
Abstract
Chronic BK polyomavirus (BKPyV) infection is recognized as a potential oncogenic factor of urothelial carcinoma (UC) in renal transplant recipients. Recent studies have reported a positive correlation among BKPyV integration, persistent overexpression of viral large T antigen (TAg), and malignancy, yet little is known about the specific integration mechanisms and the impacts of viral integration. Here, we performed whole-genome sequencing (WGS) and viral capture-based sequencing on high-grade immunohistochemically TAg-positive UCs in two renal transplant recipients. A total of 181 integration sites, including the three found by WGS, were identified by viral capture-based sequencing, indicating its enhanced sensitivity and ability in identifying low-read integration sites in subpopulations of the tumor cells. The microhomologies between human and BKPyV genomes were significantly enriched in the flanking regions of 84.5% the integration sites, with a median length of 7 bp. Notably, 75 human genes formed fusion sequences due to viral insertional integration. Among them, the expression of 15 genes were statistically associated with UC based on GEO2R expression analysis. Our results indicated a multisite and multifragment linear integration pattern and a potential microhomology or nonhomologous end joining integration mechanism at the single-nucleotide level. We put forward a potential selection mechanism driven by immunity and centered on viral integration in the carcinogenesis of BKPyV.
Similar content being viewed by others

Introduction
Oncogenesis has gradually become one of the major factors influencing the long-term survival of renal transplant recipients [1,2,3,4]. In recent years, BK polyomavirus (BKPyV) infection has been recognized as a potential risk factor for urothelial carcinoma (UC) in renal transplant recipients [5, 6]. Several studies have demonstrated that overexpression of viral large T antigen (TAg) promotes oncogenesis by inactivating both p53 and pRb [7,8,9].
Recent studies suggested a positive correlation among chronic infection of BKPyV, incidence of BKPyV integration, and persistent overexpression of viral TAg in immunosuppressed individuals [1, 10]. The immunohistochemically BKPyV TAg-positive UC has been regarded as BKPyV-associated carcinoma. Three seminal publications by Kenan et al. [11, 12] and Müller et al. [13] identified viral integration sites on host chromosomes using next-generation sequencing, providing compelling evidence of the essential role of BKPyV in the pathogenesis of high-grade carcinomas arising in the urogenital tract of kidney transplant recipients. The specific BKPyV integration mechanisms and the impacts of viral integration on the deregulation of viral oncogene expression, host gene expression, and genomic instability needs further validation, based on Wang et al.’s work [14].
In this study, we performed whole-genome sequencing (WGS) and capture-based sequencing on both tumor and uninvolved tissue of two renal transplant recipients with immunohistochemically-confirmed TAg-positive high-grade UC. Our results shed new light on the potentially critical steps in the carcinogenesis of BKPyV-associated carcinoma.
Results
Clinical cases
The research was approved by the ethical committee of Nanfang Hospital (NFEC-2020-044) and the Institutional Review Board at Massachusetts General Hospital. The study was performed in accordance with the Declaration of Helsinki. Informed consent was obtained from all subjects.
Case 1
A 42-year-old female with end-stage renal disease received a kidney transplant from a deceased young male donor who died in a motor vehicle accident at the Nanfang Hospital in Guangzhou, China, in August 2009. The immunosuppression induction included rabbit antihuman thymocyte globulin 0.1 g daily within 3 days and methylprednisolone (1 000/500/250/250 mg, daily); her maintenance immunosuppression consisted of tacrolimus, mycophenolate mofetil, and prednisone. Nine years after transplantation, in January 2018, the patient presented with gross hematuria, while the graft function was normal (serum creatinine, 0.91 mg/dL). Abdominal ultrasound and contrast-enhanced computed tomography (CT) demonstrated a 3.5 × 2.4 cm solid mass in the bladder (Fig. 1A, B). The patient underwent cystoscopy and transurethral tumor resection in January 2018. The tumor was pathologically classified as a high-grade papillary UC invading the lamina propria (Fig. 1C, D) and was staged T1N0M0 according to the AJCC Cancer Staging Manual 8th edition. Immunohistochemistry showed strong nuclear staining of polyomavirus TAg in the tumor cells (Fig. 1E, F).

