
Abstract
Breast cancer is the second leading cause of cancer-related deaths for women, due mainly to metastatic disease. Invasive tumors exhibit aberrations in recruitment and activity of immune cells, including decreased cytotoxic T cells. Restoring the levels and activity of cytotoxic T cells is a promising anticancer strategy; but its success is tumor type dependent. The mechanisms that coordinate recruitment and activity of immune cells and other stromal cells in breast cancer remain poorly understood. Using the MMTV-PyVmT/FVB mammary tumor model, we demonstrate a novel role for CCL2/CCR2 chemokine signaling in tumor progression by altering the microenvironment. Selective targeting of CCR2 in the PyVmT mammary epithelium inhibited tumor growth and invasion, elevated CD8+ T cells, decreased M2 macrophages and decreased angiogenesis. Co-culture models demonstrated these stromal cell responses were mediated by tumor-derived CCL2 and CCR2-mediated suppression of the T-cell activating cytokine, CD154. Coculture analysis indicated that CCR2-induced stromal reactivity was important for tumor cell proliferation and invasion. In breast tumor tissues, CD154 expression inversely correlated with CCR2 expression and correlated with relapse free survival. Targeting the CCL2/CCR2 signaling pathway may reprogram the immune angiogenic and microenvironments and enhance effectiveness of targeted and immunotherapies.
Similar content being viewed by others


Introduction
Breast cancer is the most common form of cancer diagnosed in women, with over 1.8 million cases diagnosed annually worldwide and is the second leading cause of cancer-related deaths for women. The majority of breast cancers are diagnosed as nonmetastatic disease [1]. Understanding the pathobiology of early breast cancer progression would lead to more effective treatment strategies to reduce patient mortality.
Invasive tumors exhibit aberrations in recruitment and activity of innate and adaptive immune cells [2]. Decreased numbers of CD8+ (cytotoxic) T cells correlate with poor patient prognosis in invasive breast cancers [3,4,5]. Decreased CD8+ T-cell activity is associated with increased tumor associated macrophages (TAMs), characterized as wound healing or M2 polarized macrophages [6]. TAMs inhibit T-cell proliferation and prevent T-cell elimination of tumor cells by expressing immunosuppressive molecules, increasing checkpoint signaling in T cells, and promoting tumor growth and survival through secretion of angiogenic and growth factors [7, 8]. The tumor vasculature limits T-cell recruitment and function by increasing expression of immunosuppressive cytokines and immune checkpoint molecules [9]. Restoring cytotoxic T-cell function could be an effective anticancer strategy but its success is tumor type-dependent [10]. The mechanisms that coordinate recruitment and activity of stromal cells in breast cancer remain poorly understood.
CCR2 is a G protein coupled receptor (GPCR) that binds to chemokines to regulate macrophage recruitment during wound healing and infection [11,12,13]. While CCR2 bind multiple chemokines, CCR2 binds strongest to CCL2. CCL2 and CCR2 knockout mice show defects in macrophage recruitment without compensatory upregulation of other chemokine ligands [14, 15]. These studies indicate a unique biological role for CCL2/CCR2 signaling in inflammation.
CCL2 and CCR2 are overexpressed in pancreatic, prostate, colon, and breast cancers [16, 17]. In breast and prostate cancer, CCL2 blockade in animal models inhibits tumor growth and metastasis associated with decreased recruitment of CCR2+ macrophages to the primary tumor [16, 18]. We recently showed that CCR2 is overexpressed in cancer cells. CCR2 knockdown in breast cancer cells inhibited tumor growth and invasion without significantly affecting the immune and angiogenic microenvironments [19, 20]. These studies were conducted in immunocompromised mice, preventing a clear assessment on the microenvironment during CCL2/CCR2-mediated tumor progression.
Using animal models, co-culture systems and patient samples, we demonstrated a novel role for epithelial CCL2/CCR2 signaling in suppressing CD154 signaling to mediate mammary tumor growth, invasion, and inflammation. These studies have important clinical implications.
Results
CCR2 knockdown inhibits mammary tumor growth, invasion, and inflammation
To assess changes in the microenvironment during CCR2-mediated tumor progression, we utilized the MMTV-PyVmT/FVB model, an immune-competent mammary tumor model [21]. To ensure consistent tumor formation, tumors were established in FVB mice via mammary intraductal injection of PyVmT mammary carcinoma cells, which mimics the development and progression of invasive ductal carcinoma in patients [22, 23]. To target CCR2 expression in mammary tumors, we delivered siRNAs complexed to TAT cell penetrating peptides through calcium cross-linking. siRNA/TAT peptide complexes penetrated tumor tissues to induce gene knockdown more efficiently than conventional polyethyleneimine particles [24,25,26]. We previously identified a formula of peptide/siRNA complexes that selectively transfect mammary carcinoma cells over stromal cells [27].
Tumors 0.4 cm in diameter were injected with control (Con-si) or CCR2 (CCR2-si) siRNA complexes once a week for 3 weeks and harvested for analysis (Fig. 1a). While there were more CCR2+ cells in the mammary epithelial population, CCR2 expression appeared higher in individual myeloid cells, as determined by flow cytometry. CCR2-si treatment decreased the number of CD24 + CCR2+ cells by 40% and did not affect the number of CCR2+ myeloid cells (Fig. 1b, Supplemental Fig. 1). CCR2 knockdown inhibited mammary tumor growth, and according to H&E stain, inhibited tumor invasion and stromal reactivity (Fig. 1c, d). CCR2 knockdown decreased tumor cell proliferation (Fig. 1e). Tumor cell survival was not affected (Fig. 1g). By flow cytometry and immunofluorescent staining, CCR2-deficient tumors showed fewer F4/80 + CD11b+ macrophages and Cd11b + Arginase I + (M2) macrophages (Fig. 2a, b) and did not affect the number of F4/80 + CCR2+ macrophages (Fig. 2c). CCR2 knockdown increased numbers of overall and activated CD8+ T cells but not CD4+ T cells (Fig. 2d, e). Furthermore, CCR2 deficiency deceased tumor angiogenesis, as indicated by VWF8 immunostaining (Fig. 2f). Overall, CCR2 knockdown in PyVmT mammary tumors inhibited mammary tumor growth and invasion, decreased M2 macrophages, elevated cytotoxic T cells and decreased angiogenesis.

a Experimental design: PyVmT mammary tumors were established though mammary intraductal injection of FVB mice. When tumors reached 0.4 cm in diameter, mice received intratumoral injections of PBS vehicle control, 10 μg TAT peptides complexed to control siRNAs (Con-si) or CCR2 siRNAs (CCR2-si), n = 7 per group. Mice were sacrificed when control tumors reached ~1.5 cm in diameter, 77 days after cellular transplantation. b Flow cytometry analysis was conducted on tumor cell suspensions for percentage of CCR2+ cells in mammary epithelial cells (CD24+) vs. myeloid cells (Cd11b+). c Tumor diameter was measured over time by caliper. d Mean tumor mass at endpoint. Representative tumor tissues shown. Mammary tumors were stained by H&E (e), or immunostained for PCNA expression (f) or cleaved caspase-3 (g). Arrowheads depict positive staining. Scale bar = 200 microns. Staining was quantified by ImageJ. Expression was normalized to hematoxylin staining. Statistical analysis was determined by two-tailed t test (b, e, f) or one way ANOVA with Bonferonni post-hoc analysis (c, d). Statistical significance was determined by p < 0.05, *p < 0.05, **p < 0.01, ns not significant. Mean + SEM are shown.

