
Abstract
Cumulative evidence suggests that microRNAs (miRNAs) promote gene expression in cancers. However, the pathophysiologic relevance of miRNA-mediated RNA activation in hepatocellular carcinoma (HCC) remains to be established. Our previous miRNA expression profiling in seven-paired HCC specimens revealed miR-93-5p as an HCC-related miRNA. In this study, miR-93-5p expression was assessed in HCC tissues and cell lines by quantitative real-time PCR and fluorescence in situ hybridization. The correlation of miR-93-5p expression with survival and clinicopathological features of HCC was determined by statistical analysis. The function and potential mechanism of miR-93-5p in HCC were further investigated by a series of gain- or loss-of-function experiments in vitro and in vivo. We identified that miR-93-5p, overexpressed in HCC specimens and cell lines, leads to poor outcomes in HCC cases and promotes proliferation, migration, and invasion in HCC cell lines. Mechanistically, rather than decreasing target mRNA levels as expected, miR-93-5p binds to the 3′-untranslated region (UTR) of mitogen-activated protein kinase kinase kinase 2 (MAP3K2) to directly upregulate its expression and downstream p38 and c-Jun N-terminal kinase (JNK) pathway, thereby leading to cell cycle progression in HCC. Notably, we also demonstrated that c-Jun, a downstream effector of the JNK pathway, enhances miR-93-5p transcription by targeting its promoter region. Besides, downregulation of miR-93-5p significantly retarded tumor growth, while overexpression of miR-93-5p accelerated tumor growth in the HCC xenograft mouse model. Altogether, we revealed a miR-93-5p/MAP3K2/c-Jun positive feedback loop to promote HCC progression in vivo and in vitro, representing an RNA-activating role of miR-93-5p in HCC development.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC), accounting for 75–85% of primary liver cancers [1], arises from a continuous accumulation of gain-of-function mutations in oncogenes and loss-of-function mutations in tumor suppressors. It is associated with a meager 5-year survival rate due to lack of early-stage diagnosis, distant metastasis, and high risk of postoperative recurrence. Notably, microRNAs (miRNAs) and miRNA-mediated regulatory networks can be promising candidates in the prevention and treatment of HCC. Despite extensive investigation on these “dark matters” [2], there is still a limited insight into the underlying pathogenic mechanisms linking miRNA to HCC.
miRNAs are a family of small non-coding RNAs of 20–24 nucleotides in length that have been traditionally thought to contribute to cancer either by inducing the sequence-specific degradation of complementary mRNA or by repressing translation. Several miRNAs are aberrantly expressed in HCC, and their deregulation is linked to malignant phenotypes of HCC, such as cell proliferation, invasiveness, and metastasis [3]. Our previous study and others demonstrated that miR-93-5p, derived from the miR-106b-25 cluster, is amplified in breast cancer [4], lung cancer [5], and HCC [6]. It could promote cell proliferation, migration, and invasion in HCC through posttranscriptional silencing of target genes, including PTEN [7], p21 [8], and PDCD4 [9]. Recently, the evidence is accumulating that miRNA can increase gene expression by binding to the sequences in the promoter [10], enhancer [11], 5′-untranslated region (UTR) [12], or 3′-UTR [13] of target genes. However, neither the pathophysiologic relevance nor the mechanism of miRNA activation has been previously investigated in HCC.
The mitogen-activated protein kinase (MAPK) pathway is one of the most frequently oncogenic pathways in human cancers [14, 15] and, therefore, is regarded as attractive therapeutic targets for treating cancers. Several miRNAs, like miR-520a-3p in lung cancer [16], miR-34c-3p in breast cancer [17], and miR-302a in HCC [18], have been proposed to negatively regulate mitogen-activated protein kinase kinase kinase 2 (MAP3K2), a component of the MAPK pathway. Nevertheless, whether MAP3K2, along with downstream signaling molecules, can be positively regulated by miRNA remains elusive.
In this study, we use an integrated method to identify MAP3K2 as a direct target of miR-93-5p in HCC. We validate the oncogenic role of miR-93-5p in a series of in vivo and in vitro studies. In addition, we also demonstrate a novel miR-93-5p-driven positive feedback loop, which, if perturbed, promotes HCC growth. Thus, our work defines a bona fide RNA-activating role of miR-93-5p in HCC oncogenesis.
Results
miR-93-5p is frequently upregulated in human HCC tissues and cell lines
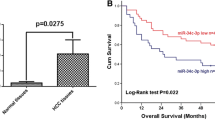
Recently, our miRNA expression profiling revealed frequent overexpression of miR-93-5p in seven-paired HCC specimens (fold change = 2.59, P < 0.001) [6], which is in accordance with a previous report [19]. To gain insight into the biological significance of miR-93-5p, we quantified the expression level of miR-93-5p and its precursor by quantitative PCR (qPCR), and analyzed its correlation with clinical parameters in 73 pairs of human HCC and matched non-tumor tissues. Mean miR-93-5p level in HCC tissues was 1.9-fold higher than that in matched non-tumor tissues (73 cases; Fig. 1a), which is similar to the data from the cancer genome atlas (TCGA; Fig. 1b). Also, relative to non-tumor tissues, miR-93-5p precursor was upregulated in paired HCC tissues (29 cases; Supplementary Fig. 1), consistent with a prior study [20]. The miR-93-5p level was positively correlated with tumor volume (χ2 = 3.963, P = 0.047), TNM stage (χ2 = 3.984, P = 0.046) and alpha-fetoprotein (AFP) level (χ2 = 7.602, P = 0.006; Supplementary Table 1). Fluorescence in situ hybridization (FISH) was subsequently carried out to determine miR-93-5p expression patterns in HCC specimens. The results demonstrated that miR-93-5p was mainly clustered in the nuclear fraction, and expressed at higher levels in HCC tissues compared with matched non-tumor tissues (Fig. 1c).

