
Abstract
Cyclic nucleotide phosphodiesterases (PDE) break down cyclic nucleotides such as cAMP and cGMP, reducing the signaling of these important intracellular second messengers. Several unique families of phosphodiesterases exist, and certain families are clinically important modulators of vasodilation. In the current work, we have summarized the body of literature that describes an emerging role for the PDE4 subfamily of phosphodiesterases in malignancy. We have systematically investigated PDE4A, PDE4B, PDE4C, and PDE4D isoforms and found evidence associating them with several cancer types including hematologic malignancies and lung cancers, among others. In this review, we compare the evidence examining the functional role of each PDE4 subtype across malignancies, looking for common signaling themes, signaling pathways, and establishing the case for PDE4 subtypes as a potential therapeutic target for cancer treatment.
Similar content being viewed by others

Introduction
There are over 20 cyclic nucleotides phosphodiesterases that are known to regulate cAMP and cGMP levels. Cyclic nucleotide phosphodiesterases work by binding to cyclic nucleotides and degrading them into non-cyclical monophosphates. The cyclic nucleotide phosphodiesterases are subdivided into 11 families, each of which contains several unique isoforms. PDE4, PDE7, and PDE8 are specific for hydrolysis of cAMP. PDE5, PDE6, and PDE9 are specific for hydrolysis of cGMP. PDE1, PDE2, PDE3, PDE10, and PDE11 are nonspecific and will hydrolyze both cAMP and cGMP [1]. The history of successfully targeting specific PDE isoforms is variable, and inhibitors for PDE3, PDE4, and PDE5 are presently available in the United States. PDE3 inhibitors are used for heart failure and peripheral artery disease, owing to their vasodilatory properties [2]. PDE5 inhibitors are used for pulmonary hypertension and erectile dyfunction, also resulting from tissue-specific vasodilatory properties [3]. PDE4 inhibitors, on the other hand, are potent inhibitors of inflammation, and they have been approved for the treatment of inflammatory diseases ranging from arthritis to chronic obstructive pulmonary disease. In addition, PDE4 inhibitors are approved as oral or topical treatments for psoriasis and atopic dermatitis, respectively. The main challenge with PDE4 inhibitors is their very narrow therapeutic index and limiting gastrointestinal tolerability due to nausea, emesis, and diarrhea [4, 5].
Because PDE4 is such an appealing clinical target for many additional non-dermatologic indications, additional research has continued to pursue strategies to widen the therapeutic index. Nearly 10 years ago, it was found that small-molecule inhibitors that only partially inhibit the activity of specific PDE4 isozymes have reduced adverse effect profiles with sustained clinical potency [6]. PDE4 enzymes are generally categorized into four subtypes (PDE4A, PDE4B, PDE4C, and PDE4D), and promising therapeutics are currently in development for neurologic disorders that target PDE4B and PDE4D [7,8,9]. The renewed interest in subtype-specific PDE4 inhibitors warrants a comprehensive investigation into the specific PDE4 subtypes and their potential role in inflammatory pathologies. The aim of the current study is to provide a review of the current literature on the four subtypes of PDE4 and their role in cancer, specifically.
Results
PDE4A in cancer
Our search results produced 17 total studies investigating the PDE4A subtype with nine of these studies examining PDE4A specifically and eight studies examining PDE4A in conjunction with at least one other PDE4 subtype. These studies were further categorized by specific type of malignancy with the following results: hematologic (n = 7), CNS and adrenocortical tumors (n = 6), lung cancer (n = 2), liver cancer (n = 1), oral cancer (n = 1), and breast cancer (n = 1). One study addressed the role of PDE4A in the context of multiple malignancies thus a total of n = 19.
CNS tumors
Five studies examined the association of PDE4A with various CNS tumors. Four studies looked at the expression levels of PDE4A in various CNS tumors. PDE4A was found to have low expression in normal pituitary tissue [10] and overexpression in medulloblastoma, glioblastoma, glioma, and somatotroph, lactotroph, corticotroph, and nonfunctioning gonadotroph adenoma cells [10,11,12,13]. No studies reported the decreased expression of PDE4 in malignant CNS tissues.
With a reported overexpression of PDE4A in CNS tumors, two studies sought to determine whether or not this increased expression resulted in increased PDE4 activity. It was found that PDE4A overexpression resulted in significantly shortened doubling times in medulloblastoma and glioblastoma cells [11] as well as hypercellular lesions with human neurofibromatosis 1-associated glioma features [12], suggesting a role for the elevation of PDE4A in tumor proliferation.
Two studies examined the interaction of PDE4A with the aryl hydrocarbon receptor-interacting protein (AIP) gene and the associated effects of disrupting this interaction. Although the function of AIP is not well described, it is thought to act as a tumor suppressor involved in the regulation of cell proliferation. Naturally occurring oncogenic variants in AIP that disrupted the PDE4A-AIP interaction played a significant role in pituitary tumorigenesis [10]. This disruption was also observed in the somatotropinoma and adrenocortical carcinoma tumor cells of a MEN1- and p53-negative mother–daughter pair with acromegaly due to somatotropinoma [14]. In these patients, a functional mutation blocking the AIP protein from interacting with PDE4A was identified in the tumoral DNA of the daughter’s somatotropinoma and the mother’s adrenocortical carcinoma.
Hematologic malignancies
Association with hematologic malignancies constitutes the most common area of research for PDE4A in cancer. Six studies investigated PDE4A expression in hematologic malignancies including B-cell chronic lymphocytic leukemia (B-CLL), chronic myelomonocytic leukemia (CML), myelodysplatic syndrome, T cell leukemia, essential thrombocytopenia, polycythemia vera, and primary myelofibrosis. Of the six studies, one saw a decrease in PDE4A expression in CD34+ granulocytes taken from patients with essential thrombocytopenia, polycythemia vera, and primary myelofibrosis [15]. In contrast, the other five studies found a distinct upregulation of PDE4A expression in chronic lymphocytic leukemia, myelodysplastic syndrome, and T-leukemic cell lines [16,17,18,19,20]. In addition, it was found that PDE4A is the most abundantly expressed PDE subtype in B-lymphoblastoid cell lines [19] and T-leukemic cell lines [19, 20]. Interestingly, quiescent isolated human peripheral blood lymphocytes express mRNA for PDE4B as the principal transcript and it is only after mitogenic stimulation with phytohemagglutinin, a known initiator of mitosis, that mRNA for PDE4A is induced [19], suggesting that PDE4A upregulation may not be an early event in carcinogenesis.
One study examining the functional role of PDE4A in hematologic malignancy investigated the apoptotic pathway following exposure to Yessotoxin, a potent inducer of apoptosis in human leukemic cell lines. They found that PDE4A is required only for non-apoptotic cell death, driven primarily by autophagy, following Yessotoxin exposure [21]. Another study found that lower PDE4A expression is indicative of a response to a hypomethylating agent in patients with myelodysplastic syndrome and CML [18]. Taken together, these data suggest that PDE4 expression may be associated with resistance to cytotoxic agents.
Lung, liver, and oral cancers
Two studies examined PDE4A in lung cancer and both studies found PDE4A expression to be upregulated in various lung cancer cell lines. One study found that PDE4A expression induces epithelial–mesenchymal transition, a critical event in the pathogenesis of organ fibrosis and cancer, in alveolar epithelial type 2 cells following stimulation with transforming growth factor (TGF)-β1. Both PDE4A mRNA and protein expression is upregulated following (TGF)-β1 stimulation, resulting in significant loss of E-cadherin [22], a process that has been linked to metastasis. Another study demonstrated increased PDE4A expression during hypoxia by hypoxia-inducible factors across a panel of lung cancer cell lines. In addition, this study linked PDE4A to tumor cell proliferation and colony formation and showed that PDE4A knockdown can reduce VEGF secretion and have antitumor effects in lung cancer xenografts [23]. A singular study in hepatocellular carcinoma identified a role for PDE4 in driving epithelial–mesenchymal transition by increasing N-cadherin and vimentin while simultaneously decreasing E-cadhein [24]. Finally, one study showed that PDE4A is downregulated in mice treated with black raspberry chemopreventive agent [25]. These highly mechanistic studies support a role for PDE4A in tumorigenesis and metastasis.