(A, B) CT images. Plain scan (A) and contrast-enhanced (B) CT show local thickening of the left posterior wall and a sessile soft-tissue mass intruding into the bladder cavity (white arrow). The surface of the tumor is not smooth and has shallow lobulation ranging 29 × 30 mm. C–F Histology and immunohistochemistry of bladder tumor. Hematoxylin and eosin (H&E) staining of the tumor tissue at ×40 (C) and ×200 (D) show high-grade urothelial carcinoma invading the lamina propria; immunohistochemistry at ×40 (E) and ×200 (F) with antibodies against SV40 TAg show strong staining in the nuclei of tumor cells.
Case 2
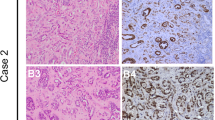
The clinical profile of this case has been previously reported in the study describing the muti-stage carcinogenesis of BKPyV [14]. A 76-year-old female with end-stage renal disease secondary to systemic lupus erythematosus received a kidney transplant in Massachusetts General Hospital in Boston in 2006. The maintenance immunosuppression consisted of tacrolimus, mycophenolate mofetil, Plavix, and prednisone. In March 2018, the patient presented with persistent Escherichia coli urosepsis, uric acidosis, and increased serum creatinine (2.6 from 1.3 mg/dL baseline). Ultrasound indicated hydronephrosis of the transplant kidney and a bladder mass. Cystoscopy showed a 2.4 × 4.2 × 4.3 cm dominant tumor arising from the right anterior bladder wall, and 5–10 additional tumors throughout the bladder. Radical cystectomy following a positive bladder biopsy revealed an invasive high-grade papillary UC invading through the muscularis propria to involve the uterus with metastasis in two lymph nodes, staged at T4N2M0 according to the 8th edition of AJCC Cancer Staging Manual (Fig. 2A, B). Immunohistochemistry showed strong polyomavirus TAg staining in the nuclei of tumor cells (Fig. 2C, D).

(A, B) Histology and immunohistochemistry of bladder tumor. Hematoxylin and eosin (H&E) staining of tumor tissue at ×200 shows high-grade urothelial carcinoma invading muscularis propria (A) and metastatic to the lymph nodes (B); immunohistochemistry with antibodies against SV40 TAg at ×200 shows strong nuclear staining in the tumor cells invading muscularis propria (C) and metastatic to lymph nodes (D).
Distribution of breakpoints in the human and BKPyV genomes
Both WGS (>30× coverage) and capture-based viral gene sequencing (>500× coverage) were conducted on tumor tissue and uninvolved tissue of two BKPyV-associated UC samples using the NovaSeq platform (Illumina Genome Network). A total of 181 BKPyV integration breakpoints were detected in the two tumor samples (Supplementary Table S1), with 7 in case 1 and 174 in case 2. Twenty of them were further verified via targeted PCR amplification and Sanger sequencing (Supplementary Fig. S1).
Case 1
We detected 682.46 million clean reads by WGS, 99.49% of which were successfully mapped to human reference sequence 19 (hg19) and BKPyV reference sequences with an average depth of 64.76×; 638 reads were successfully mapped to viral sequences, with an average depth of 17.30×. Three viral integration sites were identified by WGS. By viral capture-based BKPyV gene sequencing on the tumor tissue sample, on average, 2 780 (range 23–9 630) reads were mapped to each site with an average sequencing depth of 5711.08×. Seven integration sites, including the three viral integration sites identified by WGS, were identified from BKPyV genotype Ic (THK-9a, GenBank: AB217921) (Fig. 3A, B).

(A) Broad distribution of breakpoints mapped to BKPyV genome. Orange histograms: depth of coverage of BKPyV genome in each position; black dots: position of BKPyV insertional breakpoints; size of dots: larger dots correspond to higher number of supporting discordant reads. (B) Broad distribution of breakpoints mapped to human genome. Outer circles: human chromosomes; black dots and size of dots: the same as (A). All the data were obtained from virus capture sequencing. (A, B) are from case 1, while (C, D) are from case 2. According to the sequencing depth and coverage of the BKPyV genome, portions of the viral sequences were not detected in the sequencing. Integration sites existed at both ends of the missing segments, indicating their integration into the host chromosomes.