PyVmT mammary tumors treated with Con-si or CCR2-si were analyzed for a F4/80 + CD11b+ macrophages by flow cytometry b M2 macrophages by co-immunofluorescence staining for arginase I (green) and CD11b (red), c co-immunofluorescence staining for CCR2 (green) F4/80 (red) expression, d flow cytometry analysis for CD4 + CD8+ or CD8+CD69+ lymphocytes, e activated cytotoxic T cells by co-immunofluorescence staining for CD8 (red) and CD69 (green) expression, f angiogenesis by immunostaining for Von Willibrand Factor 8 (VWF8), arbitrary units are shown. Black arrows indicate positive staining. n = 6 tumors per group, with >15 images per tumor section. Immunostaining was quantified by ImageJ, and normalized to DAPI (b, c, e) or hematoxylin (f). Arbitrary units are shown. Statistical analysis was performed using two-tailed t test. Statistical significance was determined by p < 0.05. *p < 0.05. Mean ± SEM are shown. Scale bar = 100 microns.
Contribution of CCL2 and CD154 to CCR2-mediated tumor cell proliferation and migration
To identify inflammatory factors regulated by CCL2/CCR2 signaling, we profiled cytokine expression in MDA-MB-231 cells by antibody array. Of the cytokines and growth factors profiled, CD154 expression was most significantly affected in CCL2-deficient cells (Supplemental Table 1). CD154 is a member of the Tumor Necrosis Factor family of ligands that mediates co-stimulatory interactions between antigen presenting cells (APC) and T cells [28, 29]. CD154 was increased in CCL2-deficient MDA-MB-231 tumors and CCR2-deficient DCIS.com xenografts (Supplemental Fig. 2A, B), corresponding to decreased tumor growth and invasion reported previously [19, 30]. In PyVmT mammary tumors, CCR2-si delivery decreased CCL2 and CCR2 expression, and increased CD154 expression (Fig. 3a–c). Thus, targeting CCR2 or CCL2 expression increased CD154 expression in multiple breast tumor models.

PyVmT mammary tumors treated with Con-si or CCR2-si were immunostained for a CCR2, b CD154, or c CCL2 expression. Staining was quantified by ImageJ and normalized to hematoxylin staining (total area); arbitrary units are shown. Scale bar = 100 microns. n = 6 mice per group, with four images per sample. Statistical analysis was performed using two-tailed t test. Mean ± SEM are shown.
The functional contribution of CD154 and CCL2/CCR2 signaling was analyzed in cultured cells. CD154, but not its receptor CD40, was most highly expressed in nontransformed MCF10A cells, compared to breast cancer cells (Supplemental Fig. 3A, B). CD154 treatment inhibited wound closure of DCIS.com, MCF10CA1d, and MDA-MB-231 breast cancer cells, demonstrating tumor suppressive effects on multiple breast cancer cell lines (Supplemental Fig. 3C). CCR2 deficiency in PyVmT and DCIS.com cells elevated CD154 expression but did not affect CCL2 expression (Fig. 4a–c, Supplemental Fig. 4A, B). CCR2 knockdown did not affect proliferation (Fig. 4d) but inhibited wound closure migration, which was rescued with CD154 antibody neutralization (Fig. 4e, Supplemental Fig. 4C), and with CD154 shRNA knockdown (Fig. 4f, g). These data indicate that CCL2/CCR2 signaling in carcinoma cells regulates cell migration by suppressing CD154 expression. CCL2 knockdown in CCR2 expressing mammary carcinoma cells inhibited cell migration but not cell proliferation (Fig. 5a–c, Supplemental Fig. 4D, E). CCL2 knockdown did not affect wound closure in CCR2-deficient cells, confirming specificity for CCL2/CCR2 signaling in mammary carcinoma cells (Fig. 5d). These data indicate that tumor cell-derived CCL2 regulates cell migration through CCR2-dependent mechanisms.

PyVmT mammary carcinoma cells transfected with Con-si or CCR2-si were analyzed for CCR2 expression by flow cytometry (a) CD154 (b) or CCL2 (c) expression in conditioned medium by ELISA (c). Control or CCR2-deficient PyVmT cells were treated with or without 1 μg/ml IgG or anti-CD154 neutralizing antibodies, and were analyzed for PCNA expression by immunofluorescence staining (d), or wound closure (e). PyVmT cells were generated to stably express control shRNAs (Con-sh) or CD154 shRNAs (CD154-sh),and then were transfected with Con-si or CCR2-si. Cells were analyzed for expression of CD154 and CCR2 by immunoblot (f) or changes in wound closure (g). PCNA staining was quantified by ImageJ. Expression was normalized to DAPI, arbitrary units are shown. Statistical significance was determined by p < 0.05, *p < 0.05, ns not significant. Mean ± SEM are shown.

PyVmT cells stably expressing control (Con-sh) or two different CCL2 shRNAs (CCL2-sh1, CCL2-sh3) were analyzed for a CCL2 expression by ELISA, b PCNA expression and wound closure in Con-si (c) or CCR2-si transfected cells (d). PCNA staining was quantified by ImageJ. Expression was normalized to DAPI, arbitrary units are shown. Statistical significance was determined by p < 0.05. *p < 0.05, **p < 0.01, ns not significant. Mean ± SEM are shown.
CD154 expression is regulated at the transcriptional level [31, 32]. To understand the mechanisms through which CCR2 negatively regulated CD154 expression in mammary carcinoma cells, we screened transcription factors that modulated CD154 gene expression in immune cells, notably NF-ΚB and NFATc1 [33, 34]. CCR2 overexpression in MCF10A cells decreased CD154 and NFATc1 expression, and decreased phosphorylation of IΚBα, which inhibits NF-ΚB translocation and activity. CCR2 knockout in DCIS.com cells or CCR2 siRNA knockdown in PyVmT cells increased CD154 expression corresponding to increased NFATc1 and NF-ΚB activity (Supplemental Fig. 5A). Inhibition of NFATc1 and NF-ΚB with Cyclosporine A [35, 36] decreased CD154 expression in CCR2-deficient PyVmT cells. Targeting NF-ΚB with the IKK inhibitor, Bay11-7082 [37] decreased expression of CD154 (Supplemental Fig. 5B). Bay11-7082 decreased NFATc1 expression, consistent with studies showing that NF-ΚB regulates NFATc1 expression and activity [36, 38]. NFATc1 and NF-KB activity are regulated by SRC and PKC signaling in multiple cell types [39,40,41,42]. CCR2 regulates SRC and PKC activity in breast cancer cell lines [20]. While CCR2 inhibition with INBC3284 [43, 44] enhanced CD154 expression, inhibition of SRC with PP2 [45] and PKC with Go6983 [39] did not affect CD154 expression in PyVmT cells (Supplemental Fig. 5C). These data indicate that CCR2 suppresses CD154 gene expression in mammary carcinoma cells by inhibiting NFATc1 and NF-ΚB, independently of PKC and SRC.