Breast cancers
There is presently only a single study linking PDE4A to breast cancer. However, this study deserves mention because it linked lower expression of PDE4A to a decrease in progression-free and overall survival. This study, in stark contrast to other studies investigating PDE4A, shows that an increase in PDE4A expression is associated with better outcomes. This study found that PDE4A expression was elevated in estrogen receptor (ER) positive and progesterone receptor positive breast cancer [26], two factors correlated with improved prognosis. More studies are warranted to confirm these findings in breast cancer.
PDE4B in cancer
Our search identified 34 studies investigating the PDE4B subtype and its role in cancer. The role of PDE4B in neoplasia was investigated across multiple reports in hematological (n = 16), colon (n = 8), and lung (n = 2). Singular reports examined the role of PDE4B in cancers of the liver, kidney, oral cavity, breast, CNS, endometrium, skin, and prostate cancer.
Hematologic malignancies
Of all the subtypes and malignancies, PDE4B and its role in hematologic malignancies comprises the largest body of literature on PDE4 and cancer with a total of 16 research studies. PDE4B has been examined in the setting of various hematologic neoplasias including diffuse large B-cell lymphoma (DLBCL), acute lymphoblastic leukemia (ALL), chronic lymphoblastic leukemia (CLL), and multiple myeloma (MM).
Regardless of the specific malignancy, the agreement across multiple studies was that PDE4B expression was found to be elevated in hematologic malignancies. Increased PDE4B was observed in DLBCL [27,28,29], CD4+ lymphoid cancer cells [30], CLL [17] and B-lymphoblastoid cells [19, 16]. One study found that a single nucleotide polymorphism (SNP) (rs12142375) linked to ALL risk is also an important regulator of PDE4B expression [31], thus providing a potential genetic target between PDE4B and hematologic malignancies.
In addition, multiple studies found an inducible upregulation of PDE4B expression in hematologic malignancies [19, 29, 30, 32]. PDE4B was readily upregulated in activated human peripheral blood mononuclear cells (PBMC) following stimulation with IL-2, the primary growth factor for promoting survival and proliferation of activated T cells. This observation, combined with the finding that PDE4B is expressed in CD4+ lymphoid cancer cells, but not primary CD4+ T cells, suggests a role for PDE4B in the survival and proliferation of cancer cells [30].
It should be mentioned that PDE4B levels also appear to increase in the presence of elevated cAMP as part of a negative feedback loop. PDE4B was upregulated in T-leukemic and B-lymphoblastoid cells after mitogenic stimulation with 1-methyl-3-isobutylxanthine (IBMX) and the cAMP analog, dibutyryl cAMP (dBcAMP) [19]. Likewise, PDE4B was upregulated in MM cells following treatment with HENECA, a mitogenic A2A receptor agonist, and rolipram [29]. Rolipram is a nonselective PDE4 inhibitor. When rolipram alone was used to treat CLL cells in another study, a marked upregulation of PDE4B occurred [32], suggesting that the upregulation of PDE4B is in response to the pathologic accumulation of cAMP in lymphoid cancer cells following rolipram treatment.
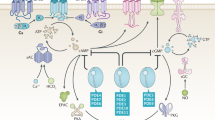
A number of studies also examined the associated effects of PDE4B expression in hematologic malignancies. Increased PDE4B expression was correlated with poorer outcomes in primary DLBCL, and an association was found between the inhibition of phosphatidylinositol 3-kinase/AKT (PI3K/AKT) pathway and cAMP-mediated apoptosis [27]. Similarly, increased PDE4B expression was associated with significantly higher microvessel density, which is an abnormal vascular network critical for tumor metabolism and metastatic potential. This was associated with poorer outcomes in DLBCL [28]. This association was reinforced when a Myc-induced lymphomagenesis in mice bearing a PDE4B-null background showed lower microvessel density compared with PDE4B wild-type mice [28]. These responses were attributed to modulation of vascular endothelial growth factor (VEGF) via PDE4B-dependent downregulation of cAMP and subsequent effects on the PI3K/AKT pathway [28]. Thus, the PDE4B/cAMP/PI3K/AKT/VEGF pathway may play an important role in the pathogenesis of hematologic malignancies (Fig. 1).

PDE4 upregulation is a late event in many cancer cells preceded by the following steps: (1) A mitogen activates cAMP in cancer cells. (2) cAMP inhibits the PI3K/AKT pathway. (3) Inhibition of the PI3K/AKT pathway results in decreased VEGF secretion. (4) Decreased VEGF leads to decreased angiogenesis. (5) Decreased angiogenesis leads to compensatory upregulation of PDE5 through HIF, and subsequent increased activity of the PI3K/AKT pathways and enhanced VEGF secretion and angiogenesis.
Four studies examined the clinical applications of these findings in the context of PDE4B, hematologic malignancies, and pharmacologic resistance. Using gene set enrichment analysis, overexpression of PDE4B in DLBCL was found to impinge on the cAMP inhibitory activities of the AKT/mTOR pathway and define glucocorticoid (GC) resistance. Furthermore, inhibition of PDE4 in a xenograft model of human lymphoma restored GC sensitivity [33]. This is supported by an in vitro study demonstrating that miR-124, an established anti-tumorigenic microRNA, influences GC-induced apoptosis in DLBCL by directly targeting PDE4B and stable expression of miR-124 in DLBCL cell lines diminishes PDE4B expression [34]. Using a genome-wide approach, an association was identified between PDE4B SNPs and risk of relapse in patients with childhood ALL [35]. Lastly, genetic inhibition of PDE4B improved the efficacy of SYK inhibitors through control of cAMP-modulated phosphorylation and activity of the tyrosine kinase SYK [36].
Colon cancers
Our systematic search identified eight studies that associated the PDE4B subtype with colon cancer. With the oldest study published in 2011, this collection represents some of the most recent literature of this scoping review. There may be a connection between the KRAS–PDE4B interaction and the development and survival of colonic cancer cells. One study showed that PDE4B expression is upregulated by oncogenic KRAS [37]. The same study analyzed public datasets and showed the higher expression of PDE4B in tumor samples from colorectal cancer patients when compared with those from healthy control. In addition, increased expression of PDE4B mRNA was found to be correlated with relapsed colorectal cancer in this public data subset [37]. In biopsies obtained from patients with and without colorectal neoplasia, both PDE activity and expression were lower overall in colorectal neoplasia while real-time qPCR analysis showed overexpression of the subtype PDE4B, suggesting that PDE4B is selectively overexpressed as a malfunctioning protein in non-neoplastic appearing colonic mucosa from colorectal neoplasia patients [38]. Indeed, PDE4B RNA appears to be one increased in both colonic adenomas and adjacent normal colonic tissues, and a protective role has been hypothesized for it in the adjacent normal tissues [39]. Additional studies also support the claim that PDE4B is signaling downstream of mutant KRAS in vitro [40, 41], and one study proved novel compounds capable of PDE4B-specific docking and inhibition to be superior to traditional chemotherapeutics such as doxorubicin in cell based assays [42]. Additional work suggested that PDE4B is also downstream of the Myc oncogene, and PDE4B works to suppress cAMP-mediated inhibition of the AKT/mTOR pathway in colon cancer models [43]. A singular study examined the development of colon cancer and the effects of diet on carcinogenesis. The protein level of PDE4B was upregulated while miR-26b, the microRNA associated with PDE4B, was downregulated in rats fed a chemoprotective diet [44]. These findings suggest that PDE4B may play a role in colon cancer, particularly in cases with KRAS activating mutations.