Case 2
We detected 712 million clean reads by WGS, 91.81% of which were successfully mapped to hg19 and viral reference sequences with an average depth of 31.53×; 85 reads were successfully mapped to pure viral sequences, with an average depth of 1.75×. No viral integration sites were identified by WGS. Data of capture-based BKPyV gene sequencing showed that 138 (range 5–10 462) reads on average mapped to each site with an average sequencing depth of 1 053.65×, while 174 integration sites from BKPyV genotype Ib-1 (Dik, GenBank: AB211369) were distributed on 22 human chromosomes (Fig. 3C, D). For control of the sequencing specificity, two separate cases of non-BKPyV-associated bladder UC (negative TAg expression by immunohistochemistry), with clinicopathological characteristics similar to cases 1 and 2 in respect to age, sex, tumor grade, and stage were sequenced by viral capture-based sequencing, and no integration site was detected. The integration data has been previously reported in the study describing the muti-stage carcinogenesis of BKPyV [14] and used here to study viral integration pattern.
BKPyV breakpoints in the cases were significantly gathered in the non-translated region of the large T gene, which indicated possibly functional TAg expression (χ2 test, P < 0.05) (Fig. 3). As to the human genome, the analysis of nucleotide sequences at the integration sites showed that BKPyV integration could occur in both intrageneric and intergeneric regions. A total of 74 genes (5 exons and 69 introns) formed fusion genes with viral genes due to viral integration. Certain regions of genes such as repeating elements, transcription factors binding sites, DNase hypersensitivity clusters, etc. were interrupted by viral integration (Supplementary Table S2). By comparing gene expression between UC and normal tissues using GEO2R expression analysis, we found that differential gene expression of 15 genes was statistically associated with UC, including genes NOTCH4, CNTNAP2, KCNQ3, DLG2, APOH, PRLR, PDE4D, SLC9A5, ARHGAP6, AMBRA1, PTPRT, PELI2, DGKB, SCN7A and NEIL2. Elements of these 15 genes involved in the integrations can be found in Supplementary Table S2 for speculating the possible change in gene expression.
Mechanisms of BKPyV genome integration at the nucleotide level
A total of 153 integration sites (84.5%, 153 of 181) of the integration sites, the franking DNA sequence share homology between the cellular DNA and viral DNA. The length of the homologous regions ranged from 3 to 16 bp, with a median of 7 bp in the homologous flanking regions, which we termed microhomologous type or type I integration sites (Fig. 4A). Exogenous gene fragment, a segment of DNA that does not match either human or BKPyV sequence was found in 6.6% (12 of 181) integration sites, which we defined as type II integration sites (Fig. 4A). In type II integration sites, the length of exogenous gene fragments ranged from 2 to 10 bp, with a median of 3.5 bp (Fig. 4B). Six sites (3.3%, 6 of 181) were not associated with any specific pattern and were temporarily classified as type III integration sites (Supplementary Table S3). The remaining 10 sites (5.5%, 10 of 181) whose length of the homologous regions was 2 bp could not be well discriminated.

(A) Alignment of the sequence around the integration site between the human genome and the BKPyV genome. Junction boundaries are shown as vertical lines. All BKPyV sequences are from the reference strand. Blue, human gene partner; red, viral gene partner; yellow, nucleotides that align to both reference sequences (microhomologies); green, nucleotides that did not align to either reference sequence (exogenous molecular scar). (B) The analysis of microhomology and exogenous fragments adjacent to integration segments. (C) Two potential viral integration mechanisms mediated by microhomology end joining (MMEJ) or nonhomologous end joining (NHEJ).