CCR2 signaling in breast cancer cells mediates endothelial recruitment and sprouting
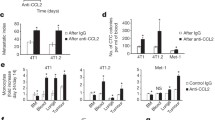
As vascular density is a hallmark of breast tumor malignancy, we examined whether CCR2 signaling in mammary carcinoma cells regulated endothelial cell branching and recruitment. Immunofluorescence staining revealed positive CCR2 expression in murine lung microvascular endothelial cells (Supplemental Fig. 6). Conditioned medium from CCR2-deficient PyVmT cells inhibited endothelial branching, which was not rescued with anti-CD154. Conditioned medium from CCL2-deficient PyVmT cells did not affect endothelial branching (Supplemental Fig. 7). Endothelial cell recruitment was determined by measuring invasion through Matrigel coated transwells in response to tumor conditioned medium. Conditioned medium from CCR2-deficient PyVmT cells inhibited endothelial invasion, which was rescued with CD154 antibody neutralization, and with CD154 shRNA knockdown. CCL2 knockdown in PyVmT cells inhibited endothelial cell invasion, which was rescued with CCL2 treatment (Fig. 6a). These data indicate that CCR2 signaling in PyVmT cells mediates endothelial cell recruitment and branching. Only endothelial cell recruitment is dependent on tumor cell-derived CCL2 and tumor cell suppression of CD154 mediated by CCR2.

Control (Con-si) or CCR2-deficient (CCR2-si) PyVmT cells were transfected with shRNAs to control (Con-sh), CD154 (CD154-sh) or CCL2 (CCL2-sh3), and analyzed for effects on endothelial cells and macrophages in the following manner: a Invasion of lung microvascular endothelial cells (LMECs) through Matrigel coated transwells for 24 h was analyzed in response to tumor conditioned medium treated with/without 1 μg/ml IgG or CD154 or 100 ng/ml CCL2. LMECs on the transwell underside were identified by DAPI staining. b Recruitment of mcherry Raw 264.7 macrophages into 3D cultures of PyVmT cells established in TheraKanTM devices, with/without IgG, anti-CD154, or CCL2 treatment for 24 h. Left panel shows experimental setup. Right panels show macrophages recruited to devices, as detected by fluorescence microscopy. c Arginase I expression in Raw 264.7 macrophages was measured in response to tumor conditioned medium, with/without IgG, CD154 or CCL2 for 24 h. Immunostaining for Arginase I was quantified by ImageJ. Expression was normalized to hematoxylin staining. Arbitrary units are shown. n = 3 experiments, two replicates per experimental group, four images per replicate. Statistical analysis was performed using one way ANOVA with Bonferroni post-hoc comparison. Statistical significance was determined by p < 0.05. *p < 0.05, **p < 0.01, n.s. not significant. Mean + SEM are shown. Scale bar = 100 microns.
CCR2 signaling in breast cancer cells mediates macrophage recruitment and polarization
We previously developed a macrophage infiltration assay using the TheraKanTM device. 3D collagen cultures are established in the device, which is placed in a larger test chamber containing immune cells. The device contains a channel opening for immune cells to migrate through in response to secreted factors from 3D cultures [30]. Here, mCherry labeled Raw 264.7 macrophages were measured for infiltration into PyVmT 3D cultures by fluorescence microscopy. PyVmT cells induced robust macrophage recruitment, which was decreased with CCR2 knockdown. Anti-CD154 treatment and CD154 shRNA knockdown in CCR2-deficient PyVmT cultures rescued macrophage infiltration. CCL2 knockdown in PyVmT cells inhibited macrophage recruitment, which was not rescued with recombinant CCL2 treatment (Fig. 6b). These data indicate that CCL2/CCR2 signaling regulates macrophage recruitment indirectly, by suppressing CD154 expression.
We examined for changes in arginase I expression, a marker for M2 macrophage polarization. Conditioned medium from CCR2-deficient PyVmT and DCIS.com cells inhibited arginase I expression in Raw 264.7 cells (Fig. 5c, Supplemental Fig. 8A). Arginase I expression was rescued with CD154 antibody neutralization and with CD154 knockdown in PyVmT cells. Arginase I expression was decreased with conditioned medium from CCL2-deficient PyVmT cells or DCIS.com cells and was not rescued with CCL2 treatment (Fig. 6c, Supplemental Fig. 8B). In summary, PyVmT cells regulate macrophage polarization through CCR2-mediated suppression of CD154.
CCR2 signaling mediates proliferation and invasion of mammary carcinoma cells through macrophage-dependent mechanisms
PyVmT:macrophage co-cultures were examined for changes in tumor cell proliferation and invasion. CCR2 knockdown in PyVmT cells reduced tumor cell proliferation, associated with decreased macrophage recruitment. PyVmT cell proliferation in CCR2-deficient cultures was rescued with anti-CD154 antibody neutralization, and with CD154 shRNA knockdown. CCL2 knockdown in PyVmT cells inhibited macrophage-mediated tumor cell proliferation. Proliferation was not rescued with CCL2 treatment (Fig. 7a). These data indicate that macrophages enhance PyVmT cell proliferation through CCR2-mediated suppression of CD154. Live imaging studies revealed increased invasion of PyVmT cells co-cultured with macrophages compared to PyVmT cells alone (Supplemental Fig. 9). CCR2 knockdown in PyVmT cells in co-cultures inhibited tumor cell invasion, which was rescued with CD154 antibody neutralization, and with CD154 shRNA knockdown. CCL2 knockdown in PyVmT cells inhibited tumor cell invasion, which was rescued with CCL2 treatment (Fig. 7b). These studies indicate that macrophages enhance PyVmT tumor cell invasion through tumor-derived CCL2- and CCR2-dependent suppression of CD154.

Control (Con-si) or CCR2-deficient (CCR2-si) PyVmT cells were transfected with shRNAs to control (Con-sh), CD154 (CD154-sh) or CCL2 (CCL2-sh3). PyVmT cells were then co-cultured with mcherry Raw 264.7 macrophages, with/without 1 μg/ml IgG or CD154 or 100 ng/ml CCL2. a Co-cultures were co-stained for PyVmT (red) and PCNA (green). PCNA and PyVmT co-staining were quantified by ImageJ. Expression was normalized to PyVmT staining. b PyVmT mammary carcinoma cells expressing GFP were co-cultured with mcherry Raw 264.7 macrophages in collagen and were subject to live imaging for 24 h. Individual PyVmT mammary carcinoma cells were measured for distance invaded by ImageJ. Panels show Euclidian displacement tracks for all cells tracked. Statistical analysis was performed using Brown-Forsythe test with Games-Howell post-hoc comparison. Statistical significance was determined by p < 0.05. *p < 0.05, **p < 0.01. Mean + SEM are shown.
CD154 expression associated with favorable prognosis in breast cancers
CCR2 expression in breast carcinomas correlate with poor patient prognosis [19]. Normal and breast tumor tissues were immunostained for CCR2 and CD154 expression. Normal breast tissues showed high CD154 expression and low CCR2. Invasive breast carcinomas showed low CD154 expression and high CCR2 expression (Fig. 8a–d). mRNA datasets revealed that CD154 expression significantly associated with relapse free patient survival in breast cancers overall, and more specifically, in luminal A/B and basal breast cancers but not HER2 overexpressing breast cancers (Fig. 8e, Supplemental Figures 10A-D). Thus, CD154 inversely correlates with CCR2 expression and is associated with favorable prognosis in breast cancer.