Other cancers
Singular and dual reports of PDE4B’s involvement in various other malignancies included cancers of the lung (n = 2), liver (n = 1), kidneys (n = 1), oral cavity (n = 1), prostate (n = 1), breast (n = 1), CNS (n = 1), endometrium (n = 1), and skin (n = 1). Similar to the previously described findings, PDE4B expression was found to be increased in non-small cell lung cancer tissues [45], human glioblastoma cell lines [13], and human gingiva-derived malignant melanoma cell lines [46]. Studies examining the functional properties of PDE4B also concurred with this oncogenic role of PDE4B. PDE4B was associated with anti-apoptotic and metastatic effects in kidney cancer [47] and endometrial cancer [48], and cell growth regulation through cAMP in malignant melanoma [46]. Further, two additional studies highlight in vitro findings that specific PDE4B inhibition is both cytotoxic in A549 lung cancer cells [49] and growth inhibitory in oral cancer cells [50].
In contrast to these studies, PDE4B was found to be downregulated in castration-resistant prostate cancer and advanced prostate cancer [51]. Taken alone, this seems like an outlier, but considered in conjunction with the singular study describing the downregulation of PDE4A in breast cancer [26], it should be noted that PDE4 subtypes may play different roles in hormonally regulated cancers and this is an interesting area for future investigation.
PDE4C in cancer
Our search identified seven studies pertaining to PDE4C and cancer. None of these studies examined the role of PDE4C in the same malignancy. The studies examined malignancies of the blood [18], skin [52], central nervous system [53], lung [45], thyroid [54], and one study focused on malignancies associated with p53 mutations in particular [55].
In high-grade glioma samples, there was hypermethylation of PDE4C promoter sites and hypomethalation in low-grade glioma [53]. This relationship is important to note as it contrasts the overexpression of PDE4A seen in CNS tumors. In other malignancies, however, PDE4C expression follows the more common trend of overexpression in patients with myelodysplastic syndrome [18] and thyroid adenomas [54]. PDE4C was also identified as a novel target gene of mutated transcription factor p53, potentially linking this subtype to a wide range of p53-associated malignancies [55].
PDE4D in cancer
Our search results produced 44 studies examining PDE4D and its role in cancer making PDE4D the most examined PDE4 subtype in this study with a wide distribution of focus covering hematologic (n = 4), lung (n = 5), prostate (n = 7), skin (n = 4), head and neck (n = 6), CNS (n = 3), colon and gastric (n = 6), breast (n = 4), bladder (n = 1), pancreatic (n = 1), and ovarian (n = 1) malignancies. One study also examined PDE4D across solid tumors in general.
Hematologic malignancies
Four studies focused on PDE4D and hematologic malignancies. Two of these studies examined baseline PDE4D expression in hematologic malignancy. Interestingly, a 30-fold decrease in PDE4D mRNA was observed in cells taken from patients with chronic lymphocytic leukemia compared with PBMC from healthy adults [56] while PDE4D was one of the most abundantly expressed PDEs in Jurkat T-leukemic cell lines [20] suggesting a discrepancy in expression between hematologic malignancies.
Two studies also examined PDE4D expression following various mitogenic and pharmacologic stimulation. An increase in PDE4D mRNA expression was seen in human peripheral blood cells following stimulation with phytohemagglutinin (a mitogen used to trigger T-lymphocyte cell division) and in B-lymphobolastoid cell lines following incubation with dBcAMP and IBMX to mimic phytohemagglutinin’s mitogenic effects [19]. In addition, PDE4D expression was upregulated in ALL cells but not B-CLL cells following increased activation of adenylyl cyclase using forskolin [32]. Together, these findings may suggest an important role for PDE4D in rapid cell division.
Lung cancers
PDE4D appears to play an important role in the development and progression of lung cancer. PDE4D expression was found to be regulated by the mutant form of serine/threonine kinase 11/liver kinase B1, a kinase present in lung cancer [57] and is positively correlated with mTOR expression [58]. Increased PDE4D expression was also noted in response to hypoxia in 8/10 lung cancer cell lines [23] and TGF-beta1 stimulation to mimic epithelial–mesenchymal transition, a critical event in the pathogenesis of organ fibrosis and cancer [22]. In addition, PDE4D expression was linked to tumor cell proliferation [23, 57], tumor cell differentiation [59], and loss of E-cadherin [22]. Together, these studies form a strong foundation connection PDE4D to the both the development and progression of lung cancer making it a prime target for future investigations.
Prostate cancer
The seven studies produced by our search suggest a role for PDE4D in prostate cancer, but it appears that specific subtypes of PDE4D may play individual roles in prostate cancer. PDE4D7 was found to be highly expressed in androgen-sensitive prostate cancer cells and starkly downregulated in androgen-insensitive cells, connecting a low PDE4D7 expression with poorer outcomes [60, 61]. In addition, PDE4D7 is upregulated in primary human prostate cancer while PDE4D5 and PDE4D9 are downregulated [62, 63]. Studies that examined PDE4D in general found an overall overexpression in prostate cancer [64] and found that PDE4D inhibition lead to growth inhibition [64, 65], increased apoptosis, and decreased proliferation and migration of prostate cancer cells [65]. Two studies also examined promotor sites and found that the CpG site of PDE4D is differentially methylated in prostate cancer [66] and noted an increased promoter methylation of PDE4D5 [62] suggesting potential markers for disease staging and/or targeted treatment. Together, these studies show an upregulation of PDE4D in prostate cancer but also highlight the potential of specific PDE4D subtypes in classification, prognostication, and treatment of prostate cancer.
Skin cancers
PDE4D was found to be overexpressed in BRAF-mutated melanoma cell lines and this PDE4D overexpression promotes invasion through its interaction with focal adhesion kinase via the scaffolding protein RACK1 [67]. The PDE4D gene was also found to be negatively associated with survival in patients with metastatic melanoma lesions using gene expression profiling [68]. PDE4D also appears to play an important role mechanistically in the development of skin cancer by regulating cell growth through cAMP in human gingiva-derived malignant melanoma cell lines [46]. Interestingly, PDE4D expression was also found to be upregulated in melanocytes by MSH/cAMP/MITF pathway, leading to reduced melanocyte pigmentation [69]. Inhibition of PDE4D in vivo, likewise, resulted in an enhancement of skin pigmentation. However, the changes induced by topical inhibition of PDE4D in these mice did not exhibit melanocytic features.
Head and neck cancers
With a total of six studies, head and neck cancers are the third-most investigated malignancy with respect to PDE4D, although the role of PDE4D in the preogression of head and neck cancers is far from clear. In nasopharyngeal carcinoma, PDE4D expression was found to be overexpressed in vitro and in vivo via western blot [70]. Lentiviral knockdown of PDE4D also led to the downregulation of AKT signaling pathway via EGFR, resulting in cell cycle arrest in G0/G1 phase. Contrary to this work, chromosomal profiling of abnormalities using high-density SNP arrays in 101 patient samples with esophageal carcinoma at various stages showed a positive correlation between disease progression and recurrent loss of chromosome regions, resulting in disruption of single genes including PDE4D in losses from chromosome 5q [71]. However, the effects resulting from the disruption of PDE4D in these patients are not characterized. A separate whole genome SNP array of 23 primary esophageal adenocarcinoma samples identified 126 homozygous deletions that included known tumor suppressor genes such as CDKN2A (p16) and SMAD4, as well as gains in proto-oncogenes including MYC and BCL9, and this work also detected a homozygous deletion of PDE4D at 5q [72]. These findings in esophageal adenocarcinoma and nasopharyngeal carcinoma highlight the possible multiple roles of PDE4D in cancer progression. This is further supported by contradictory data about PDE4D SNPs and esophageal squamous cell carcinoma in genotype studies with individuals of diverse ancestries [73].