Discussion
BKPyV is ubiquitous, with 70% of children seropositive to BKPyV by the age of 10 [9]. The epidemiological survey showed a worldwide seroprevalence in adults of 75% (46–94%) and of 30–90% in the United States and Europe, respectively [13, 15, 16]. BKPyV infection is usually asymptomatic as the virus remains latent in the urothelium [12]. However, BKPyV can be reactivated and causes a series of urinary system diseases in the setting of immunosuppression [17, 18]. In allogeneic hematopoietic cell transplant recipients, BKPyV reactivation primarily causes hemorrhagic cystitis [19]. Additionally, BKPyV has been associated with urinary tumors, usually high grade, in immunosuppressed patients [20,21,22,23]. Recent studies suggested the persistent expression of TAg encoded by BKPyV and viral integration contribute to oncogenesis [11,12,13, 20], but the exact integration pattern and carcinogenic mechanism remain unknown.
In this study, we performed and compared WGS and viral capture-based sequencing techniques on immunohistochemically TAg-positive high-grade UC in two female renal transplant recipients. In most genomic studies, sequencing depth of WGS stays at around 30–50×. In this study, the sequencing depth is 30× in case 1 and 65× in case 2. Three viral integration sites were found in case 1 and none in case 2. WGS can only determine the respectively frequent viral integration sites but fail to identify those with low reads. Many potential viral integration information may be lost. By contrast, the viral capture-based sequencing technology is set to design probes for viral genome and screen the amplified integrated fragments after library-building. It has a major advantage on increasing the sequencing depth and identifying viral integration sites, particularly those in subpopulations of tumor cells [24, 25]. In this study, the depth of capture targeted sequencing depth was 5 711 and 1 053, respectively, in the two cases, enabling us to identify 178 extra viral integration sites, in addition to the three identified by WGS, in these two tumors.
The graft function of the case 1 patient maintained relatively well, except for mild gross hematuria after carcinogenesis, but for case 2, transplant kidney hydronephrosis and lymph node metastasis occurred due to high-volume bladder tumors. By sequencing data, we mapped 181 BKPyV integration sites between the two patients, with 7 sites in case 1 tumor mainly in chromosome 20 and chromosome 3, and 174 sites in case 2 tumor distributed widely across all chromosomes. There appears to be a correlation between the number of integration sties in the tumors and clinical progression in these two patients. Therefore, we postulate that the frequency of BKPyV integration may impact the tumor aggressiveness.
Distinct from published studies, 181 different insertional breakpoints mapped broadly across both human and BKPyV genomes were observed, which could not be explained by the previously proposed linear integration model [11,12,13]. Therefore, we proposed a new integration-oncogenesis pattern (Fig. 5). The BKPyV-related tumor contained a heterogenous population of tumor cells with various viral integration patterns and integration sites, representing different clones of the tumor. Clonal proliferation of the tumor cells generated a series of subgroups that diverged in number and integration patterns, leading to the divergence of sequencing depths and supporting reads at each integration site (Fig. 5).

BKPyV-associated carcinoma is initially transformed from BKPyV-integrated urothelial cells. After the production of tumor cell, its monoclonal proliferation accompanied by viral integration, results in the presence of several tumor cell subgroups with different integration patterns.
Microhomology is a series of nucleotide sequences (<70 bp) that are identical at the junctions of the two genomic segments [26, 27]. A significant enrichment of microhomologies was shown in the flanking regions of type I integration sites, which might be generated by DNA double-strand break repair pathway in the phase of the cell cycle [28, 29]. We observed microhomologies in the flanking regions at most integration sites (84.5%) in our study. Immunosuppressants can arrest cells in the G0/G1 phase, preventing them entering S phase [29, 30]. The replication fork stalled at the DNA lesion in host cells harboring double-strand break repair, and replication was temporarily paused. Repair protein and regulatory protein in the G0/G1 phase contributed to the initiation of the repair mechanisms, such as fork stalling and template switching and microhomology-mediated break-induced replication [28, 31]. Type I integration might be achieved by that BKPyV coopted the microhomology-mediated end joining (MMEJ) DNA repair pathway to fuse its sequence into the host genome by aligning the microhomologies flanking the breakpoint.