Normal breast (n = 35) or IDC tissues (n = 48) were immunostained for CCR2 or CD154. a Representative images for normal, invasive, and control-stained tumors. b CD154 and c CCR2 expression was quantified by ImageJ and shown in bar-and-whisker plots. Whiskers indicate min and max values. Box indicates upper and lower quartile range. Line indicates median. d Scatter plot of CCR2 and CD154 expression in patient samples; Spearman’s ρ shown. e CD154 (CD40LG 207892_at) was probed for expression in KM-Plotter in breast cancer for changes in relapse free survival (RFS). Statistical analysis was performed using Mann–Whitney test (b, c), Spearman test (d) or Log rank test (e). HR hazard ratio. Statistical significance was determined by p < 0.05. f Proposed model for how CC2L2/CCR2 modulation of the tumor microenvironment contributes to tumor growth and invasion: CCL2 derived from breast cancer cells promotes recruitment and polarization of macrophages. CCL2/CCR2 signaling suppresses CD154 expression in breast cancer cells to facilitate endothelial cell recruitment and angiogenesis. Suppression of CD154 also results in macrophage recruitment and polarization, which in turn inhibits cytotoxic T-cell activity in breast tumors.
Discussion
Overall, CCR2 signaling in breast cancer cells regulates breast tumor growth and invasion by mediating tumor angiogenesis, recruiting M2 macrophages, and suppressing CD8+ cytotoxic T-cell activation. These stromal responses are facilitated by CCR2 suppression of CD154 and secretion of CCL2 by carcinoma cells (Fig. 8f).
Our studies demonstrate that autocrine CCR2 signaling suppresses CD154 expression in mammary carcinoma cells by inhibiting NF-KB and NFATc1. While PKC and SRC regulate NF-KB and NFATc1 activity, these pathways were not important for CD154 expression in PyVmT mammary carcinoma cells. CCR2 activates p42/44MAPK, SMAD3 and RHOA in breast cancer cells. However, these pathways are regulated by paracrine CCL2/CCR2 signaling, not autocrine CCL2/CCR2 signaling mechanisms [46]. Little is known about autocrine CCR2 signaling in breast cancer cells. GPCRs also activate PLC, p38MAPK and PI-3 kinase pathways to mediate NF-KB or NFATc1 activity [47,48,49]. Autocrine CCR2 signaling may regulate these pathways to suppress transcriptional activation of CD154. Candidate screening or unbiased proteomics profiling methods may identify upstream regulators of NF-ΚB and NFATc1 in mammary carcinoma cells. CD154 mRNA stability may also be regulated through binding of polypyrimidine tract-binding protein complexes to the CD154 3’ untranslated region [50,51,52]. Future biochemical studies may be conducted to characterize the mechanisms through which CCR2 negatively regulates CD154 expression in mammary carcinoma cells.
Targeting CCR2 expression decreased overall CCL2 expression in mammary tumors, but not in cultured cells. CCL2 is expressed in stromal cells and tumor epithelium [53]. Targeting tumor-derived CCR2 in vivo could have suppressed recruitment of CCL2 expressing stromal cells, contributing to decreased CCL2 bioavailability in the tumor. Alternatively, the reduction of stromal reactivity in CCR2-deficient tumors would decrease expression of factors sustaining CCL2 expression in the tumor, including IL6 and IL1B [54, 55]. Although CCR2 regulates CCL2 expression through a positive feedback loop in immune cells [56], CCL2 expression is not dependent on CCR2 expression in tumor cells and may be regulated through complex interactions between the stroma and epithelium.
Invasion involves cell-mediated breakdown of extracellular matrices and alterations in cell adhesion and cell migration [57]. Here, PyVmT cells alone could significantly migrate in wound closure assays but showed significantly lower levels of invasion through collagen compared to co-culture with macrophages. These data indicate that tumor cells require interactions with stromal cells to invade and is consistent with intravital imaging studies detecting tumor cell complexes with macrophages during invasion and metastasis [58, 59]. We found that macrophage induced tumor cell invasion was negatively regulated by CD154, associated with decreased arginase I expression. CD154 signaling in immune cells activate multiple signaling pathways including p42/44MAPK and NF-ΚB to modulate cell adhesion molecules [60, 61] and expression of TNF, IL6 and IL8, which may regulate changes in the actin cytoskeleton and expression of proteases in macrophages and tumor cells [62].
CCL2/CCR2 signals to endothelial cells and delivery of CCL2 induces angiogenesis during wound healing in vivo [13]. Here, we show that CCR2 signaling in breast cancer cells facilitates endothelial cell recruitment and branching. Endothelial cell recruitment but not branching was dependent on CCL2 expression and suppression of CD154. These data suggest that CCR2 signaling regulates expression of other factors that facilitate endothelial branching. While no significant changes in VEGF or IL6 were detected with CCL2 signaling [30], CCL2/CCR2 signaling could regulate other angiogenic factors such as FGF, HGF, or EGF, or suppress expression of angiogenic inhibitors such as angiostatins and endostatins [63, 64] to enable endothelial branching and tumor angiogenesis.
How would CCR2 signaling in carcinoma cells mediate the relationships between the vasculature and immune cells? Tumors require a vascular network to deliver nutrients and grow. At the same time, immune cells also require blood vessels to reach the tumor. Tumors coopt blood vessels for their own advantage by inducing a network of leaky and unstable blood vessels that induce an acidic and hypoxic environment show to inhibit cytotoxic T cells and recruit M2 macrophages. Tumor endothelial cells express immune checkpoint molecules such as PD-L1 and pro-apoptotic molecules such as FasL to inhibit activity and survival of cytotoxic T cells [9, 65]. In vitro studies suggest that CCR2 signaling enhances endothelial cell recruitment and induces sprouting while enhancing M2 macrophage recruitment and activity. CCR2 signaling in breast cancer cells may suppress cytotoxic T-cell activity though co-option of the endothelium and enhancing recruitment of macrophages and suppression APC function through increased CCL2 expression and suppression of CD154. As breast tumors are often poor in T-cell infiltrates, targeting CCR2 could restore an immunogenic microenvironment by preventing tumor cell cooption of endothelium and macrophages.
Checkpoint inhibitors and chimeric antigen receptor T-cell therapies show promising results for melanomas, lung cancers, and acute leukemias but not for breast cancer [66,67,68,69]. Oncogenic mechanisms such as CCR2 may prevent effectiveness of immunotherapies in breast cancer. Pharmacologic inhibitors to CCR2 or CCR2/CCR5 dual antagonists show promising results in clinical trials for liver disease [70] and pancreatic cancer [71]. Currently, candidates for CCR2 targeting are identified by the levels of CCR2 in immune infiltrating cells and are not screened for CCR2 expression in carcinoma cells. We show here that CCR2 signaling in carcinoma cells influences the angiogenic and immune microenvironments by suppressing CD154. CCR2 overexpression in carcinoma cells could be used to identify tumor types receptive to CCR2 targeting. Thus, targeting CCR2 could have wide reaching effects on cancer that could be translated to the clinic.