Analysis of miRNA expression on thyroid adenoma found direct repression of PDE4D mRNA by two miRNA at the 3′UTR region [74]. Downregulation of PDE4D miRNA may explain the low cAMP levels in thyroid adenoma, as well as their relatively benign characteristics. Similarly, an increase in PDE4D expression activity was found in 18 primary thyroid adenoma samples with mutant TSH receptor Gsa, while maintained in normal thyroid tissues [54].
CNS
Similar to PDE4A findings in CNS tumors, in which PDE4A overexpression resulted in increased growth of medulloblastoma and glioblastoma cells, expression of PDE4D was found to enhance Hedgehog signaling in medulloblastoma via direct interaction with Neuropilins and subsequent indirect inhibition of protein kinase A [75]. Inhibition of PDE4D in mouse allograft model led to the suppression of Hedgehog and the inhibition of medulloblastoma growth. Likewise, in glioblastoma, treatment with a novel PDE4D inhibitor, rolipram, led to cell cycle arrest and differentiation of glioblastoma derived cells [76]. This effect may be mediated via the upregulation of cAMP/CREB signaling pathway due to the loss of PDE4D activity. In addition, a study of DNA alterations 30 glioma patients via AP-PCR found that altered PDE4D was associated with high levels of chromosomal instability and in both primary and secondary glioblastoma [77]. The presence of PDE4D alterations were also associated more commonly with wild-type p53 and p16, suggesting that decreased cAMP concentration may play a role in tumor suppression mechanism of p53 and p16. However, the phenotypic traits of these PDE4D alterations were not characterized. Interestingly, these PDE4D alterations were not found to have a significant impact on patient survival.
Colon and gastric cancers
In colon cancer cell lines, an underexpression of a specific p53 induced microRNA, miR-129-5p, was found to cause an elevated expression of PDE4D, resulting in oncogenic activity [78]. Treatment with miR-129-5p suppressed expression of PDE4D, leading to cAMP/BIM mediated colon cancer cell cycle arrest and apoptosis. Similarly, other studies found that, growth and survival in colon cancer cells are dependent on a PDE4D [79,80,81]. PDE4D expression has also been linked to co-expression with another novel oncogene, PIWIL1, in gastric cancer cells [82]. Interestingly, a loss of PDE4D in a colon adenocarcinoma cell line, SW480, demonstrated an acquired resistance against triapine, an anticancer iron-chelator belonging to the thiosemicarbazone class [83]. This chemotherapy resistance is associated with an upregulation of cAMP and Epac/Rap1 signaling pathway, which has a role in metastasis and metabolism in solid tumors, and it is in stark contrast another study that described increased sensitivity to doxorubicin in gastric cells with miR-494 expression and silencing of PDE4D [84]. Thus, PDE4D may be associated with both oncogenic and chemosensitvity functions in gastric and colon cancers.
Breast, bladder, and pancreatic cancers
In a large-scale exome-wide analysis of 8287 subjects for rare variants with minor allele frequency in women of African ancestry in the African American Breast Cancer Epidemiology and Risk Consortium [85], PDE4D was found to be associated with ER negative and progesterone, estrogen, and human epidermal growth factor receptor negative (triple negative) breast cancers, but does not show an overall increased risk for breast cancer. In a separate whole genome parallel sequencing of BRCA1 mutation positive breast cancer with either ER+ or ER-expression, recurrent homozygous deletion of PDE4D was found in the ER negative subset of BRCA1 positive breast cancers [86]. In addition, a PDE4D SNP was found to be associated with higher acini count per terminal duct lobular units, an established breast cancer risk factor, in a pooled analysis of 872 women [87] and a PDE4D-CCNB1 fusion gene was identified in two endometreial tumors [88]. PDE4D expression was also associated with CD177 co-expression and neutrophil recruitment into pancreatic cancers, corresponding with poor prognosis [89]. A more mechanistic study showed that PDE4D inhibition may provide a means to overcome tamoxifen resistance in ER positive breast cancer models [90]. Conversely, low PDE4D levels were correlated with poor prognosis in bladder cancer patients [91].
Discussion
Although there is some evidence that PDE4A may be involved in the regulation of tumor suppressor genes, the more abundant theme is that PDE4A upregulation is a response to mitogenic stimulation in tumors and hematologic malignancies. In fact, in solid tumors, data suggest that PDE4A may be involved in VEGF-mediated angiogenesis and later epithelial–mesenchymal transition and metastasis.
There is a large body of literature supporting a role for PDE4B in hematologic malignancies. The nonspecific PDE4 inhibitor roflumilast has entered early phase I clinical trials for B-cell malignancies [92]. The purported mechanism for these agents is through renewed suppression of AKT-mediated pathways by cAMP. There was also substantial evidence linking PDE4B upregulation to cancers of the colon, perhaps as a downstream signaling effect of KRAS mutation.
The literature supporting a role for PDE4C in cancer is both scant and contradictory. Given the lack of clinically available tools for targeting PDE4C, and the paucity of data supporting a role for PDE4C-mediated oncogenesis, this seems like the PDE4 subtype that is least worth pursing for future studies.
PDE4D is the most heavily studied of all the PDE4 subtypes. While there is conflicting data about the role of PDE4D in hematologic malignancies, the data are more clear with PDE4D in solid tumors. There is strong evidence that PDE4D contributed to tumor progression in lung, CNS, and skin cancers, while specific isoforms of PDE4D play opposing roles in prostate cancer.
While the focus of this work was to provide a scoping review of the published literature on PDE4 subtypes in cancer, it is worth briefly discussing that we excluded 93 works for their use of nonspecific PDE4 subtype inhibitors (Fig. 2) and these works may include helpful studies that act through many of the pathways described herein. Indeed, combination therapies of nonselective PDE4 inhibitors with other antineoplastic agents have been tested in wide variety of pre-clinical cancer models to target the pathways described here. Examples include the use of rolipram with bevacizumab, an inhibitor of VEGF, in glioblastoma models [93, 94] and roflumilast with idelalisib, a PI3K inhibitor, in DLBCL and CLL [95]. Like the many other targeted therapies that may be used successfully in combination, the synergistic effects of PDE4 inhibition with concomintant inhibition of other mitogen-stimulated pathways should not be overlooked as a therapeutic strategy.

The articles were sorted into three categories: PDE4A, PDE4B, PDE4C, PDE4D, PDE4A/B, PDE4A/C, PDE4A/D, PDE4B/C, PDE4B/D, PDE4C/D, and PDE4A/B/C/D. Articles that cited PDE4 and cancer without referencing specific subtypes and articles that used nonspecific PDE4 inhibitors for pharmacologic knockdown of PDE4 were categorized as “Unspecified”. Articles that examined PDE4 outside the context of cancer were placed in the “Not Relevant” category. For the purposes of constructing a meaningful data analysis, papers from each of the categories with two unique PDE subtypes were added to the individual subtype categories. This led to a final classification of papers as PDE4A (n = 17), PDE4B (n = 34), PDE4C (n = 7), and PDE4D (n = 44).
One concern with the use of PDE4 inhibitors for the treatment of cancer is their immunosuppressive effects. Sustained signaling through cAMP is generally immunosuppressive. Specifically, cAMP signaling can lead to the inhibition of NF-κB, a potent pro-inflammatory mediator, as well as downregulation of TNF-α, IFN-γ, and IL-17 [96]. With the advent of cancer treatment strategies that harness the immune system, such as checkpoint inhibitors and adoptive cell transfer, PDE4 inhibitors should be investigated with caution. It is unclear what effect these inhibitors may have on immunogenic treatments.