Another DNA repair pathway, the nonhomologous end joining (NHEJ) mechanism, is error-prone and could result in the addition or deletion of DNA sequences at the repair junction [32, 33]. The exogenous sequence at type II integration site might be derived from NHEJ-mediated insertion. It is possible at the type II and type III integration sites, BKPyV may use another DNA repair pathway as NHEJ, leaving a fragment of exogenous sequence between the fusion gene (Fig. 4C and Supplementary Table S3).
We previously reported a patient with BKPyV-associated sarcomatoid UC in the graft kidney who recovered 8 months after surgical resection of allograft and cessation of immunosuppressants [1]. Over 80 cases of BKPyV-associated urinary carcinoma in transplant recipients have been reported to date. Michel et al. suggested that immunosuppression was a major risk factor for the development of malignancy in transplant recipients [34]. BKPyV-associated carcinomas appear to be characterized by a higher frequency of upper urinary tract involvement, high grade, and variant histology compared to these in non-immunosuppressed patients [35]. Therefore, BKPyV-associated malignancy is associated with immunosuppression. The immunosuppressive therapy triggers lymphocyte clearance, cellular immunity and humoral immune failure, permits the survival of BKPyV-infected host cells, and leads to the structural alterations in host chromosomes. After that, BKPyV has the chance to integrate into the chromosomes of host cells [36,37,38].
Based on our finding of viral integration affecting gene expression and Kenan’s previous model of BKPyV pathogenic process [11, 12, 20], we proposed a mechanism driven by immunity and emphasizing the role of viral integration during oncogenesis (Fig. 6). The diversity of viral integration patterns resulted in a heterogeneity of the host cells; some host cells with growth advantages gained by viral integration at certain genes would survive during immune suppression, providing opportunities for neoplastic transformation. Recovery of immunity will break the subtle balance of the tumor microenvironment and affect the survival of tumor cells.

Based on the results in this study, we proposed a potential pathogenesis pattern in immunocompromised patients. The patient was initially infected by BKPyV during childhood and then formed adaptive immunity while the virus entered the latency period with no significant viral replication. BKPyV reactivation and productive infection occur in settings of immunosuppression. The host-infected cells release mature daughter virions, resulting in cell death or cell lysis. The virus invades the urothelium, causing local inflammation and fibrotic scar formation, resulting in further BKPyV viruria or viremia, hemorrhagic cystitis, BKPyV-associated nephropathy (BKVAN) or other complications, which finally leads to irreversible renal dysfunction or graft loss. At the same time, the BKPyV genome is fragmented by continuous replication of the virus, and some sites in the host genome are broken by external factors, such as viral infection and immunosuppression. As viral infection progresses, BKPyV may coopt MMEJ repair pathways or NHEJ repair pathways to fuse itself into the broken host genome. At the preliminary stage, virus fragments with different lengths integrate into various parts of the human chromosomes. Multiple integration patterns lead to the heterogeneity of host cells, resulting in the production of a series of subgroups with difference.
In summary, through DNA integration in the host cell chromosome, BKPyV can disrupt host gene expression and alter gene structure by forming fusion genes and this process may contribute to the viral tumorigenesis. The frequency of BKPyV integration may impact tumor aggressiveness and disease course. Therefore, timely monitoring and reducing BKPyV load in immunocompromised populations is an effective way to prevent BKPyV-associated carcinoma.
Materials and methods
Histopathology
Histological examination and standard diagnostic immunohistochemistry for the characterization of the tumor were performed following routine pathology procedures. Immunohistochemical detection of polyomavirus-associated antigens was performed on formalin-fixed paraffin-embedded tissue, with mouse monoclonal antibodies directed against the SV40 TAg (ab16879, Abcam, Cambridge, MA).
BKPyV load measurement
BKPyV genome loads in blood and urine specimens were tested by quantitative real-time PCR using a BKPyV PCR Kit (SinoMD Gene Co. Ltd, Beijing, China) with TaqMan fluorescence probe techniques; the assay was performed on the ABI 7500 System (Applied Biosystems, CA, USA).