a miR-93-5p level in paired HCC and non-tumor specimens (n = 73). *P < 0.05 by Student’s t test. b Relative miR-93-5p level in HCC tissues (n = 372) and partially paired non-tumor tissues (n = 50) obtained from TCGA. ****P < 0.0001 by Student’s t test. c Fluorescence in situ hybridization images showing miR-93-5p expression in human HCC and non-tumor tissues. Scale bars represent 25 μm. d Kaplan–Meier curves for overall survival curve (n = 344) or e disease-free survival curve (n = 365) stratified according to high or low miR-93-5p expression in HCC patients. The median expression level of miR-93-5p derived from TCGA was used as the cutoff value. P values calculated by the log-rank test. f miR-93-5p expression in HCC cell lines (including SK-Hep-1, HepG2, BEL-7402, Hep3B, MHCC-97L, MHCC-97H, and HCC-LM3) compared with normal hepatocytes L02 as revealed by qPCR. Data represent mean ± s.d. in a, b, and f. DAPI 4′,6-diamidino-2-phenylindole.
As higher tumor volume, TNM stage, and AFP level contribute to HCC-related mortality, we speculated that upregulated miR-93-5p may be linked to HCC survival. Kaplan–Meier survival analysis in TCGA cohort showed that HCC cases with high miR-93-5p were correlated with shorter overall survival (P = 0.045; Fig. 1d) [21] and disease-free survival (P = 0.011; Fig. 1e) than those with low miR-93-5p in the first 3 years. Thus, highly expressed miR-93-5p is associated with poor HCC prognosis.
Furthermore, consistent with human data, the expression level of miR-93-5p in all tested HCC cell lines, including SK-Hep-1, HepG2, BEL-7402, Hep3B, MHCC-97L, MHCC-97H, and HCC-LM3, was higher than that in normal hepatocyte L02 (Fig. 1f). Collectively, these results suggest that upregulated miR-93-5p expression is a frequent event and may play an oncogenic role in HCC.
miR-93-5p modulates cell proliferation, migration, and invasion in vitro
To investigate the oncogenic role of miR-93-5p in HCC, we performed both inhibition studies using miR-93-5p inhibitor and overexpression studies using miR-93-5p mimics (Supplementary Fig. 2a). We confirmed that inhibition of miR-93-5p decreased cell proliferation and blocked G1 to S phase transition in MHCC-97H and HCC-LM3 cells, as shown by CCK8 and fluorescence-activated cell sorting (FACS) assays, while overexpression of miR-93-5p had the opposite effect in HepG2 and SK-Hep-1 cells (Fig. 2a, b).

a CCK8 assay for viability, b FACS for cell cycle progression, c wound-healing assay, and d transwell assay for migration or invasion in MHCC-97H and HCC-LM3 cells transfected with miR-93-5p inhibitor or NC, along with HepG2 and SK-Hep-1 cells transfected with mimics or NC. Data represent mean ± s.d. (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001 by Student’s t test. NC negative control.
To further characterize miR-93-5p effect on cell proliferation through CCK8, colony formation and BrdU assays, HCC cells stably transfected with recombinant lentiviral particles containing miR-93-5p sponge or miR-93-5p were carried out (Supplementary Fig. 2b). Knockdown of miR-93-5p obviously decreased cell growth and colony formation capability, along with cells in S phase as a consequence of G1 phase block in MHCC-97H cells. Conversely, overexpression of miR-93-5p markedly increased these parameters in SK-Hep-1 cells (Supplementary Fig. 3). Thus, miR-93-5p increased cell proliferation through facilitating G1 to S phase transition. Both the results from transient and persistent alteration were consistent with the clinicopathologic role of miR-93-5p in HCC tissues.
Also, we investigated the role of miR-93-5p on cell migration and invasion, the critical determinants of malignant progression and metastasis. We found that miR-93-5p inhibitor decreased migration and invasion using wound-healing and Transwell assays in MHCC-97H and HCC-LM3 cells, whilst overexpression of miR-93-5p increased migration and invasion in HepG2 and SK-Hep-1 cells (Fig. 2c, d). These results support a functional role for miR-93-5p in mediating cell proliferation, migration, and invasion in HCC cells, and suggest a mechanism by which overexpression of miR-93-5p may contribute to HCC progression.
miR-93-5p directly activates MAP3K2 expression by targeting its 3′-UTR
To explore the molecular mechanism by which miR-93-5p promotes HCC growth, we identified candidate targets of miR-93-5p using online prediction tools, including miRDB, TargetScan, and miRTarBase (Supplementary Fig. 4). Validation by qPCR identified MAP3K2 as the primary target. Inhibition of miR-93-5p reduced the mRNA level of MAP3K2 by 23% in MHCC-97H cells and 49% in HCC-LM3 cells, while overexpression of miR-93-5p upregulated MAP3K2 by nearly threefold in HepG2 and SK-Hep-1 cells (Fig. 3a).

a MAP3K2 mRNA expression in HCC cells transfected with miR-93-5p inhibitor or mimics, and corresponding NC. Data represent mean ± s.d. (n = 3), *P < 0.05, **P < 0.01 by Student’s t test. b Scatter plot showing a positive correlation of miR-93-5p and MAP3K2 in 34 pairs of human HCC specimens by qPCR. P = 0.0006 by Spearman rank correlation. c Ectopic miR-93-5p regulated protein expression of MAP3K2, and its downstream targets (including MKK4, p-MKK4, p38, p-p38, JNK, p-JNK, ATF2, p-ATF2, c-Jun, p-c-Jun, and p21) in four HCC cell lines by Western blot. d miR-93-5p and its five putative complementary binding sites on MAP3K2 promoter, 5′-UTR, CDS 1, CDS 2, and 3′-UTR, as well as mutant 3′-UTR with a mutated binding sequence in miR-93-5p seed site. e Dual-luciferase reporter assay showing the effect of overexpressed miR-93-5p on MAP3K2 promoter/5′-UTR or CDS/3′-UTR reporter activity in SK-Hep-1 cells. f Dual-luciferase reporter assay showing the effect of overexpressed miR-93-5p on MAP3K2 CDS or 3′-UTR reporter activity in SK-Hep-1 and HEK-293T cells. g Effect of overexpressed miR-93-5p on mutant and wild-type 3′-UTR reporter activity in SK-Hep-1 cells, as well as effect of repressed miR-93-5p on mutant and wild-type 3′-UTR reporter activity in MHCC-97H cells. Data represent mean ± s.d. (n = 3), *P < 0.05, **P < 0.01, ****P < 0.0001 by one-way ANOVA. NC negative control, mut mutant, wt wild-type.