Strengths and limitations
Potent, marketed PDE4 inhibitors are available for clinical proof-of-concept studies in cancer as in the case of the published clinical Phase 0/1 biomarker and pharmacodynamic study of roflumilast in patients with advanced B-Cell hematologic malignancies (NCT01888952) [92] and as in an on-going clinical study of roflumilast in combination with standard chemotherapy for high-risk DLBCL (NCT03458546). The underlying therapeutic hypothesis is that overexpression of PDE4B contributes to oncogenesis through downregulation of intracellular cAMP and that this can be opposed by pharmacological inhibition of the cyclic nucleotide phosphodiesterase. The limitation of marketed PDE4 inhibitors is their poor gastrointenstinal tolerability with escalation of dose which may preclude achieving sufficient inhibition of specific PDE4 subtypes for demonstration of clinical benefit [5]. Alternatively, subtype selective PDE4 inhibitors may show better tolerability [6], but to date it has only been possible to target PDE4B [9, 97] or PDE4D [6, 98] with subtype selective inhibitors. A major limitation in structure-based drug design is the absolute amino acid sequence conservation of the PDE4 active site across PDE4 subtypes [99]. Subtype selective PDE4 inhibitors, in contrast, bind in allosteric sites located on regulatory domains that open and close across the PDE4 active site, and these binding sites differ in amino acid sequence between PDE4 subtypes [6, 9, 98]. In addition, specific isoforms protein products translated from the four PDE4 genes have been shown to have unique cellular localizations and functions [100]. It is plausible that these unique proteins may account for some of the discrepencies in PDE4 subtype functions observed and reported here. Future studies should report PDE4 isoform specificity wherever possible to determine the clinical relevance of PDE4 isoform compartmentalization and local cAMP gradients within cancer cells.
Methods
Search strategy
In order to systematically evaluate the role of PDE4 specific subtypes in cancer, we searched MEDLINE via PubMed for studies that mentioned PDE4 and its association with cancer. Our specific advanced search terms included: “PDE4 and Cancer” OR “PDE4A and Cancer” OR “PDE4B and Cancer” OR “PDE4C and Cancer” OR “PDE4D and Cancer”. Our search results provided 297 papers as of November 1, 2019.
Screening criteria
Two investigators reviewed each initial study to determine whether it was specific for one or more PDE4 subtypes. The articles were sorted into the following 13 categories (Fig. 2): PDE4A (n = 9), PDE4B (n = 26), PDE4C (n = 3), PDE4D (n = 36), PDE4A/B (n = 3), PDE4A/C (n = 1), PDE4A/D (n = 3), PDE4B/C (n = 1), PDE4B/D (n = 3), PDE4C/D (n = 1), PDE4A/B/C/D (n = 1), unspecified (n = 93), and not relevant (n = 117). Articles that cited PDE4 and cancer without referencing specific subtypes and articles that used nonspecific PDE4 inhibitors for the pharmacologic knockdown of PDE4 were categorized as “Unspecified”. Articles that examined PDE4 outside the context of cancer were placed in the “Not Relevant” category. Disagreements between reviewers were resolved by re-evaluation and discussion with a third reviewer.
Data consolidation
For the purposes of constructing a meaningful data analysis, papers from each of the categories with two unique PDE subtypes were added to the individual subtype categories. This led to a final classification of papers as PDE4A (n = 17), PDE4B (n = 34), PDE4C (n = 7), and PDE4D (n = 44).
Conclusions
The largest current bodies of research with PDE4 subtypes in cancer surround PDE4A, PDE4B, and PDE4D. The general trend seems to be that PDE4 subtypes correspond to poor prognosis in many tumor types, although there are a small number of studies contradicting this across each of the subtypes. PDE4A and PDE4B have the largest bodies of evidence supporting their utlity as targets for hematologic malignancies, where PDE4D may play a larger role in solid tumors. Regardless of the type of malignancy, there is strong evidence supporting PDE4 inhibition as a viable way to potentiate cAMP-mediated inhibition of AKT pathways. PDE4 may be a viable target for anticancer therapies, and this warrants future studies into specific subtypes across malignancies.
References
Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. 2014;13:290.
Nanayakkara S, Mak V, Crannitch K, Byrne M, Kaye DM. Extended release oral milrinone, CRD-102, for advanced heart failure. Am J Cardiol. 2018;122:1017–20.
Das A, Durrant D, Salloum FN, Xi L, Kukreja RC. PDE5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol Ther. 2015;147:12–21.
Zebda R, Paller AS. Phosphodiesterase 4 inhibitors. J Am Acad Dermatol. 2018;78:S43–52.
Li H, Zuo J, Tang W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front Pharmacol. 2018;9. https://doi.org/10.3389/fphar.2018.01048.
Burgin AB, Magnusson OT, Singh J, Witte P, Staker BL, Bjornsson JM, et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol. 2010;28:63–70.
Zhang C, Xu Y, Zhang H-T, Gurney ME, O’Donnell JM. Comparison of the pharmacological profiles of selective PDE4B and PDE4D inhibitors in the central nervous system. Sci Rep. 2017;7:40115.
Hagen TJ, Mo X, Burgin AB, Fox D, Zhang Z, Gurney ME. Discovery of triazines as selective PDE4B versus PDE4D inhibitors. Bioorg Med Chem Lett. 2014;24:4031–4.
Fox D, Burgin AB, Gurney ME. Structural basis for the design of selective phosphodiesterase 4B inhibitors. Cell Signal. 2014;26:657–63.
Bolger GB, Bizzi MF, Pinheiro SV, Trivellin G, Smoot L, Accavitti M-A, et al. cAMP-specific PDE4 phosphodiesterases and AIP in the pathogenesis of pituitary tumors. Endocr Relat Cancer. 2016;23:419–31.
Goldhoff P, Warrington NM, Limbrick DDJ, Hope A, Woerner BM, Jackson E, et al. Targeted inhibition of cyclic AMP phosphodiesterase-4 promotes brain tumor regression. Clin Cancer Res. 2008;14:7717–25.
Warrington NM, Gianino SM, Jackson E, Goldhoff P, Garbow JR, Piwnica-Worms D, et al. Cyclic AMP suppression is sufficient to induce gliomagenesis in a mouse model of neurofibromatosis-1. Cancer Res. 2010;70:5717–27.
Moon E-Y, Lee G-H, Lee M-S, Kim H-M, Lee J-W. Phosphodiesterase inhibitors control A172 human glioblastoma cell death through cAMP-mediated activation of protein kinase A and Epac1/Rap1 pathways. Life Sci. 2012;90:373–80.
Toledo RA, Mendonca BB, Fragoso MCBV, Soares IC, Almeida MQ, Moraes MB, et al. Isolated familial somatotropinoma: 11q13-loh and gene/protein expression analysis suggests a possible involvement of aip also in non-pituitary tumorigenesis. Clinical. 2010;65:407–15.
Cokic VP, Mossuz P, Han J, Socoro N, Beleslin-Cokic BB, Mitrovic O, et al. Microarray and proteomic analyses of myeloproliferative neoplasms with a highlight on the mTOR signaling pathway. PLoS ONE. 2015;10:e0135463.
Sarfati M, Mateo V, Baudet S, Rubio M, Fernandez C, Davi F, et al. Sildenafil and vardenafil, types 5 and 6 phosphodiesterase inhibitors, induce caspase-dependent apoptosis of B-chronic lymphocytic leukemia cells. Blood. 2003;101:265–9.
Kim DH, Lerner A. Type 4 cyclic adenosine monophosphate phosphodiesterase as a therapeutic target in chronic lymphocytic leukemia. Blood. 1998;92:2484–94.