Materials and methods
Animal surgery
For CCR2 siRNA delivery, Female FVB mice 8 weeks old were purchased from Charles River. Mammary intraductal injections were performed as described [22, 72]. For CCL2 siRNA delivery, female Balb/c mice 8 weeks old were purchased from Charles River. 100,000 4T1 cells were injected into the mammary fat pad. Animals were monitored twice weekly for tumor formation by palpation and measurement by caliper. Individual mice were assigned to experimental groups by simple randomization and blinded to the principal investigator until statistical analysis. Tumors 0.4 cm in diameter were injected with: vehicle or 10 μg/100 μl of siRNA complexes. Injections were performed in four different areas of the tumor, using a 27 gauge needle. Injections were performed once/week for 3 weeks. Animal monitoring continued until control tumors reached 1.5 cm in diameter, the maximum allowable size set by institutional guidelines. Mice were excluded from injection and analysis if no tumors formed throughout study.
Cell culture
Raw 264.7 were purchased from ATCC. Raw 264.7 mcherry+ cells were generated by lentiviral transduction using pSico-Ef1a-mCherry plasmid (Addgene). PyVmT carcinoma cells were isolated and characterized previously [73]. DCIS.com CRISPR ablated for CCR2 expression were generated and characterized previously [19]. Lung microvascular endothelial cells were generously provided by and characterized by Dana Brantley-Sieders [74] (Vanderbilt University) and cultured in EBMTM media (Lonza cat no.CC-3121). All other cell lines were cultured in DMEM/10% FBS/1% penicillin-streptomycin (Cellgro cat no.30-004-CI). Cells were cultured 6 months or less at a time and tested for mycoplasma after thawing using the MycoAlert™ Plus Mycoplasma Detection Kit (Lonza cat no.LT07-701).
shRNA/siRNA
PyVmT and DCIS.com cells expressing EGFP control, CCL2 or CD154 shRNAs were generated by retroviral transduction using methods described [19, 30, 75]. Targeting sequences for EGFP control, murine and human CCR2 and CCL2 shRNAs and siRNAs were described previously [20, 75]. The CD154 targeting sequence was 5′-GAAGACTCCCAGCGTCAGCT-3′.
Preparation of siRNA complexes
TAT peptides were synthesized and complexed to siRNAs in 45 μl sterile deionized water containing 75 mM CaCl2 at N/P = 5 as described [30].
Generation of conditioned media
Five lakhs cells were seeded in 10 cm plates for 24 h, transfected with siRNA complexes for 24 h, and incubated in 7 ml DMEM/1% FBS for 24 h. Medium was centrifuged to eliminate debris and filtered through 0.45 micron PES membranes.
Wound closure assay
Two lakhs cells/well were seeded in 24-well plates, transfected with siRNA complexes in growth media for 24 h. Cells were serum starved for 24 h and scratched in DMEM/1% FBS with or without 3 μg/ml CD154, 1 μg/ml anti-CD154 (BD Biosciences cat no.552559) or control mouse IgG (Millipore cat no.12-371). Images were captured at ×10 magnification at 0 and 24 h. Wound closure was measured using ImageJ MRI_Wound_Healing_Tool.ijm.
Flow cytometry
Tissues were processed using methods described previously [30]. Two lakhs cells were stained for 30 min (1:100) to antibodies: CCR2-PE (RnD Systems cat no. FAB5538P), CD24-Brilliant Violet (BD Pharmingen cat no. 562563), F4/80-PE (Serotec cat no. MCA497PE), CD69-FITC (Biolegend cat no.104505), CD4-PE-Cy5 (BD Pharmingen cat no. 553654), CD8-APC (Biolegend cat no. 100711), CD11b-APC-Cy7 (BD Pharmingen cat no. 557657), human anti-CD40-FITC (R&D Systems, cat no. MAB6321) or murine anti-CD40-FITC (BD Biosciences cat no. 12040181). Cells were analyzed using a BD LSR II flow cytometer. Compensation controls were performed to minimize spectral overlap artifacts. Isotype control antibodies were utilized for background correction.
Histology/Immunohistochemistry
Tissues were processed, stained by H&E, or immunostained for: cleaved caspase-3, PCNA, Von Willebrand Factor 8, CCL2, CCR2 and CD154 using procedures described [30, 46]. Specificity of CCL2 antibodies was tested through immunostaining of CCL2-deficient 4T1 mammary tumors (Supplemental Fig. 11). CD154 expression was detected using anti-CD154 (BD Biosciences cat no.552559) and rabbit biotinylated antibodies bound to streptavidin-peroxidase (Vector Laboratories, cat no.PK-6100). Five images/field at ×20 magnification were captured using the EVOS FL Auto Imaging System. Expression was quantified using ImageJ as described [76].
Immunofluorescence
Cells were fixed in neutral formalin buffer and permeabilized with methanol. 3D cultures or carcinoma tissues were immunostained using procedures described [77]. Samples were incubated with antibodies (1:100) to: CCR2-FITC (Biolegend cat no. 150607), F4/80 (Abcam, cat no. ab6640), CD8 (Biolegend cat no.100715), CD69-FITC (Biolegend cat no.104505), CD11b-APC-Cy7 (BD Pharmingen cat. no.557657), Arginase-1 (Santa Cruz Biotechnology cat no.sc20150), PyVmT (Abcam cat no. 73989) or PCNA (Biolegend cat no.307902) at 4 °C overnight. CD8 and F4/80 were detected with anti-rat-Alexa Fluor-568 (Invitrogen cat no.A-11077). Arginase I was detected with anti-rabbit-Alexa Fluor-488 (Invitrogen cat no.A-11034). PyVmT was detected with anti-mouse Alexa Fluor-568 (Invitrogen cat no. A-11004). PCNA was detected with anti-mouse-Alexa Fluor-488 (Invitrogen cat no. A-11001). Samples were counterstained with DAPI and mounted using PBS:glycerol. Five images/field were captured at ×10 magnification using the EVOS FL Auto Imaging System (Invitrogen). Expression was quantified by ImageJ as described [76].
Cytokine/growth-factor array
Two lakhs MDA-MB-231 cells were seeded in six-well plates for 24 h, and transfected with TAT peptides complexed to 1 μg siRNAs as described [30]. Cells were incubated with 3 ml of serum-free DMEM for 24 h. Media was analyzed by Proteome Profiler Human Cytokine XL array containing 102 targets in duplicate (R&D Biosystems cat no. ARY022). Luminescent spots were imaged by film exposure. Mean pixel density was extracted in ImageJ via GelAnalyzer plugin.
Immunoblot
The following chemicals were obtained: Cyclosporin A (TCI Chemical cat no.C2408), Bay11-7082 (Enzo cat no. E1279), PP2 (Tocris cat no. 1407), Gö 6983 (Cayman Chemical cat no. 13311), INCB3284 (Cayman Chemical cat no. 1163). Two lakhs cells seeded in 24-well plates were treated with/without inhibitors. Protein cell lysates were prepared and immunoblotted using procedures described [46]. Membranes were blotted with antibodies to: CD154 (Santa Cruz Biotechnology cat no. sc978), NFATc1 (Biolegend, cat no.649601), phospho-IKBa (ser32) (Cell Signaling Technology cat no. 9246), IΚBα or ACTIN (Sigma, cat no. A5441).