DNA extraction and whole-genomic library preparation
Formalin-fixed paraffin-embedded samples with over 80% tumor content were collected for isolation of genomic DNAs with QIAamp MinElute Virus Spin Kit (57704, Qiagen, Germany). The whole-genomic library containing the target genes was established using Library Construction Kit (MyGenostics Inc, Chongqing, China). Briefly, 3–5 μg of genomic DNA was sheared into 150 bp fragments by ultrasonication, which were then end blunted, “A” tailed, adapter ligated and amplified for ten cycles by PCR. One microliter of prepared library samples was quantified using a Nanodrop 2000. Three microliters of prepared library samples were visualized on a 1% agarose gel, and the size of the electrophoresis fragment was 300–500 bp.
Capture targeted gene regions and sequencing
BKPyV probes were designed according to the full-length genome of pre-evaluated BKPyV types by MyGenostics (MyGenostics Inc, Chongqing, China). The reaction system consisted of the DNA library constructed, 1 000 ng; blocking agent BL 12 μL; virus DNA probe, 5 μL; and hybridization buffer, 19 μL. Enrichment products of the library were adsorbed onto the beads via biotin and streptavidin magnetic beads, and uncaptured DNA fragments were removed by washing. Then, the eluted targeted gene fragments were enriched by 18 cycles of PCR, purified and sequenced using the Illumina NovaSeq 6000 sequencer (Illumina Inc., San Diego, CA).
Specifically, the following genotypes of the BKPyV were used for sequence alignment and viral genotyping.
AB211369.1 BK polyomavirus DNA, complete genome, isolate: Dik;
AB217919.1 BK polyomavirus DNA, complete genome, isolate: TW-3a;
AB217920.1 BK polyomavirus DNA, complete genome, isolate: TW-8a;
AB217921.1 BK polyomavirus DNA, complete genome, isolate: THK-9a;
DQ305492.1 BK polyomavirus strain UT, complete genome;
DQ989796.1 BK polyomavirus isolate PittVR2, complete coding regions;
DQ989798.1 BK polyomavirus isolate PittVM3, complete coding regions;
DQ989802.1 BK polyomavirus isolate PittNP4, complete coding regions;
DQ989804.1 BK polyomavirus isolate PittNP5, complete coding regions;
DQ989806.1 BK polyomavirus isolate PittVR8, complete coding regions;
DQ989812.1 BK polyomavirus isolate PittVR4, complete coding regions;
DQ989813.1 BK polyomavirus isolate PittNP1, complete coding regions;
KY487998.1 Human polyomavirus 1 strain BK-2, complete genome;
M23122.1 BK polyomavirus strain AS, complete genome.
Bioinformatics analysis
Low-quality, short sequences, and duplicate reads were filtered using the Trim Galore program in quality control, and then 3′/5′ adapters were trimmed using the Cutadapt program. Reads with a sequencing quality greater than 20 and read lengths greater than 80 bp were retained. Illumina clean reads were aligned to each BKPyV reference genome with Burrows-wheeler Aligner (BWA), and the data were screened with an average sequencing depth of more than 10 and over 4× coverage of more than 50%.
For the potential BKPyV integrated sites in human genome, we aligned the Illumina clean reads with the reference human genome hg19 using the BWA program. In addition, recalibration and realignment were also performed using the GATK software. The pair-end reads are uniquely mapped with one end to the one human chromosome and the BKPyV reference genome, and the read pair was identified as a discordant read pair. If a specific position has more than three discordant read pairs, it will be considered as a potential BKPyV integration site. The breakpoints of the integrated sites were identified using the BreakDancer program. The annotation of integrated sites was according to the human hg19 and BKPyV annotation information (NCBI genome database). The coverage was calculated based on reported reads. Coverage = reads × 100 bp/genome size of the BKPyV genome.
Detection of BKPyV DNA signals
If a paired-end read was not able to map to the human genome, but was able to map to BKPyV genome, it would be reported as a signal of BKPyV DNA for subsequent analysis. The coverage, depth, and mapping ratio on the BKPyV genome were calculated based on these reported reads. A sample was considered BKPyV-positive if coverage of the sample on BKPyV genome was higher than 50% of the BKPyV genome.