Chamseddine AN, Cabrero M, Wei Y, Ganan-Gomez I, Colla S, Takahashi K, et al. PDE4 differential expression is a potential prognostic factor and therapeutic target in patients with myelodysplastic syndrome and chronic myelomonocytic leukemia. Clin Lymphoma Myeloma Leuk. 2016;16:S67–73.
Jiang X, Paskind M, Weltzien R, Epstein PM. Expression and regulation of mRNA for distinct isoforms of cAMP-specific PDE-4 in mitogen-stimulated and leukemic human lymphocytes. Cell Biochem Biophys. 1998;28:135–60.
Dong H, Zitt C, Auriga C, Hatzelmann A, Epstein PM. Inhibition of PDE3, PDE4 and PDE7 potentiates glucocorticoid-induced apoptosis and overcomes glucocorticoid resistance in CEM T leukemic cells. Biochem Pharm. 2010;79:321–9.
Fernandez-Araujo A, Alfonso A, Vieytes MR, Botana LM. Yessotoxin activates cell death pathways independent of Protein Kinase C in K-562 human leukemic cell line. Toxicol Vitr. 2015;29:1545–54.
Kolosionek E, Savai R, Ghofrani HA, Weissmann N, Guenther A, Grimminger F, et al. Expression and activity of phosphodiesterase isoforms during epithelial mesenchymal transition: the role of phosphodiesterase 4. Mol Biol Cell. 2009;20:4751–65.
Pullamsetti SS, Banat GA, Schmall A, Szibor M, Pomagruk D, Hanze J, et al. Phosphodiesterase-4 promotes proliferation and angiogenesis of lung cancer by crosstalk with HIF. Oncogene. 2013;32:1121–34.
Peng Y, Li Y, Tian Y, Ao G. PDE4a predicts poor prognosis and promotes metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. J Cancer. 2018;9:2389–96.
Knobloch TJ, Ryan NM, Bruschweiler-Li L, Wang C, Bernier MC, Somogyi A et al. Metabolic regulation of glycolysis and AMP activated protein kinase pathways during black raspberry-mediated oral cancer chemoprevention. Metabolites. 2019;9. https://doi.org/10.3390/metabo9070140.
Wittliff JL, Sereff SB, Daniels MW. Expression of genes for methylxanthine pathway-associated enzymes accompanied by sex steroid receptor status impacts breast carcinoma progression. Horm Cancer. 2017;8:298–313.
Smith PG, Wang F, Wilkinson KN, Savage KJ, Klein U, Neuberg DS, et al. The phosphodiesterase PDE4B limits cAMP-associated PI3K/AKT-dependent apoptosis in diffuse large B-cell lymphoma. Blood. 2005;105:308–16.
Suhasini AN, Wang L, Holder KN, Lin A-P, Bhatnagar H, Kim S-W, et al. A phosphodiesterase 4B-dependent interplay between tumor cells and the microenvironment regulates angiogenesis in B-cell lymphoma. Leukemia. 2016;30:617–26.
Rickles RJ, Pierce LT, Giordano TP3rd, Tam WF, McMillin DW, Delmore J, et al. Adenosine A2A receptor agonists and PDE inhibitors: a synergistic multitarget mechanism discovered through systematic combination screening in B-cell malignancies. Blood. 2010;116:593–602.
Nagy ZS, Ross JA, Rodriguez G, Balint BL, Szeles L, Nagy L, et al. Genome wide mapping reveals PDE4B as an IL-2 induced STAT5 target gene in activated human PBMCs and lymphoid cancer cells. PLoS ONE. 2013;8:e57326.
Liu S, Liu Y, Zhang Q, Wu J, Liang J, Yu S, et al. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 2017;18:194.
Tiwari S, Dong H, Kim EJ, Weintraub L, Epstein PM, Lerner A. Type 4 cAMP phosphodiesterase (PDE4) inhibitors augment glucocorticoid-mediated apoptosis in B cell chronic lymphocytic leukemia (B-CLL) in the absence of exogenous adenylyl cyclase stimulation. Biochem Pharm. 2005;69:473–83.
Kim S-W, Rai D, Aguiar RCT. Gene set enrichment analysis unveils the mechanism for the phosphodiesterase 4B control of glucocorticoid response in B-cell lymphoma. Clin Cancer Res. 2011;17:6723–32.
Kim J, Jeong D, Nam J, Aung TN, Gim J-A, Park KU, et al. MicroRNA-124 regulates glucocorticoid sensitivity by targeting phosphodiesterase 4B in diffuse large B cell lymphoma. Gene. 2015;558:173–80.
Yang JJ, Cheng C, Devidas M, Cao X, Campana D, Yang W, et al. Genome-wide association study identifies germline polymorphisms associated with relapse of childhood acute lymphoblastic leukemia. Blood. 2012;120:4197–204.
Kim S-W, Rai D, McKeller MR, Aguiar RCT. Rational combined targeting of phosphodiesterase 4B and SYK in DLBCL. Blood. 2009;113:6153–60.
Tsunoda T, Ota T, Fujimoto T, Doi K, Tanaka Y, Yoshida Y, et al. Inhibition of phosphodiesterase-4 (PDE4) activity triggers luminal apoptosis and AKT dephosphorylation in a 3-D colonic-crypt model. Mol Cancer. 2012;11:46.
Mahmood B, Damm MMB, Jensen TSR, Backe MB, Dahllof MS, Poulsen SS, et al. Phosphodiesterases in non-neoplastic appearing colonic mucosa from patients with colorectal neoplasia. BMC Cancer. 2016;16:938.
Pleiman JK, Irving AA, Wang Z, Toraason E, Clipson L, Dove WF et al. The conserved protective cyclic AMP-phosphodiesterase function PDE4B is expressed in the adenoma and adjacent normal colonic epithelium of mammals and silenced in colorectal cancer. PLoS Genet. 2018;14. https://doi.org/10.1371/journal.pgen.1007611.
Nishi K, Luo H, Ishikura S, Doi K, Iwaihara Y, Wills L, et al. Apremilast induces apoptosis of human colorectal cancer cells with mutant KRAS. Anticancer Res. 2017;37:3833–9.
Lapa GB, Tsunoda T, Shirasawa S, Baryshnikova MA, Evseev GG, Afanasyeva DA, et al. Synthesis of new congeners of 1-methyl-3-aminoisoquinolines, evaluation of their cytotoxic activity, in silico and in vitro study of their molecular targets as PDE4B. Chem Biol Drug Des. 2016;87:575–82.
Mareddy J, Nallapati SB, Anireddy J, Devi YP, Mangamoori LN, Kapavarapu R, et al. Synthesis and biological evaluation of nimesulide based new class of triazole derivatives as potential PDE4B inhibitors against cancer cells. Bioorg Med Chem Lett. 2013;23:6721–7.
Kim DU, Kwak B, Kim SW. Phosphodiesterase 4B is an effective therapeutic target in colorectal cancer. Biochem Biophys Res Commun. 2019;508:825–31.
Shah MS, Schwartz SL, Zhao C, Davidson LA, Zhou B, Lupton JR, et al. Integrated microRNA and mRNA expression profiling in a rat colon carcinogenesis model: effect of a chemo-protective diet. Physiol Genomics. 2011;43:640–54.
He R-Q, Li X-J, Liang L, Xie Y, Luo D-Z, Ma J, et al. The suppressive role of miR-542-5p in NSCLC: the evidence from clinical data and in vivo validation using a chick chorioallantoic membrane model. BMC Cancer. 2017;17:655.
Narita M, Murata T, Shimizu K, Nakagawa T, Sugiyama T, Inui M, et al. A role for cyclic nucleotide phosphodiesterase 4 in regulation of the growth of human malignant melanoma cells. Oncol Rep. 2007;17:1133–9.