Next, we measured the correlation between MAP3K2 mRNA level and miR-93-5p by qPCR in 34 pairs of HCC and matched non-tumor specimens. A positive correlation of MAP3K2 mRNA level with miR-93-5p expression was seen in almost all 34 cases of human HCC tissues (P = 0.0006; Fig. 3b). Moreover, Western blot consistently confirmed the same result at the protein level in four HCC cell lines (Fig. 3c). These results indicate that miR-93-5p is involved in the activation of MAP3K2, but whether miR-93-5p directly or indirectly targets MAP3K2 remains unknown.
Since direct upregulation of genes by miRNAs is rare, we first sought to identify a potential intermediate that mediates the effect of miR-93-5p on MAP3K2. Presumably, the candidate intermediate should be directly targeted by miR-93-5p and function as upstream of the MAPK pathway. An extensive search through several protein databases (including IntAct, STRING, and MINT) resulted in a set of putative intermediates, including Ras, Rho, PTPNs, PTPRs, TRIM32, HCCS, GNA12, TOLLIP, CASPs, and XIAP (Supplementary Fig. 5a). However, none of these prospective candidates was bona fide target of miR-93-5p in the validation assay (Supplementary Fig. 5b, c).
Previous studies indicated that miRNAs could modulate gene expression through their direct interaction with the complementary sequences in the promoter [10], 5′-UTR [12], coding sequence (CDS) [22], and 3′-UTR [13] of targets. Notably, the suggestion of direct interaction between miR-93-5p and MAP3K2 was also supported by argonaute-crosslinking and immunoprecipitation (AGO-CLIP) assay (Supplementary Table 2) [23, 24]. A search in the UCSC genome browser and miRBase database revealed five putative complementary binding sites on MAP3K2 promoter, 5′-UTR, CDS, and 3′-UTR of miR-93-5p, wherein the 3′-UTR of MAP3K2 best matches the miR-93-5p sequence (Fig. 3d). We next determined the exact binding sequence using dual-luciferase reporter assay. First, we constructed vectors containing either MAP3K2 promoter/5′-UTR or CDS/3′-UTR, each of which was co-transfected with miR-93-5p mimics in SK-Hep-1 cells. miR-93-5p significantly increased MAP3K2 CDS/3′-UTR-driven luciferase activity, but not MAP3K2 promoter/5′-UTR or control, suggesting that the binding site is located within CDS or 3′-UTR of MAP3K2 (Fig. 3e). Next, we looked at the luciferase activity of CDS or 3′-UTR in both SK-Hep-1 and HEK-293T cells. miR-93-5p only stimulated MAP3K2 3′-UTR − induced luciferase activity in both cell types, further narrowing down the binding site within MAP3K2 3′-UTR (Fig. 3f). Finally, to pinpoint the exact binding sequence in the MAP3K2 3′-UTR, we constructed a mutant 3′-UTR that abrogates its complementarity with miR-93-5p (Fig. 3d). Importantly, miR-93-5p mimics failed to induce luciferase activity of mutant 3′-UTR in SK-Hep-1 cells. Consistently, miR-93-5p inhibitor failed to reduced luciferase activity of mutant 3′-UTR in MHCC-97H cells, thus defining the 3′-UTR of MAP3K2 as the target of miR-93-5p. Moreover, altered miR-93-5p expression modulated the luciferase activity of wild-type 3′-UTR in both SK-Hep-1 and MHCC-97H cells (Fig. 3g). These results indicate that miR-93-5p directly regulates MAP3K2 through its partial binding to the 3′-UTR of MAP3K2. Also, the binding sequence, 5′-GCACT TT-3′ (265–271), in MAP3K2 3′-UTR is highly conserved between human, mouse, chimpanzee, and sheep (Supplementary Fig. 6).
MAP3K2 phosphorylates and activates MAPK kinases (MKKs), while the latter in turn phosphorylate and activate MAPKs. Activated MAPKs, including p38 and c-Jun N-terminal kinase (JNK), then phosphorylate various substrates, including c-Jun and activating transcription factor 2 (ATF2) [25]. To identify possible downstream effectors of MAP3K2 responsive to miR-93-5p, the total protein level, and the phosphorylation state of downstream targets in the MAPK signaling pathway were analyzed by western blot. The results showed that perturbation of miR-93-5p dramatically affects the phosphorylation level of MKK4, p38, JNK, ATF2, and c-Jun without changing the total protein level, suggesting that miR-93-5p modulates the p38 and JNK pathways through kinase function of MAP3K2. Moreover, miR-93-5p negatively modulated the protein level of p21, which is reported as a target of c-Jun and plays a role in G1 to S phase arrest (Fig. 3c) [26]. Taken together, these results reveal that miR-93-5p regulates the MAP3K2/p38-JNK/p21 signaling pathway by binding to the 3′-UTR of MAP3K2, thereby affecting the proliferation, migration, and invasion of HCC.
miR-93-5p acts through MAP3K2 to promote HCC cell proliferation
To clarify the tumorigenic role of MAP3K2 in HCC, we performed either knockdown of MAP3K2 in MHCC-97H and HCC-LM3 cells or overexpression of MAP3K2 in HepG2 and SK-Hep-1 cells. Knockdown of MAP3K2 reduced cell proliferation and blocked G1 to S phase transition in MHCC-97H and HCC-LM3 cells, while overexpression of MAP3K2 promoted G1 to S phase transition in HepG2 and SK-Hep-1 cells (Fig. 4a, b). We also found that knockdown of MAP3K2 decreased migration and invasion via wound-healing and transwell assays in MHCC-97H and HCC-LM3 cells (Supplementary Fig. 7).

a CCK8 assay for viability in MHCC-97H and HCC-LM3 cells transfected with siRNA against MAP3K2, and its NC. b FACS for cell cycle progression in MHCC-97H and HCC-LM3 cells transfected with siRNA against MAP3K2 and its NC, in HepG2 and SK-Hep-1 cells transfected with plasmid carrying MAP3K2 and its NC. c Ectopic MAP3K2 modulated protein level of MAP3K2, MKK4, p-MKK4, JNK, p-JNK, p38, p-p38, c-Jun, p-c-Jun, and p21 in HCC cells. d MAP3K2 expression in 15 pairs of human HCC and non-tumor tissues. e Effects of ectopic miR-93-5p acting through MAP3K2 on cell cycle and f on protein level of MAP3K2, p-JNK, p-c-Jun, and p21 in HCC cells. Data represent mean ± s.d. (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by Student’s t test. NC negative control, siMAP3K2 small interfering RNA against MAP3K2, T and P tumor and paratumor tissue.