References
Fu F, Deng W, Yu S, Liu Y, Yu L, Liu R, et al. Occurrence and regression of BK polyomavirus associated carcinoma: a clinical and next-generation sequencing study. Clin Sci. 2018;132:1753–63.
Rosales BM, De La Mata N, Vajdic CM, Kelly PJ, Wyburn K, Webster AC. Cancer mortality in kidney transplant recipients: an Australian and New Zealand population-based cohort study, 1980–2013. Int J Cancer. 2020;146:2703–11.
O’Neill JP, Sexton DJ, O’Leary E, O’Kelly P, Murray S, Deady S, et al. Post-transplant malignancy in solid organ transplant recipients in Ireland The Irish Transplant Cancer Group. Clin Transplant. 2019;33:e13669.
Awan AA, Niu J, Pan JS, Erickson KF, Mandayam S, Winkelmayer WC, et al. Trends in the CAuses of Death among Kidney Transplant Recipients in the United States (1996–2014). AM J Nephrol. 2018;48:472–81.
Papadimitriou JC, Randhawa P, Rinaldo CH, Drachenberg CB, Alexiev B, Hirsch HH. BK polyomavirus infection and renourinary tumorigenesis. AM J Transplant. 2016;16:398–406.
Starrett GJ, Buck CB. The case for BK polyomavirus as a cause of bladder cancer. Curr Opin Virol. 2019;39:8–15.
Saribas AS, Coric P, Hamazaspyan A, Davis W, Axman R, White MK, et al. Emerging from the unknown: structural and functional features of agnoprotein of polyomaviruses. J Cell Physiol. 2016;231:2115–27.
Houben R, Angermeyer S, Haferkamp S, Aue A, Goebeler M, Schrama D, et al. Characterization of functional domains in the Merkel cell polyoma virus Large T antigen. Int J Cancer. 2015;136:E290–300.
Hesbacher S, Pfitzer L, Wiedorfer K, Angermeyer S, Borst A, Haferkamp S, et al. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget. 2016;7:32956–68.
Elfadawy N, Yamada M, Sarabu N. Management of BK polyomavirus infection in kidney and kidney-pancreas transplant recipients: a review article. Infect Dis Clin North AM. 2018;32:599–613.
Kenan DJ, Mieczkowski PA, Latulippe E, Cote I, Singh HK, Nickeleit V. BK polyomavirus genomic integration and large T antigen expression: evolving paradigms in human oncogenesis. AM J Transplant. 2017;17:1674–80.
Kenan DJ, Mieczkowski PA, Burger-Calderon R, Singh HK, Nickeleit V. The oncogenic potential of BK-polyomavirus is linked to viral integration into the human genome. J Pathol. 2015;237:379–89.
Muller DC, Ramo M, Naegele K, Ribi S, Wetterauer C, Perrina V, et al. Donor-derived, metastatic urothelial cancer after kidney transplantation associated with a potentially oncogenic BK polyomavirus. J Pathol. 2018;244:265–70.
Wang YC, Liu YN, Deng WF, Fu FX, Yan SS, Yang HW, et al. Viral integration in BK polyomavirus-associated urothelial carcinoma in renal transplant recipients: multistage carcinogenesis revealed by next-generation virome capture sequencing. Oncogene. 2020;39:5734–42.
Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, et al. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis. 2009;199:837–46.
Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. Plos Pathog. 2009;5:e1000363.
Pinto M, Dobson S. BK and JC virus: a review. J Infect. 2014;68:S2–8.
Trofe-Clark J, Sawinski D. BK and other polyomaviruses in kidney transplantation. Semin Nephrol. 2016;36:372–85.
Lee YJ, Zheng J, Kolitsopoulos Y, Chung D, Amigues I, Son T, et al. Relationship of BK polyoma virus (BKV) in the urine with hemorrhagic cystitis and renal function in recipients of T Cell-depleted peripheral blood and cord blood stem cell transplantations. Biol Blood Marrow Transplant. 2014;20:1204–10.