ELISA
Condition medium were analyzed for human CCL2 (Peprotech, cat no. 900-K31) or murine CCL2 (Peprotech, cat no. 900-K126) using commercial ELISA protocols. For CD154 ELISA, high-protein binding plates were coated with 1 μg/ml capture CD154 antibody (BD Bioscience cat no.552559) overnight. Plates were blocked in PBS/10% BSA for 2 h. Samples were diluted 1:1 in PBS/1% BSA/Tween-20 and plated at 100 μl/well for 24 h at 4 °C. Wells were incubated with 0.5 μg/ml biotinylated anti-CD154 (BD Biosciences, cat no. 552560) for 2 h, followed by streptavidin-peroxidase (Vector Laboratories, cat no. PK-6100) for 30 min. Reactions were catalyzed using tetramethylbenzidine substrate (Pierce cat no. 34028) and read at OD450nM using a BioTek Microplate Reader.
Endothelial branching/recruitment assay
Both assays were conducted using procedures described previously [78]. Briefly, for the branching assay, 10,000 endothelial cells were seeded/well in 96 Matrigel coated plates and incubated for 24 h. 4 images/well at 10× magnification were captured using the EVOS FL Auto Imager. Branchpoints were manually counted. Briefly, for the recruitment assay, 75,000 endothelial cells were plated on the topside of Matrigel coated transwells and incubated for 8 h. Cells on the topside were removed by cotton swab. Cells on the underside were labeled with DAPI. 4 images/well were captured at 10x magnification using the EVOS FL Auto imager and measured by ImageJ.
Macrophage infiltration assay
TheraKan devicesTM were manufactured by Fennik Life Sciences (Kansas City, KS). Macrophage infiltration was determined using methods described [30]. Briefly, 100,000 breast cancer cells were cultured in each device containing 275 μl rat tail collagen (Corning cat no. 354231). Devices were incubated with 500,000 Raw 264.7 mCherry+ cells in six well dishes. Devices were imaged at ×10 magnification using an EVOS FL Auto imager for up to 48 h.
For live imaging assays, PyVmT cells were transfected with siRNA complexes for 48 h, and then resuspended in a 1:1 mixture of growth-factor-reduced Matrigel to rat tail collagen at 1 × 106 cells/ml on ice. Forty microliters matrices was spotted in 24-well plates and incubated at 37 °C for 30 min. Two lakhs raw macrophages was added per well, with/without 100 ng/ml CCL2 or 1 μg/ml CD154. Plates were imaged at 15 min intervals for up to 18 h using an EVOS FL Auto imager. Images were analyzed with Fiji TrackMate plugin, with >100 cells tracked per group.
Biospecimens
Tissue microarrays comprised of de-identified 35 invasive ductal carcinoma (n = 4 stage 1, n = 28 stage 2, n = 6 stage 3), 35 matching normal adjacent breast tissues, and 5 normal breast tissue samples in duplicate cores (1.5 mm diameter) were obtained from US biomax (cat no. BR802b). Tissue microarrays of 33 invasive ductal carcinoma samples and 40 normal breast tissues were obtained by the Biospecimen core repository and characterized previously [79]. All cases were from female patients; median age = 45.
Dataset analysis
mRNA breast cancer datasets were accessed on www.kmplot.com on October 12, 2018 to examine CD154 gene expression (207892_at) with the following parameters: relapse free survival, basal = 879, luminal A (n = 2504), Luminal B (n = 1425), HER2+ (n = 335).
Statistical analysis
In vitro experiments were conducted with triplicate samples and performed a minimum of three times. For animal experiments, sample size analysis was based off studies of tumor volume using PS Power and Sample Size Program Ver3.0. It indicated that we needed a minimum of n = 6/group to reach a sufficient power of 0.90 with alpha = 0.05. Statistical analysis was performed using Graphpad Prism software. Kolmogorov–Smirnov test of normality distribution was performed. Kruskall–Wallis test with Dunn’s post-hoc comparison, Mann–Whitney or Spearman correlation tests were used for data with nonnormal distribution. The Student’s two-tailed t test was used for two groups of data normally distributed. One way ANOVA with Bonferroni’s post-hoc comparison was used for >2 groups of data normally distributed. Alternatively, data were analyzed using Brown–Forsythe test with Game–Howell post-hoc comparison. Data met the assumptions of variance between groups. Estimate of variation within data groups for in vitro and in vivo experiments was <25%.*p < 0.05, **p < 0.01, ***p < 0.001, ns not significant. Data are presented as Mean ± SEM or min–max values with upper and lower quartile ranges.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. 2018;68:394–424.
Ribatti D. The concept of immune surveillance against tumors. The first theories. Oncotarget. 2017;8:7175–80.
Al-Saleh K, Abd El-Aziz N, Ali A, Abozeed W, Abd El-Warith A, Ibraheem A, et al. Predictive and prognostic significance of CD8(+) tumor-infiltrating lymphocytes in patients with luminal B/HER 2 negative breast cancer treated with neoadjuvant chemotherapy. Oncol Lett. 2017;14:337–44.
Liu S, Lachapelle J, Leung S, Gao D, Foulkes WD, Nielsen TO. CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res. 2012;14:R48.
Seo AN, Lee HJ, Kim EJ, Kim HJ, Jang MH, Lee HE, et al. Tumour-infiltrating CD8+ lymphocytes as an independent predictive factor for pathological complete response to primary systemic therapy in breast cancer. Br J Cancer. 2013;109:2705–13.
Richardsen E, Uglehus RD, Johnsen SH, Busund LT. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res. 2015;35:865–74.
Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117:1583–91.
Zhao X, Qu J, Sun Y, Wang J, Liu X, Wang F, et al. Prognostic significance of tumor-associated macrophages in breast cancer: a meta-analysis of the literature. Oncotarget. 2017;8:30576–86.
Georganaki M, van Hooren L, Dimberg A. Vascular targeting to increase the efficiency of immune checkpoint blockade in cancer. Front Immunol. 2018;9:3081.
Law AM, Lim E, Ormandy CJ, Gallego-Ortega D. The innate and adaptive infiltrating immune systems as targets for breast cancer immunotherapy. Endocr Relat Cancer. 2017;24:R123–R144.
Balkwill FR. The chemokine system and cancer. J Pathol. 2012;226:148–57.
Palomino DC, Marti LC. Chemokines and immunity. Einstein. 2015;13:469–73.
Ridiandries A, Tan JTM, Bursill CA. The role of chemokines in wound healing. Int J Mol Sci. 2018;19, pii: E3217.
Kurihara T, Bravo R. Cloning and functional expression of mCCR2, a murine receptor for the C-C chemokines JE and FIC. J Biol Chem. 1996;271:11603–7.
Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–26.
Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. 2016;7:28697–710.
Yao M, Yu E, Staggs V, Fan F, Cheng N. Elevated expression of chemokine C-C ligand 2 in stroma is associated with recurrent basal-like breast cancers. Modern Pathol. 2016;29:810–23.
Borsig L, Wolf MJ, Roblek M, Lorentzen A, Heikenwalder M. Inflammatory chemokines and metastasis-tracing the accessory. Oncogene. 2014;33:3217–24.