In addition, we inspected components in the p38 and JNK pathways regulated by MAP3K2 by western Blot. MAP3K2 positively modulated the phosphorylation state rather than the total protein level of MKK4, JNK, p38, and c-Jun, while p21 protein level was negatively controlled by MAP3K2 (Fig. 4c). We next assessed the MAP3K2 protein level in 15 pairs of human HCC tissues. Compared with non-tumor tissues, MAP3K2 was significantly overexpressed in HCC tissues (Fig. 4d). These data were consistent with the function of miR-93-5p, based on which we infer that miR-93-5p-activated MAP3K2 facilitates cell proliferation, migration, and invasion via regulating the p38-JNK/p21 pathway in HCC.
Next, we looked at whether miR-93-5p induced HCC cell proliferation is mediated by MAP3K2. We treated MHCC-97H cells with miR-93-5p inhibitor or lentiviral particles carrying miR-93-5p sponge, and then with MAP3K2 expression plasmid. Restoration of MAP3K2 activated the JNK pathway, and abrogated G1 to S phase arrest and suppression of proliferation induced by inhibition of miR-93-5p in MHCC-97H cells. Moreover, we treated SK-Hep-1 cells with miR-93-5p mimics or lentiviral particles carrying miR-93-5p, and then with small interfering RNA against MAP3K2. Consistently, downregulation of MAP3K2 inhibited the JNK pathway, and impeded G1 to S phase progression and proliferation capability stimulated by miR-93-5p overexpression in SK-Hep-1 cells (Fig. 4e, f and Supplementary Fig. 8). Therefore, miR-93-5p functions through MAP3K2 to modulate the JNK pathway and HCC cell proliferation, reinforcing that MAP3K2 is a direct target of miR-93-5p.
c-Jun enhances miR-93-5p transcription by targeting its promoter
c-Jun is the major downstream substrate of the JNK pathway that has been proposed to play a critical role in tumorigenesis [25]. It is also reported that TGF-β1 activates c-Jun to promote the transcription of miR-106b in breast cancer [27]. As a member of the miR-106b-25 cluster, miR-93-5p may also be regulated by c-Jun. We used the UCSC database to identify the promoter region of miR-93-5p, and the JASPAR database to predict possible c-Jun binding motif on the miR-93-5p promoter. Three putative binding regions were revealed, site 1: 5′-GGAAT GATCC CAG-3′ (−2426 to −2414), site 2: 5′-TCCAC GAGGT CAT-3′ (−2149 to −2137) and site 3: 5′-GTAGT GATCT CAG-3′ (−1291 to −1279; Fig. 5a).

a Three putative binding motifs of c-Jun on the miR-93-5p promoter and their mutation. b Ectopic c-Jun level positively regulated miR-93-5p expression in MHCC-97H and HepG2 cells. c Effect of ectopic c-Jun level on protein expression of MAP3K2, JNK, and p-JNK in MHCC-97H and HepG2 cells. d The amplification of three putative binding sites was confirmed by PCR after ChIP in HepG2 cells. e Luciferase reporter activity of wild-type or mutant promoter vectors in HEK-293T and HepG2 cells transfected with c-Jun plasmid or its control. Data represent mean ± s.d. (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by Student’s t test. mut mutant, si-con small interfering RNA control, si-c-Jun small interfering RNA against c-Jun.
Next, we probed whether c-Jun plays a role in modulating miR-93-5p. Indeed, suppression of c-Jun decreased miR-93-5p expression in MHCC-97H cells, while overexpression of c-Jun increased miR-93-5p expression in HepG2 cells (Fig. 5b). Moreover, downstream targets of miR-93-5p, including MAP3K2 and p-JNK, were also positively influenced by c-Jun in MHCC-97H and HepG2 cells (Fig. 5c).
To further illustrate whether c-Jun promotes miR-93-5p expression through its interaction with three putative binding motifs, we performed chromatin immunoprecipitation (ChIP) in HepG2 cells, which indicated that c-Jun is recruited to all three binding regions (Fig. 5d). We further conducted dual-luciferase reporter assay in HEK-293T and HepG2 cells. c-Jun overexpression enhanced the luciferase activity of wild-type miR-93-5p promoter, and the effect was significantly attenuated when site 1, site 2, or site 3 was mutated in HepG2 cells. However, the effect of site 1 mutation was retained in HEK-293T cells (Fig. 5e), indicating that site 2 and site 3 are conserved functional motifs in response to c-Jun activation. In conclusion, c-Jun activates miR-93-5p transcription through its direct interaction with the promoter region. Hence, a positive feedback loop including miR-93-5p, MAP3K2, and c-Jun is established.
miR-93-5p promotes HCC cell proliferation by targeting MAP3K2 in vivo
To confirm the observations from in vitro studies, we examined the in vivo relevance of miR-93-5p-mediated regulation of HCC growth by using a xenograft mouse model bearing tumors originating from MHCC-97H and SK-Hep-1 cells. MHCC-97H and SK-Hep-1 cells were stably transfected with recombinant lentiviral particles containing miR-93-5p sponge and miR-93-5p, respectively. In line with our in vitro studies, treatment with miR-93-5p sponge markedly decreased tumor growth, tumor weight, and tumor volume, while overexpression of miR-93-5p had the reverse effect (Fig. 6a). At the molecular level, miR-93-5p sponge suppressed miR-93-5p expression and inactivated the MAP3K2/p-JNK/p21 pathway, while overexpression of miR-93-5p enhanced miR-93-5p expression and activated the MAP3K2/p-JNK/p21 pathway (Fig. 6b). Furthermore, immunohistochemistry (IHC) analysis showed that miR-93-5p sponge reduced the level of proliferating cell nuclear antigen (PCNA), Ki-67, MAP3K2, p-JNK, and increased the level of p21, whereas overexpressed miR-93-5p displayed the opposite pattern (Fig. 6c). Collectively, these data support a crucial role for miR-93-5p in promoting HCC growth via the MAP3K2/JNK/p21 pathway in vivo.