Holloway DT, Kon M, DeLisi C. In silico regulatory analysis for exploring human disease progression. Biol Direct. 2008;3:24.
Dong P, Xiong Y, Yue J, Xu D, Ihira K, Konno Y, et al. Long noncoding RNA NEAT1 drives aggressive endometrial cancer progression via miR-361-regulated networks involving STAT3 and tumor microenvironment-related genes. J Exp Clin Cancer Res. 2019;38. https://doi.org/10.1186/s13046-019-1306-9.
Praveena KSS, Durgadas S, Suresh Babu N, Akkenapally S, Ganesh Kumar C, Deora GS, et al. Synthesis of 2,2,4-trimethyl-1,2-dihydroquinolinyl substituted 1,2,3-triazole derivatives: their evaluation as potential PDE 4B inhibitors possessing cytotoxic properties against cancer cells. Bioorg Chem. 2014;53:8–14.
Babu PV, Mukherjee S, Deora GS, Chennubhotla KS, Medisetti R, Yellanki S, et al. Ligand/PTC-free intramolecular Heck reaction: synthesis of pyrroloquinoxalines and their evaluation against PDE4/luciferase/oral cancer cell growth in vitro and zebrafish in vivo. Org Biomol Chem. 2013;11:6680–5.
Kashiwagi E, Shiota M, Yokomizo A, Itsumi M, Inokuchi J, Uchiumi T, et al. Downregulation of phosphodiesterase 4B (PDE4B) activates protein kinase A and contributes to the progression of prostate cancer. Prostate. 2012;72:741–51.
Obernolte R, Ratzliff J, Baecker PA, Daniels DV, Zuppan P, Jarnagin K, et al. Multiple splice variants of phosphodiesterase PDE4C cloned from human lung and testis. Biochim Biophys Acta. 1997;1353:287–97.
Bao Z, Feng Y, Wang H, Zhang C, Sun L, Yan Z, et al. Integrated analysis using methylation and gene expression microarrays reveals PDE4C as a prognostic biomarker in human glioma. Oncol Rep. 2014;32:250–60.
Persani L, Lania A, Alberti L, Romoli R, Mantovani G, Filetti S, et al. Induction of specific phosphodiesterase isoforms by constitutive activation of the cAMP pathway in autonomous thyroid adenomas. J Clin Endocrinol Metab. 2000;85:2872–8.
Garritano S, Inga A, Gemignani F, Landi S. More targets, more pathways and more clues for mutant p53. Oncogenesis. 2013;2:e54.
Zhang L, Murray F, Zahno A, Kanter JR, Chou D, Suda R, et al. Cyclic nucleotide phosphodiesterase profiling reveals increased expression of phosphodiesterase 7B in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2008;105:19532–7.
He N, Kim N, Song M, Park C, Kim S, Park EY, et al. Integrated analysis of transcriptomes of cancer cell lines and patient samples reveals STK11/LKB1-driven regulation of cAMP phosphodiesterase-4D. Mol Cancer Ther. 2014;13:2463–73.
Karachaliou N, Codony-Servat J, Teixidó C, Pilotto S, Drozdowskyj A, Codony-Servat C, et al. BIM and mTOR expression levels predict outcome to erlotinib in EGFR-mutant non-small-cell lung cancer. Sci Rep. 2015;5:17499.
Baty F, Klingbiel D, Zappa F, Brutsche M. High-throughput alternative splicing detection using dually constrained correspondence analysis (DCCA). J Biomed Inf. 2015;58:175–85.
Henderson DJP, Houslay MD, Bangma CH, Hoffmann R. Creating a potential diagnostic for prostate cancer risk stratification (InformMDxTM) by translating novel scientific discoveries concerning cAMP degrading phosphodiesterase-4D7 (PDE4D7). Clin Sci. 2019;133:269–86.
Henderson DJP, Byrne A, Dulla K, Jenster G, Hoffmann R, Baillie GS, et al. The cAMP phosphodiesterase-4D7 (PDE4D7) is downregulated in androgen-independent prostate cancer cells and mediates proliferation by compartmentalising cAMP at the plasma membrane of VCaP prostate cancer cells. Br J Cancer. 2014;110:1278–87.
Bottcher R, Dulla K, van Strijp D, Dits N, Verhoef EI, Baillie GS, et al. Human PDE4D isoform composition is deregulated in primary prostate cancer and indicative for disease progression and development of distant metastases. Oncotarget. 2016;7:70669–84.
Van Strijp D, De Witz C, Heitkötter B, Huss S, Bögemann M, Baillie GS et al. The association of the long prostate cancer expressed PDE4D transcripts to poor patient outcome depends on the tumour’s TMPRSS2-ERG fusion status. Prostate Cancer. 2019;2019. https://doi.org/10.1155/2019/8107807.
Rahrmann EP, Collier LS, Knutson TP, Doyal ME, Kuslak SL, Green LE, et al. Identification of PDE4D as a proliferation promoting factor in prostate cancer using a Sleeping Beauty transposon-based somatic mutagenesis screen. Cancer Res. 2009;69:4388–97.
Powers GL, Hammer KDP, Domenech M, Frantskevich K, Malinowski RL, Bushman W, et al. Phosphodiesterase 4D inhibitors limit prostate cancer growth potential. Mol Cancer Res. 2015;13:149–60.
Geybels MS, Alumkal JJ, Luedeke M, Rinckleb A, Zhao S, Shui IM, et al. Epigenomic profiling of prostate cancer identifies differentially methylated genes in TMPRSS2:ERG fusion-positive versus fusion-negative tumors. Clin Epigenetics. 2015;7:128.
Delyon J, Servy A, Laugier F, Andre J, Ortonne N, Battistella M, et al. PDE4D promotes FAK-mediated cell invasion in BRAF-mutated melanoma. Oncogene. 2017;36:3252–62.
Bogunovic D, O’Neill DW, Belitskaya-Levy I, Vacic V, Yu Y-L, Adams S, et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Natl Acad Sci USA. 2009;106:20429–34.
Khaled M, Levy C, Fisher DE. Control of melanocyte differentiation by a MITF-PDE4D3 homeostatic circuit. Genes Dev. 2010;24:2276–81.
Xu T, Wu S, Yuan Y, Yan G, Xiao D. Knockdown of phosphodiesterase 4D inhibits nasopharyngeal carcinoma proliferation via the epidermal growth factor receptor signaling pathway. Oncol Lett. 2014;8:2110–6.
Gu J, Ajani JA, Hawk ET, Ye Y, Lee JH, Bhutani MS, et al. Genome-wide catalogue of chromosomal aberrations in barrett’s esophagus and esophageal adenocarcinoma: a high-density single nucleotide polymorphism array analysis. Cancer Prev Res. 2010;3:1176–86.
Nancarrow DJ, Handoko HY, Smithers BM, Gotley DC, Drew PA, Watson DI, et al. Genome-wide copy number analysis in esophageal adenocarcinoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2008;68:4163–72.
Bye H, Prescott NJ, Lewis CM, Matejcic M, Moodley L, Robertson B, et al. Distinct genetic association at the PLCE1 locus with oesophageal squamous cell carcinoma in the South African population. Carcinogenesis. 2012;33:2155–61.
Floor SL, Tresallet C, Hebrant A, Desbuleux A, Libert F, Hoang C, et al. microRNA expression in autonomous thyroid adenomas: Correlation with mRNA regulation. Mol Cell Endocrinol. 2015;411:1–10.
Ge X, Milenkovic L, Suyama K, Hartl T, Purzner T, Winans A et al. Phosphodiesterase 4D acts downstream of Neuropilin to control Hedgehog signal transduction and the growth of medulloblastoma. Elife. 2015;4. https://doi.org/10.7554/eLife.07068.