Brummer G, Acevedo DS, Hu Q, Portsche M, Fang WB, Yao M, et al. Chemokine signaling facilitates early-stage breast cancer survival and invasion through fibroblast-dependent mechanisms. Mol Cancer Res. 2018;16:296–308.
Yao M, Fang W, Smart C, Hu Q, Huang S, Alvarez N, et al. CCR2 chemokine receptors enhance growth and cell-cycle progression of breast cancer cells through SRC and PKC activation. Mol Cancer Res. 2019;17:604–17.
Guy C, Cardiff R, Muller W. Induction of mammary tumors by expression a polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–61.
Behbod F, Kittrell FS, LaMarca H, Edwards D, Kerbawy S, Heestand JC, et al. An intraductal human-in-mouse transplantation model mimics the subtypes of ductal carcinoma in situ. Breast Cancer Res. 2009;11:R66.
Scribner KC, Behbod F, Porter WW. Regulation of DCIS to invasive breast cancer progression by Singleminded-2s (SIM2s). Oncogene. 2012;32:2631–9.
Baoum A, Xie SX, Fakhari A, Berkland C. "Soft" calcium crosslinks enable highly efficient gene transfection using TAT peptide. Pharm Res. 2009;26:2619–29.
Khondee S, Baoum A, Siahaan TJ, Berkland C. Calcium condensed LABL-TAT complexes effectively target gene delivery to ICAM-1 expressing cells. Mol Pharm. 2011;8:788–98.
Pickel L, Matsuzuka T, Doi C, Ayuzawa R, Maurya DK, Xie SX, et al. Overexpression of angiotensin II type 2 receptor gene induces cell death in lung adenocarcinoma cells. Cancer Biol Ther. 2010;9:277–85.
Fang WB, Yao M, Jokar I, Alhakamy N, Berkland C, Chen J, et al. The CCL2 chemokine is a negative regulator of autophagy and necrosis in luminal B breast cancer cells. Breast Cancer Res Treat. 2015;150:309–20.
Hassan GS, Stagg J, Mourad W. Role of CD154 in cancer pathogenesis and immunotherapy. Cancer Treat Rev. 2015;41:431–40.
Rakhmilevich AL, Alderson KL, Sondel PM. T-cell-independent antitumor effects of CD40 ligation. Int Rev Immunol. 2012;31:267–78.
Fang WB, Yao M, Brummer G, Acevedo D, Alhakamy N, Berkland C. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget. 2016;7:49349–67.
Cron RQ. CD154 transcriptional regulation in primary human CD4 T cells. Immunologic Res. 2003;27:185–202.
Franca TT, Barreiros LA, Al-Ramadi BK, Ochs HD, Cabral-Marques O, Condino-Neto A. CD40 ligand deficiency: treatment strategies and novel therapeutic perspectives. Expert Rev Clin Immunol. 2019;15:529–40.
Crist SA, Sprague DL, Ratliff TL. Nuclear factor of activated T cells (NFAT) mediates CD154 expression in megakaryocytes. Blood. 2008;111:3553–61.
Srahna M, Remacle JE, Annamalai K, Pype S, Huylebroeck D, Boogaerts MA, et al. NF-kappaB is involved in the regulation of CD154 (CD40 ligand) expression in primary human T cells. Clin Exp Immunol. 2001;125:229–36.
Boss V, Abbott KL, Wang XF, Pavlath GK, Murphy TJ. The cyclosporin A-sensitive nuclear factor of activated T cells (NFAT) proteins are expressed in vascular smooth muscle cells. Differential localization of NFAT isoforms and induction of NFAT-mediated transcription by phospholipase C-coupled cell surface receptors. J Biol Chem. 1998;273:19664–71.
Meyer S, Kohler NG, Joly A. Cyclosporine A is an uncompetitive inhibitor of proteasome activity and prevents NF-kappaB activation. FEBS Lett. 1997;413:354–8.
Mori N, Yamada Y, Ikeda S, Yamasaki Y, Tsukasaki K, Tanaka Y, et al. Bay 11-7082 inhibits transcription factor NF-kappaB and induces apoptosis of HTLV-I-infected T-cell lines and primary adult T-cell leukemia cells. Blood. 2002;100:1828–34.
Martinez-Martinez S, Gomez del Arco P, Armesilla AL, Aramburu J, Luo C, Rao A, et al. Blockade of T-cell activation by dithiocarbamates involves novel mechanisms of inhibition of nuclear factor of activated T cells. Mol Cell Biol. 1997;17:6437–47.
Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80.
Kim JH, Kim K, Jin HM, Song I, Youn BU, Lee SH, et al. Negative feedback control of osteoclast formation through ubiquitin-mediated down-regulation of NFATc1. J Biol Chem. 2010;285:5224–31.
Moscat J, Diaz-Meco MT, Rennert P. NF-kappaB activation by protein kinase C isoforms and B-cell function. EMBO Rep. 2003;4:31–36.
Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, et al. Protein kinase C theta affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J Exp Med. 2003;197:1525–35.
Brodmerkel CM, Huber R, Covington M, Diamond S, Hall L, Collins R, et al. Discovery and pharmacological characterization of a novel rodent-active CCR2 antagonist, INCB3344. J Immunol. 2005;175:5370–8.
Xue CB, Feng H, Cao G, Huang T, Glenn J, Anand R, et al. Discovery of INCB3284, a potent, selective, and orally bioavailable hCCR2 antagonist. ACS Med Chem Lett. 2011;2:450–4.
Brandvold KR, Steffey ME, Fox CC, Soellner MB. Development of a highly selective c-Src kinase inhibitor. ACS Chem Biol. 2012;7:1393–8.
Fang WB, Jokar I, Zou A, Lambert D, Dendukuri P, Cheng N. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J Biol Chem. 2012;287:36593–608.
Dusaban SS, Purcell NH, Rockenstein E, Masliah E, Cho MK, Smrcka AV, et al. Phospholipase C epsilon links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc Natl Acad Sci USA. 2013;110:3609–14.
Slone S, Anthony SR, Wu X, Benoit JB, Aube J, Xu L, et al. Activation of HuR downstream of p38 MAPK promotes cardiomyocyte hypertrophy. Cell Signal. 2016;28:1735–41.
Yang M, He RL, Benovic JL, Ye RD. beta-Arrestin1 interacts with the G-protein subunits beta1gamma2 and promotes beta1gamma2-dependent Akt signalling for NF-kappaB activation. Biochem J. 2009;417:287–96.
Barnhart B, Kosinski PA, Wang Z, Ford GS, Kiledjian M, Covey LR. Identification of a complex that binds to the CD154 3' untranslated region: implications for a role in message stability during T cell activation. J Immunol. 2000;165:4478–86.
Ford GS, Barnhart B, Shone S, Covey LR. Regulation of CD154 (CD40 ligand) mRNA stability during T cell activation. J Immunol. 1999;162:4037–44.
Hamilton BJ, Genin A, Cron RQ, Rigby WF. Delineation of a novel pathway that regulates CD154 (CD40 ligand) expression. Mol Cell Biol. 2003;23:510–25.
Yoshimura T. The production of monocyte chemoattractant protein-1 (MCP-1)/CCL2 in tumor microenvironments. Cytokine. 2017;98:71–78.