a Xenograft tumors were generated from MHCC-97H cells transfected with lentiviral antagonizing miR-93-5p and SK-Hep-1 cells transfected with lentiviral overexpressing miR-93-5p. Tumors on the 18-day were shown. Meanwhile, tumor weight was calculated, and tumor volume was recorded at the end. mean ± s.d. (n = 4), *P < 0.05, **P < 0.01 by Student’s t test. b miR-93-5p expression and protein levels of MAP3K2, p-JNK, and p21 in various groups of xenograft tumors. c Representative H&E and IHC staining of PCNA, Ki-67, MAP3K2, p-JNK, and p21 of xenograft tumors were displayed. ×200, Scale bars represent 100 μm. d Schematic of the miR-93-5p/MAP3K2/MAPKs/p21 regulatory loop in perpetuating HCC proliferation, where miR-93-5p induced MAP3K2 promoted MAPKs, including p38 and JNK, followed by c-Jun activation and p21 repression, resulted in enhanced G1 to S phase transition. In return, activated c-Jun further enhanced miR-93-5p transcription. HE hematoxylin-eosin, PCNA proliferating cell nuclear antigen.
Discussion
The onset and development of HCC is a multistep process involving the progressive accumulation of molecular alterations and cellular events. There is emerging evidence supporting the importance of miRNAs and miRNA-mediated regulatory networks in providing a supportive role to promote or to prevent HCC onset and development. Our previous miRNA expression profiling in seven-paired HCC specimens suggested that miR-93-5p is an HCC-related miRNA [6]. Intriguingly, our present study corroborates a novel, but still poorly understood, phenomenon termed as RNA activation (RNAa) for miR-93-5p that has been ignored thus far in human HCC. Furthermore, we identify that miR-93-5p forms a regulatory feedback loop with MAP3K2 and its downstream genes, including MKK4, JNK, and c-Jun, to regulate cell proliferation, migration, and invasion in HCC using a combination of in vitro and in vivo methods.
miR-93-5p regulates diverse cellular pathways via regulation of multiple target genes, including PTEN [4], RhoC [28], and PDCD4 [29]. miR-93-5p, together with miR-106b and miR-25, belongs to an oncogenic miR-106b-25 cluster. Elevated expression of these miRNAs has been observed in gastric cancer [30], breast cancer [31], and prostate cancer [32]. Despite expression of the miR-106b-25 cluster is significantly increased in HCC [6], we find that only miR-93-5p, but not miR-106b − 5p or miR-25 − 3p (Supplementary Fig. 9), markedly upregulates MAP3K2 protein expression in HCC cells, which is an upstream kinase of the MAPK signaling pathway and acts as an oncogene through modulating cellular proliferation, apoptosis, and metastasis [33].
One of the key questions addressed here is whether miR-93-5p-mediated HCC progression is directly related to the induction of MAP3K2 expression. This finding is buttressed by five lines of evidence. Firstly, there is a miR-93-5p binding site in the 3′-UTR of MAP3K2. MAP3K2 3′-UTR-driven luciferase activity, rather than MAP3K2 promoter, 5′-UTR and CDS regions, is specifically responsive to miR-93-5p. Secondly, transfection of miR-93-5p increases MAP3K2 mRNA and protein levels, while inhibition of miR-93-5p has the opposite effect. Thirdly, restoration of MAP3K2 partially recovers the phenotype and protein levels of the JNK pathway suppressed by miR-93-5p inhibition, while suppression of MAP3K2 impedes these induced by miR-93-5p overexpression. Fourthly, several studies using AGO-CLIP assay verify the direct crosstalk between miR-93-5p and MAP3K2. Finally, the binding sequence of miR-93-5p seed site in MAP3K2 3′-UTR is highly conserved in many species, including human, chimpanzee, mouse, sheep, cat, and X. tropicalis. All the evidence presented above supports that miR-93-5p directly targets 3′-UTR of MAP3K2.
RNAa, wherein miRNA unconventionally activates gene transcription or translation, has been reported for STAT3 in ovarian cancer [10], p21 in renal cell carcinoma [34], and CCNB1 in prostate adenocarcinoma [35]. However, to date, there has been no evidence of it involved in HCC. Moreover, several lines of evidence revealed that miRNA directly promotes gene expression through binding to its promoter, enhancer, 5′-UTR or 3′-UTR. The 3′-UTR contains sequences or structural regions affecting mRNA translation and stability, such as AU-rich elements (AREs), miRNA response elements (MREs), and poly(A) tail [13, 36]. A previous study suggested that miR369-3 directly targeted the ARE in TNF-α mRNA to promote translation with the help of AGO and FXR1 [13]. In our study, there is a potential ARE sequence, 5′-AATTT TTTAT ATT-3′ (172–184), within 100 bp upstream of the target sequence, 5′-GCACT TT-3′ (265–271), in 3′-UTR of MAP3K2 mRNA identified by AREsite2 database [37]. Through binding to the target sequence, miR-93-5p may influence the neighbor ARE to modulate ARE-binding proteins, like TTP, HuR, and AUF1 [37], thus reinforcing the activation of MAP3K2. Also, regulation complex may directly bind to the target sequence in an ARE-independent manner. AGO, a potent component in the RNA-induced silencing complex, is also reported to participate in RNAa by directly binding to enhancer [11] and promoter [10] of target genes. Besides, several miRNAs directly upregulated target mRNA translation via binding to its 3′-UTR in a GRSF1-dependent manner [38, 39]. Thus, further investigations are warranted to explore the exact mechanism of the 3′-UTR-induced activation in miR-93-5p-driven HCC.
In addition, we observed that miR-93-5p activates MAP3K2 and its downstream JNK/c-Jun pathway, which subsequently weakens p21 transcription to promote hepatoma cells G1 to S phase transition. The JNK pathway also reported to enhance G1 to S phase transition through reducing p53 [40] and inducing cyclin D1 [41]. Moreover, miR-93-5p can directly inhibit p21 via binding to p21 mRNA 3′-UTR [42]. To conclude, these mechanisms all demonstrate that miR-93-5p exerts an effect on HCC proliferation. Moreover, activator protein (AP)-1 complex, composed of c-Jun and c-Fos heterodimers, has involved in cancer cells apoptosis, differentiation, and metastasis, suggesting that miR-93-5p influences HCC migration and invasion via c-Jun [25]. However, no obvious antiapoptotic effect of miR-93-5p was detected in MHCC-97H and SK-Hep-1 cells (Supplementary Fig. 10).