Kang T-W, Choi SW, Yang S-R, Shin T-H, Kim H-S, Yu K-R, et al. Growth arrest and forced differentiation of human primary glioblastoma multiforme by a novel small molecule. Sci Rep. 2014;4:5546.
Milinkovic V, Bankovic J, Rakic M, Stankovic T, Skender-Gazibara M, Ruzdijic S, et al. Identification of novel genetic alterations in samples of malignant glioma patients. PLoS ONE. 2013;8:e82108.
Cao B, Wang K, Liao J-M, Zhou X, Liao P, Zeng SX et al. Inactivation of oncogenic cAMP-specific phosphodiesterase 4D by miR-139-5p in response to p53 activation. Elife. 2016;5. https://doi.org/10.7554/eLife.15978.
McEwan DG, Brunton VG, Baillie GS, Leslie NR, Houslay MD, Frame MC. Chemoresistant KM12C colon cancer cells are addicted to low cyclic AMP levels in a phosphodiesterase 4-regulated compartment via effects on phosphoinositide 3-kinase. Cancer Res. 2007;67:5248–57.
Kim DU, Nam J, Cha MD, Kim SW. Inhibition of phosphodiesterase 4D decreases the malignant properties of DLD-1 colorectal cancer cells by repressing the AKT/mTOR/Myc signaling pathway. Oncol Lett. 2019;17:3589–98.
Chen L, Gao H, Liang J, Qiao J, Duan J, Shi H, et al. miR-203a-3p promotes colorectal cancer proliferation and migration by targeting PDE4D. Am J Cancer Res. 2018;8:2387–401.
Araújo T, Khayat A, Quintana L, Calcagno D, Mourão R, Modesto A, et al. Piwi like RNA-mediated gene silencing 1 gene as a possible major player in gastric cancer. World J Gastroenterol. 2018;24:5338–50.
Miklos W, Heffeter P, Pirker C, Hager S, Kowol CR, van Schoonhoven S, et al. Loss of phosphodiesterase 4D mediates acquired triapine resistance via Epac-Rap1-Integrin signaling. Oncotarget. 2016;7:84556–74.
Peng Q-P, Du D-B, Ming Q, Hu F, Wu Z-B, Qiu S. MicroRNA 494 increases chemosensitivity to doxorubicin in gastric cancer cells by targeting phosphodiesterases 4D. Cell Mol Biol. 2018;64:62–66.
Haddad SA, Ruiz-Narvaez EA, Haiman CA, Sucheston-Campbell LE, Bensen JT, Zhu Q, et al. An exome-wide analysis of low frequency and rare variants in relation to risk of breast cancer in African American Women: the AMBER Consortium. Carcinogenesis. 2016;37:870–7.
Natrajan R, Mackay A, Lambros MB, Weigelt B, Wilkerson PM, Manie E, et al. A whole-genome massively parallel sequencing analysis of BRCA1 mutant oestrogen receptor-negative and -positive breast cancers. J Pathol. 2012;227:29–41.
Bodelon C, Oh H, Chatterjee N, Garcia-Closas M, Palakal M, Sherman ME, et al. Association between breast cancer genetic susceptibility variants and terminal duct lobular unit involution of the breast. Int J Cancer. 2017;140:825–32.
Agostini A, Brunetti M, Davidson B, Göran Tropé C, Heim S, Panagopoulos I, et al. Identification of novel cyclin gene fusion transcripts in endometrioid ovarian carcinomas. Int J Cancer. 2018;143:1379–87.
Wang Y, Fang T, Huang L, Wang H, Zhang L, Wang Z, et al. Neutrophils infiltrating pancreatic ductal adenocarcinoma indicate higher malignancy and worse prognosis. Biochem Biophys Res Commun. 2018;501:313–9.
Mishra RR, Belder N, Ansari SA, Kayhan M, Bal H, Raza U, et al. Reactivation of cAMP pathway by PDE4D inhibition represents a novel druggable axis for overcoming tamoxifen resistance in er-positive breast cancer. Clin Cancer Res. 2018;24:1987–2001.
Qiang Z, Zhou ZY, Peng T, Jiang PZ, Shi N, Njoya EM et al. Inhibition of TPL2 by interferon-α suppresses bladder cancer through activation of PDE4D. J Exp Clin Cancer Res. 2018;37. https://doi.org/10.1186/s13046-018-0971-4.
Kelly K, Mejia A, Suhasini AN, Lin A-P, Kuhn J, Karnad AB, et al. Safety and pharmacodynamics of the PDE4 inhibitor roflumilast in advanced B-cell malignancies. Clin Cancer Res. 2017;23:1186–92.
Ramezani S, Vousooghi N, Ramezani Kapourchali F, Yousefzadeh-Chabok S, Reihanian Z, Alizadeh AM, et al. Rolipram optimizes therapeutic effect of bevacizumab by enhancing proapoptotic, antiproliferative signals in a glioblastoma heterotopic model. Life Sci. 2019;239. https://doi.org/10.1016/j.lfs.2019.116880.
Ramezani S, Vousooghi N, Kapourchali FR, Hadjighasem M, Hayat P, Amini N, et al. Rolipram potentiates bevacizumab-induced cell death in human glioblastoma stem-like cells. Life Sci. 2017;173:11–19.
Cooney JD, Lin AP, Jiang D, Wang L, Suhasini AN, Myers J, et al. Synergistic targeting of the regulatory and catalytic subunits of pi3kd in mature b-cell malignancies. Clin Cancer Res. 2018;24:1103–13.
Sakkas LI, Mavropoulos A, Bogdanos DP. Phosphodiesterase 4 inhibitors in immune-mediated diseases: mode of action, clinical applications, current and future perspectives. Curr Med Chem. 2017;24. https://doi.org/10.2174/0929867324666170530093902.
Naganuma K, Omura A, Maekawara N, Saitoh M, Ohkawa N, Kubota T, et al. Discovery of selective PDE4B inhibitors. Bioorg Med Chem Lett. 2009;19:3174–6.
Gurney ME, Nugent RA, Mo X, Sindac JA, Hagen TJ, Fox D, et al. Design and synthesis of selective phosphodiesterase 4D (PDE4D) Allosteric inhibitors for the treatment of fragile X syndrome and other brain disorders. J Med Chem. 2019;62:4884–901.
Wang H, Peng M-S, Chen Y, Geng J, Robinson H, Houslay MD, et al. Structures of the four subfamilies of phosphodiesterase-4 provide insight into the selectivity of their inhibitors. Biochem J. 2007;408:193–201.
Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91–100.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MG is an employee of Tetra Discovery Partners, Inc. that has a financial interest in the discovery and development of PDE4B and PDE4D allosteric inhibitors for the treatment of Fragile X Syndrome, Alzheimer’s disease, and other CNS disorders.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hsien Lai, S., Zervoudakis, G., Chou, J. et al. PDE4 subtypes in cancer. Oncogene 39, 3791–3802 (2020). https://doi.org/10.1038/s41388-020-1258-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-1258-8
- Springer Nature Limited
This article is cited by
-
Elevated PDE4C level serves as a candidate diagnostic biomarker and correlates with poor survival in thyroid carcinoma
Scientific Reports (2024)
-
Loss of miR-26b-5p promotes gastric cancer progression via miR-26b-5p-PDE4B/CDK8-STAT3 feedback loop
Journal of Translational Medicine (2023)
-
IgG immune complex-induced acute lung injury is ameliorated by cAMP via down-regulation of C/EBP- and AP-1-mediated transcriptions
Journal of Inflammation (2023)
-
Toxic PARP trapping upon cAMP-induced DNA damage reinstates the efficacy of endocrine therapy and CDK4/6 inhibitors in treatment-refractory ER+ breast cancer
Nature Communications (2023)
-
Novel drug-target interactions via link prediction and network embedding
BMC Bioinformatics (2022)