D. et al. Long-term expanding human airway organoids for disease modeling. Embo J. 2019;38:e100300.
Kim M, Mun H, Sung CO, Cho EJ, Jeon HJ, Chun SM, et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat Commun. 2019;10:3991.
Shi R, Radulovich N, Ng C, Liu N, Notsuda H, Cabanero M, et al. Organoid cultures as preclinical models of non-small cell lung cancer. Clin Cancer Res. 2020;26:1162–74.
Fumagalli A, Drost J, Suijkerbuijk SJ, van Boxtel R, de Ligt J, Offerhaus GJ, et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc Natl Acad Sci USA. 2017;114:E2357–64.
Roper J, Tammela T, Cetinbas NM, Akkad A, Roghanian A, Rickelt S, et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat Biotechnol. 2017;35:569–76.
Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. 2015;21:256–62.
Barkauskas CE, Chung MI, Fioret B, Gao X, Katsura H, Hogan BL. Lung organoids: current uses and future promise. Development. 2017;144:986–97.
Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA. 2009;106:12771–5.
Lazarus KA, Hadi F, Zambon E, Bach K, Santolla MF, Watson JK, et al. BCL11A interacts with SOX2 to control the expression of epigenetic regulators in lung squamous carcinoma. Nat Commun. 2018;9:3327.
Yuki K, Cheng N, Nakano M, Kuo CJ. Organoid models of tumor immunology. Trends Immunol. 2020;41:652–64.
Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Disco. 2018;8:196–215.
Aref AR, Campisi M, Ivanova E, Portell A, Larios D, Piel BP, et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip. 2018;18:3129–43.
Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J. et al. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175:1972–88.e1916.
Dijkstra KK, Monkhorst K, Schipper LJ, Hartemink KJ, Smit EF, Kaing S, et al. Challenges in establishing pure lung cancer organoids limit their utility for personalized medicine. Cell Rep. 2020;31:107588.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
KKW is a founder and equity holder of G1 Therapeutics and has consulting and sponsored research with: AstraZeneca, Janssen, Pfizer/Array Biopharma, Novartis, Merck, Zentalis; as well as sponsored research (only) with: Takeda, BMS, Mirati, Alkermes, Merus, Amgen, Ansun Biopharma, Enliven Therapeutics, Tvardi Therapeutics, Delfi Diagnostics, and Dracen Pharmaceuticals. No potential conflicts of interest were disclosed by the other authors.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pan, Y., Han, H., Labbe, K.E. et al. Recent advances in preclinical models for lung squamous cell carcinoma. Oncogene 40, 2817–2829 (2021). https://doi.org/10.1038/s41388-021-01723-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-021-01723-7
- Springer Nature Limited
This article is cited by
-
HTR2B as a novel biomarker of chronic obstructive pulmonary disease with lung squamous cell carcinoma
Scientific Reports (2024)
-
USP13 drives lung squamous cell carcinoma by switching lung club cell lineage plasticity
Molecular Cancer (2023)
-
Identification of adenoid subtype characterized with immune-escaped phenotype in lung squamous carcinoma based on transcriptomics
Experimental Hematology & Oncology (2022)