As a downstream transcription factor in the JNK pathway, c-Jun enhances miR-106b transcription by targeting its promoter in breast cancer [27]. Our study illustrates that c-Jun targets miR-93-5p promoter to activate its transcription, which yields a miR-93-5p/MAP3K2/c-Jun positive feedback loop. Thus, miR-93-5p induced c-Jun enhances the carcinogenesis role of miR-93-5p in HCC. Given that miR-106b and miR-93-5p comprised miR-106b-25 cluster, another cluster member miR-25, and its host gene MCM7 may also be regulated by c-Jun.
In conclusion, miR-93-5p exerts its functional role in regulating MAP3K2 expression through a positive feedback circuitry. Furthermore, the positive feedback regulation of miR-93-5p on MAP3K2, by targeting downstream oncogenic p-MKK4, p-JNK, and p-c-Jun, is distinguishable from other miRNAs that target MAP3K2 by negatively regulating MAP3K2 expression (Fig. 6d). With the identification of more miR-93-5p targets in HCC, we believe that this study will lead to a better understanding of the mechanisms involved in miR-93-5p-mediated development and progression of this deadly disease, and offers the implications for designing novel therapeutic interventions based on the components of the loop in HCC.
Materials and methods
Clinical specimens and cell lines
Human HCC and adjacent non-tumor tissues were collected from patients undergoing hepatectomy in 2014 at Zhongshan Hospital of Fudan University. The specimens were histopathologically confirmed as HCC. Approval of the study was obtained from the Ethics Committee of Zhongshan Hospital, Fudan University. Written informed consent was obtained from all individuals.
Human HCC cell lines, including HepG2, BEL-7402, Hep3B, MHCC-97L, MHCC-97H, and HCC-LM3, along with normal hepatocyte L02, were provided by Liver Cancer Institute, Zhongshan Hospital of Fudan University. SK-Hep-1 and HEK-293T cell lines were obtained from Cell Bank, Chinese Academy of Sciences (Shanghai, China). All cells were incubated in Dulbecco’s modified eagle medium (Corning, NY) with 10% fetal bovine serum (Corning) and 5% CO2 at 37 °C.
Fluorescence in situ hybridization (FISH)
FISH assay was performed with the ISH kit from Qiagen (Valencia, CA). The slides were deparaffinized, rehydrated, processed by H2O2, digested by proteinase K, and then incubated with 100 nM 5′ and 3′ digoxigenin-labeled meRCURY LNA miRNA detection probe (Qiagen) at 51 °C for 1 h. After blocked by 1% bovine serum albumin (Sigma-Aldrich, St. Louis, MO) for 30 min, the tissues were incubated with horseradish peroxidase-conjugated anti-digoxigenin antibody (Abcam, Cambridge, UK; Supplementary Table 3) at 4 °C overnight. Fluorescence was detected after the tyramide-signal amplification reaction for 15 min.
Cell transfection
miR-93-5p inhibitor or mimics (RiboBio, Guangzhou, China), and small interfering RNA against MAP3K2 or c-Jun (GenePharma, Shanghai, China) were transiently transfected using INTERFERin (Polyplus-transfection, Illkirch, France; Supplementary Table 4). Plasmid vector overexpressed MAP3K2 or c-Jun (GenePharma) was transiently transfected using Lipofectamine 3000 (Invitrogen, Carlsbad, CA). Stably transfected cells were established by lentiviral particles carrying miR-93-5p or miR-93-5p sponge (GenePharma).
Cell function assays
Cell proliferation was detected by Cell counting kit 8 (CCK8; Beyotime, Shanghai, China), BrdU (CST, Boston, MA) and colony formation assays [34]. To measure cell cycle, cells stained with propidium iodide (PI) were analyzed on a FACSAriall (BD Biosciences, San Jose, CA) and data were analyzed by FlowJo (Tree Star, Inc., Ashland, OR). To measure cell apoptosis, cells stained with annexin V/PI were analyzed by FACS (BD Biosciences). Cell invasion or migration ability was assessed by established Transwell (Corning) assay covered with or without Matrigel (BD Biosciences) [43]. Cell migration was also valued by wound-healing assay [44].
Real-time quantitative PCR
Total RNA was extracted using RNAiso Plus (TaKaRa, Dalian, China). Total RNA products for miRNA analysis were reversed using the miRcute Plus miRNA First-Strand cDNA Synthesis Kit, and real-time qPCR was conducted using the miRcute Plus miRNA qPCR Detection Kit (SYBR Green; TIANGEN, Beijing, China). Total RNA products for mRNA analysis were reversed using the PrimeScript RT reagent Kit with gDNA Eraser, and qPCR was conducted using SYBR Premix Ex Taq (TaKaRa). The reverse transcription was performed using the ProFlex PCR system and qPCR was performed using the 7500 real-time PCR system (Applied Biosystems, Waltham, MA). PCR analysis of miR-93-5p precursor was performed as described previously [20]. Primers for miR-93-5p and U6 were obtained from TIANGEN. Other primer sequences were listed in Supplementary Table 5.
Western blot
Western blot was performed as described previously [45]. Cells or tissues were lysed in RIPA (Beyotime) supplemented with Protease Inhibitor Cocktail (Sigma-Aldrich). Proteins were separated by 10% SDS-PAGE (Beyotime), transferred to polyvinylidene fluoride membrane (Millipore, Bedford, MA), blocked by 5% bovine serum albumin (Sigma-Aldrich) for an hour, and immunoblotted with primary antibodies overnight at 4 °C, followed by corresponding secondary antibodies (Supplementary Table 3) for an hour at room temperature. The membrane was detected by enhanced chemiluminescence (Millipore). Western blot bands were quantified by relative densitometry using Image J (version 1.50i), and normalized to the loading control and then to the negative control.
ChIP
ChIP was conducted following the manufacture’s instruction (Millipore). HepG2 cells transfected with a plasmid carrying c-Jun or control were treated with formaldehyde to cross-link protein to DNA. Then cross-link chromatin was sheared into 200–1000-bp fragments by sonication, followed by immunoselection using a c-Jun antibody (CST; Supplementary Table 3). After cross-links reverse, the purified DNA was detected by PCR and then by sequencing. PCR primers for sites 1, 2, and 3 (Supplementary Table 5) were used to detect enrichment of c-Jun-binding regions on the miR-93-5p promoter.