Nickeleit V, Singh HK, Kenan DJ, Mieczkowski PA. The two-faced nature of BK polyomavirus: lytic infection or non-lytic large-T-positive carcinoma. J Pathol. 2018;246:7–11.
Roberts IS, Besarani D, Mason P, Turner G, Friend PJ, Newton R. Polyoma virus infection and urothelial carcinoma of the bladder following renal transplantation. BRJ Cancer. 2008;99:1383–6.
Gupta G, Kuppachi S, Kalil RS, Buck CB, Lynch CF, Engels EA. Treatment for presumed BK polyomavirus nephropathy and risk of urinary tract cancers among kidney transplant recipients in the United States. AM J Transplant. 2018;18:245–52.
Starrett GJ, Buck CB. The case for BK polyomavirus as a cause of bladder cancer. Curr Opin Virol. 2019;39:8–15.
Li W, Zeng X, Lee NP, Liu X, Chen S, Guo B, et al. HIVID: an efficient method to detect HBV integration using low coverage sequencing. Genomics. 2013;102:338–44.
Groves IJ, Coleman N. Human papillomavirus genome integration in squamous carcinogenesis: what have next-generation sequencing studies taught us? J Pathol. 2018;245:9–18.
Morrical SW. DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb Perspect Biol. 2015;7:a16444.
Ottaviani D, LeCain M, Sheer D. The role of microhomology in genomic structural variation. Trends Genet. 2014;30:85–94.
Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet. 2015;47:158–63.
Yi K, Ju YS. Patterns and mechanisms of structural variations in human cancer. Exp Mol Med. 2018;50:98.
Zhou X, Workeneh B, Hu Z, Li R. Effect of immunosuppression on the human mesangial cell cycle. Mol Med Rep. 2015;11:910–6.
Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41:849–53.
Chang H, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18:495–506.
Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211.
Michel OR, Wolff DJ, Schandl CA, Drabkin HA. Urothelial carcinoma of donor origin in a kidney transplant patient. J Immunother Cancer. 2016;4:63.
Sirohi D, Vaske C, Sanborn Z, Smith SC, Don MD, Lindsey KG, et al. Polyoma virus-associated carcinomas of the urologic tract: a clinicopathologic and molecular study. Mod Pathol. 2018;31:1429–41.
Cangemi M, Montico B, Fae DA, Steffan A, Dolcetti R. Dissecting the multiplicity of immune effects of immunosuppressive drugs to better predict the risk of de novo malignancies in solid organ transplant patients. Front Oncol. 2019;9:160.
Billups K, Neal J, Salyer J. Immunosuppressant-driven de novo malignant neoplasms after solid-organ transplant. Prog Transplant. 2015;25:182–8.
McCabe MT, Low JA, Imperiale MJ, Day ML. Human polyomavirus BKV transcriptionally activates DNA methyltransferase 1 through the pRb/E2F pathway. Oncogene. 2006;25:2727–35.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (81500573), the Science Foundation of Guangdong Province (2020A1515010674), the Science and Technology Planning Project of Guangzhou (201803010109), the President Funding of Nanfang Hospital (2018B009, 2018C003) and College Students’ Innovative Entrepreneurial Training Plan Program (201812121148, X202012121239, 202012121046).
Author information
Authors and Affiliations
Contributions
CLW and YM designed and directed the study. YJ, YZ, WFD, and YCW wrote the manuscript. RJL, YNL, NE, and JG performed sample preparation, sequencing and statistical analysis and validation. MHL, RML, and BWG performed experimental work and analyzed the data. YCH, JFM, JPZ, and NL took charge for data visualization. HQN, JX, MLB, and RBC revised the manuscript. All authors read and approved the final version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Jin, Y., Zhou, Y., Deng, W. et al. Genome-wide profiling of BK polyomavirus integration in bladder cancer of kidney transplant recipients reveals mechanisms of the integration at the nucleotide level. Oncogene 40, 46–54 (2021). https://doi.org/10.1038/s41388-020-01502-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-01502-w
- Springer Nature Limited