Lee EG, Mickle-Kawar BM, Gallucci RM. IL-6 deficiency exacerbates skin inflammation in a murine model of irritant dermatitis. J Immunotoxicol. 2013;10:192–200.
Rollins BJ, Yoshimura T, Leonard EJ, Pober JS. Cytokine-activated human endothelial cells synthesize and secrete a monocyte chemoattractant, MCP-1/JE. Am J Pathol. 1990;136:1229–33.
Tylaska LA, Boring L, Weng W, Aiello R, Charo IF, Rollins BJ, et al. Ccr2 regulates the level of MCP-1/CCL2 in vitro and at inflammatory sites and controls T cell activation in response to alloantigen. Cytokine. 2002;18:184–90.
Stuelten CH, Parent CA, Montell DJ. Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat Rev Cancer. 2018;18:296–312.
Clawson GA, Matters GL, Xin P, Imamura-Kawasawa Y, Du Z, Thiboutot DM, et al. Macrophage-tumor cell fusions from peripheral blood of melanoma patients. PLoS ONE 2015;10:e0134320.
Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–56.
Pearson LL, Castle BE, Kehry MR. CD40-mediated signaling in monocytic cells: up-regulation of tumor necrosis factor receptor-associated factor mRNAs and activation of mitogen-activated protein kinase signaling pathways. Int Immunol. 2001;13:273–83.
Qian Y, Zhao Z, Jiang Z, Li X. Role of NF kappa B activator Act1 in CD40-mediated signaling in epithelial cells. Proc Natl Acad Sci USA. 2002;99:9386–91.
Yao M, Brummer G, Acevedo D, Cheng N. Cytokine regulation of metastasis and tumorigenicity. Adv Cancer Res. 2016;132:265–367.
Hida K, Maishi N, Torii C, Hida Y. Tumor angiogenesis-characteristics of tumor endothelial cells. Int J Clin Oncol. 2016;21:206–12.
Li T, Kang G, Wang T, Huang H. Tumor angiogenesis and anti-angiogenic gene therapy for cancer. Oncol Lett. 2018;16:687–702.
Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018;9:115.
Arasanz H, Lacalle A, Lecumberri MJ, de Lascoiti AF, Blanco-Luquin I, GatoCanas M, et al. Immunotherapy in malignant melanoma: recent approaches and new perspectives. Melanoma Manag. 2017;4:39–48.
Blum S, Martins F, Lubbert M. Immunotherapy in adult acute leukemia. Leuk Res. 2017;60:63–73.
Du L, Herbst RS, Morgensztern D. Immunotherapy in lung cancer. Hematol Oncol Clin North Am. 2017;31:131–41.
Vonderheide RH, LoRusso PM, Khalil M, Gartner EM, Khaira D, Soulieres D, et al. Tremelimumab in combination with exemestane in patients with advanced breast cancer and treatment-associated modulation of inducible costimulator expression on patient T cells. Clin Cancer Res. 2010;16:3485–94.
Lefebvre E, Gottwald M, Lasseter K, Chang W, Willett M, Smith PF, et al. Pharmacokinetics, safety, and CCR2/CCR5 antagonist activity of cenicriviroc in participants with mild or moderate hepatic impairment. Clin Transl Sci. 2016;9:139–48.
Linehan D, Smith Noel M, Hezel AF, Wang-Gillam A, Eskens F, Sleijfer S, et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: Correlation with blood monocyte counts. J Clin Oncol. 2018;36:92.
Valdez KE, Fan F, Smith W, Allred DC, Medina D, Behbod F. Human primary ductal carcinoma in situ (DCIS) subtype-specific pathology is preserved in a mouse intraductal (MIND) xenograft model. J Pathol. 2011;225:565–73.
Fang WB, Jokar I, Chytil A, Moses HL, Abel T, Cheng N. Loss of one Tgfbr2 allele in fibroblasts promotes metastasis in MMTV: polyoma middle T transgenic and transplant mouse models of mammary tumor progression. Clin Exp Metastasis. 2011;28:351–66.
Brantley-Sieders DM, Dunaway CM, Rao M, Short S, Hwang Y, Gao Y, et al. Angiocrine factors modulate tumor proliferation and motility through EphA2 repression of Slit2 tumor suppressor function in endothelium. Cancer Res. 2011;71:976–87.
Hembruff SL, Jokar I, Yang L, Cheng N. Loss of transforming growth factor-beta signaling in mammary fibroblasts enhances CCL2 secretion to promote mammary tumor progression through macrophage-dependent and -independent mechanisms. Neoplasia. 2010;12:425–33.
Yao M, Smart C, Hu Q, Cheng N. Continuous delivery of neutralizing antibodies elevate CCL2 levels in mice bearing MCF10CA1d breast tumor xenografts. Transl Oncol. 2017;10:734–43.
Artym VV, Matsumoto K. Imaging cells in three-dimensional collagen matrix. Curr Protoc Cell Biol. Chapter 10, 2010.
Youngblood V, Wang S, Song W, Walter D, Hwang Y, Chen J, et al. Elevated Slit2 activity impairs VEGF-induced angiogenesis and tumor neovascularization in EphA2-deficient endothelium. Mol Cancer Res. 2015;13:524–37.
Connor CS, Kimler BF, Mammen JM, McGinness MK, Wagner JL, Alsop SM, et al. Impact of neoadjuvant chemotherapy on axillary nodal involvement in patients with clinically node negative triple negative breast cancer. J Surg Oncol. 2015;111:198–202.
Funding
This work was supported by funds from the American Cancer Society (RSG-13-182-01-CSM), Susan G. Komen Foundation (CCR13261859), and by the National Institutes of Health (R01CA172764) to NC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
NC and WF are co-founders of Fennik Life Sciences, which is commercializing the TheraKanTM device.
Ethics statement
Written informed consent for tissue collection was obtained by the Biospecimen Core Repository according to Human Research Protection Program at the University of Kansas Medical Center (KUMC). Tissue samples were de-identified by the Biospecimen Core Repository prior to distribution. Medical records were used in compliance with KUMC and National Cancer Institute regulations. These regulations are aligned with the World Medical Association Declaration of Helsinki. Animals were maintained at KUMC in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care. Animal experiments were performed under an approved Institutional Animal Care and Use Committee protocol.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Brummer, G., Fang, W., Smart, C. et al. CCR2 signaling in breast carcinoma cells promotes tumor growth and invasion by promoting CCL2 and suppressing CD154 effects on the angiogenic and immune microenvironments. Oncogene 39, 2275–2289 (2020). https://doi.org/10.1038/s41388-019-1141-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-019-1141-7
- Springer Nature Limited
This article is cited by
-
Targeting macrophages: a novel treatment strategy in solid tumors
Journal of Translational Medicine (2022)
-
Role of CC-chemokine ligand 2 in gynecological cancer
Cancer Cell International (2022)
-
CC chemokine receptor 2 (CCR2) expression promotes diffuse large B-Cell lymphoma survival and invasion
Laboratory Investigation (2022)
-
Targeting CCL2-CCR4 axis suppress cell migration of head and neck squamous cell carcinoma
Cell Death & Disease (2022)
-
M2 macrophage-induced lncRNA PCAT6 facilitates tumorigenesis and angiogenesis of triple-negative breast cancer through modulation of VEGFR2
Cell Death & Disease (2020)