IHC
IHC was performed using EnVision Detection Systems (Dako, Carpinteria, CA). The primary antibodies used are shown in Supplementary Table 3.
Plasmid construction and luciferase reporter assay
MAP3K2 promoter/5′-UTR was cloned into pGL3-Basic luciferase reporter vector (Promega, Madison, WI). CDS/3′-UTR, CDS, 3′-UTR, wild-type 3′-UTR, and mutant 3′-UTR of MAP3K2 were separately cloned into pmirGLO-Basic luciferase reporter vectors (Promega). Wide-type miR-93-5p promoter and miR-93-5p promoter with mutant binding site 1, site 2, or site 3 were separately inserted into PHY-803 luciferase reporter vectors (Promega). Luciferase activity was detected using the Dual-Luciferase Reporter Assay System (Promega).
HCC xenograft models
Animal experiments were approved by the Animal Ethics Committee of Zhongshan Hospital, Fudan University, and carried out according to the “Guide for the Care and Use of Laboratory Animals” (NIH publication 86-23, revised 1985). Mouse experiments were performed as described previously [46]. Four- to five-week-old male nude mice (BALB/c) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China) and maintained in pathogen-free conditions with autoclaved water and irradiated food. Mice were completely randomly allocated into four groups with four mice per group, that is, miR-93-5p overexpression, miR-93-5p knockdown and corresponding control groups. miR-93-5p overexpressed SK-Hep-1 cells or miR-93-5p knockdown MHCC-97H cells and controls were subcutaneously injected at a density of 5 × 106 cells per mouse. During the 18-day growth, mice body weight and tumor size were measured. Tumor volume = (length × width2)/2.
Statistical analysis
All data were analyzed using IBM SPSS Statistics V20.0 (SPSS Inc., Chicago, IL) or GraphPad Prism 7 (GraphPad Software, San Diego, CA) and presented as mean ± standard deviation (s.d.), repeated from three independent experiments. Survival data were calculated using Kaplan–Meier’s and log-rank tests. Frequencies of categorical variables were analyzed by the Chi-squared test. The mutual association was detected by Spearman rank correlation. Continuous parametric data were analyzed by Student’s t test or one-way ANOVA. A two-tailed P < 0.05 represented statistically significant.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Johnson JM, Edwards S, Shoemaker D, Schadt EE. Dark matter in the genome: evidence of widespread transcription detected by microarray tiling experiments. Trends Genet. 2005;21:93–102.
Sadri Nahand J, Bokharaei-Salim F, Salmaninejad A, Nesaei A, Mohajeri F, Moshtzan A, et al. microRNAs: Key players in virus-associated hepatocellular carcinoma. J Cell Physiol. 2019;234:12188–225.
Li N, Miao Y, Shan Y, Liu B, Li Y, Zhao L, et al. MiR-106b and miR-93 regulate cell progression by suppression of PTEN via PI3K/Akt pathway in breast cancer. Cell Death Dis. 2017;8:e2796.
Du L, Zhao Z, Ma X, Hsiao TH, Chen Y, Young E, et al. miR-93-directed downregulation of DAB2 defines a novel oncogenic pathway in lung cancer. Oncogene. 2014;33:4307–15.
Zhu HR, Huang RZ, Yu XN, Shi X, Bilegsaikhan E, Guo HY, et al. Microarray expression profiling of microRNAs reveals potential biomarkers for hepatocellular carcinoma. Tohoku J Exp Med. 2018;245:89–98.
Ohta K, Hoshino H, Wang J, Ono S, Iida Y, Hata K, et al. MicroRNA-93 activates c-Met/PI3K/Akt pathway activity in hepatocellular carcinoma by directly inhibiting PTEN and CDKN1A. Oncotarget. 2015;6:3211–24.
Xue XF, Wang XN, Zhao YB, Hu RK, Qin L. Exosomal miR-93 promotes proliferation and invasion in hepatocellular carcinoma by directly inhibiting TIMP2/TP53INP1/CDKN1A. Biochem Biophys Res Commun. 2018;502:515–21.
Ji CM, Liu H, Yin Q, Li H, Gao H. miR-93 enhances hepatocellular carcinoma invasion and metastasis by EMT via targeting PDCD4. Biotechnol Lett. 2017;39:1621–9.
Chaluvally-Raghavan P, Jeong KJ, Pradeep S, Silva AM, Yu S, Liu W, et al. Direct upregulation of STAT3 by microRNA-551b-3p deregulates growth and metastasis of ovarian cancer. Cell Rep. 2016;15:1493–504.
Xiao M, Li J, Li W, Wang Y, Wu FZ, Xi YP, et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. Rna Biol. 2017;14:1326–34.
Orom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5′ UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460–71.
Vasudevan S, Tong YC, Steitz JA. Switching from repression to activation: MicroRNAs can up-regulate translation. Science. 2007;318:1931–4.
Delire B, Starkel P. The Ras/MAPK pathway and hepatocarcinoma: pathogenesis and therapeutic implications. Eur J Clin Investig. 2015;45:609–23.
Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–49.
Yu J, Tan QY, Deng B, Fang CS, Qi D, Wang RW. The microRNA-520a-3p inhibits proliferation, apoptosis and metastasis by targeting MAP3K2 in non-small cell lung cancer. Am J Cancer Res. 2015;5:802–11.
Wu J, Li WZ, Huang ML, Wei HL, Wang T, Fan J, et al. Regulation of cancerous progression and epithelial-mesenchymal transition by miR-34c-3p via modulation of MAP3K2 signaling in triple-negative breast cancer cells. Biochem Biophys Res Commun. 2017;483:10–6.
Wang M, Lv G, Jiang C, Xie S, Wang G. miR-302a inhibits human HepG2 and SMMC-7721 cells proliferation and promotes apoptosis by targeting MAP3K2 and PBX3. Sci Rep. 2019;9:2032.
Fang L, Deng Z, Shatseva T, Yang J, Peng C, Du WW, et al. MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting integrin-beta8. Oncogene. 2011;30:806–21.
Li Y, Tan W, Neo TW, Aung MO, Wasser S, Lim SG, et al. Role of the miR-106b-25 microRNA cluster in hepatocellular carcinoma. Cancer Sci. 2009;100:1234–42.
Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46:D956–63.
Niu Y, Jin Y, Deng SC, Deng SJ, Zhu S, Liu Y, et al. MiRNA-646-mediated reciprocal repression between HIF-1alpha and MIIP contributes to tumorigenesis of pancreatic cancer. Oncogene. 2018;37:1743–58.
Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–7.
Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39:D202–9.
Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–20.
Wang CH, Tsao YP, Chen HJ, Chen HL, Wang HW, Chen SL. Transcriptional repression of p21((Waf1/Cip1/Sdi1)) gene by c-jun through Sp1 site. Biochem Biophys Res Commun. 2000;270:303–10.
Gong C, Qu S, Liu B, Pan S, Jiao Y, Nie Y, et al. MiR-106b expression determines the proliferation paradox of TGF-beta in breast cancer cells. Oncogene. 2015;34:84–93.
Chen X, Chen S, Xiu YL, Sun KX, Zong ZH, Zhao Y. RhoC is a major target of microRNA-93-5P in epithelial ovarian carcinoma tumorigenesis and progression. Mol Cancer. 2015;14:11.
Chen C, Zheng Q, Kang W, Yu C. Long non-coding RNA LINC00472 suppresses hepatocellular carcinoma cell proliferation, migration and invasion through miR-93-5p/PDCD4 pathway. Clin Res Hepatol Gastroenterol. 2019;43:436–45.
Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I, et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13:272–86.
Guarnieri AL, Towers CG, Drasin DJ, Oliphant MUJ, Andrysik Z, Hotz TJ, et al. The miR-106b-25 cluster mediates breast tumor initiation through activation of NOTCH1 via direct repression of NEDD4L. Oncogene. 2018;37:3879–93.
Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal. 2010;3:ra29.
Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405.
Wang C, Tang K, Li Z, Chen Z, Xu H, Ye Z. Targeted p21(WAF1/CIP1) activation by miR-1236 inhibits cell proliferation and correlates with favorable survival in renal cell carcinoma. Urol Oncol. 2016;34:59.e23–34.
Huang V, Place RF, Portnoy V, Wang J, Qi Z, Jia Z, et al. Upregulation of Cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res. 2012;40:1695–707.
Pichon X, Wilson LA, Stoneley M, Bastide A, King HA, Somers J, et al. RNA binding protein/RNA element interactions and the control of translation. Curr Protein Pept Sci. 2012;13:294–304.
Fallmann J, Sedlyarov V, Tanzer A, Kovarik P, Hofacker IL. AREsite2: an enhanced database for the comprehensive investigation of AU/GU/U-rich elements. Nucleic Acids Res. 2016;44:D90–5.
Sun Q, Yang Z, Li P, Wang X, Sun L, Wang S, et al. A novel miRNA identified in GRSF1 complex drives the metastasis via the PIK3R3/AKT/NF-kappaB and TIMP3/MMP9 pathways in cervical cancer cells. Cell Death Dis. 2019;10:636.
Yang Z, Sun Q, Guo J, Wang S, Song G, Liu W, et al. GRSF1-mediated MIR-G-1 promotes malignant behavior and nuclear autophagy by directly upregulating TMED5 and LMNB1 in cervical cancer cells. Autophagy. 2019;15:668–85.
Das M, Jiang F, Sluss HK, Zhang C, Shokat KM, Flavell RA, et al. Suppression of p53-dependent senescence by the JNK signal transduction pathway. Proc Natl Acad Sci USA. 2007;104:15759–64.
Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R, et al. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology. 2003;37:824–32.
He Y, Yu B. MicroRNA-93 promotes cell proliferation by directly targeting P21 in osteosarcoma cells. Exp Ther Med. 2017;13:2003–11.
Wong CC, Wong CM, Tung EK, Au SL, Lee JM, Poon RT, et al. The microRNA miR-139 suppresses metastasis and progression of hepatocellular carcinoma by down-regulating Rho-kinase 2. Gastroenterology. 2011;140:322–31.
Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–33.
Yu XN, Zhang GC, Sun JL, Zhu HR, Shi X, Song GQ, et al. Enhanced mLST8 expression correlates with tumor progression in hepatocellular carcinoma. Ann Surg Oncol. 2020;27:1546–57.
Yu XN, Deng Y, Zhang GC, Liu J, Liu TT, Dong L, et al. Sorafenib-conjugated zinc phthalocyanine-based nanocapsule for trimodal therapy in an orthotopic hepatocellular carcinoma xenograft mouse model. ACS Appl Mater Interfaces. 2020;12:17193–206.
Acknowledgements
This work was partly supported by the National Natural Science Foundation of China (grant numbers 81402273, 81672720, 81672334, and 81700550), and by the National Clinical Key Special Subject of China.
Author information
Authors and Affiliations
Contributions
XS, TTL, and XNY data curation, methodology, and writing original draft; AB, HRZ, HYG, GCZ, and EB data curation, formal analysis, and writing review; JLS and GQS investigation, resources, and writing review; SQW and LD investigation, resources, visualization, and writing review; MO conceptualization, and writing review and editing; XZS and JMZ conceptualization, funding acquisition, supervision, and writing review and editing.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Shi, X., Liu, TT., Yu, XN. et al. microRNA-93-5p promotes hepatocellular carcinoma progression via a microRNA-93-5p/MAP3K2/c-Jun positive feedback circuit. Oncogene 39, 5768–5781 (2020). https://doi.org/10.1038/s41388-020-01401-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-01401-0
- Springer Nature Limited
This article is cited by
-
MiRNA-93: a novel signature in human disorders and drug resistance
Cell Communication and Signaling (2023)
-
Non-coding RNA methylation modifications in hepatocellular carcinoma: interactions and potential implications
Cell Communication and Signaling (2023)
-
Bladder Cancer Progression Is Suppressed Through the Heart and Neural Crest Derivatives Expressed 2-Antisense RNA 1/microRNA-93-5p/Defective in Cullin Neddylation 1 Domain Containing 3 Axis
Applied Biochemistry and Biotechnology (2023)
-
Circular RNA MTCL1 promotes advanced laryngeal squamous cell carcinoma progression by inhibiting C1QBP ubiquitin degradation and mediating beta-catenin activation
Molecular Cancer (2022)