
Abstract
The assembly of the death-inducing signaling complex (DISC) and death effector domain (DED) filaments at CD95/Fas initiates extrinsic apoptosis. Procaspase-8 activation at the DED filaments is controlled by short and long c-FLIP isoforms. Despite apparent progress in understanding the assembly of CD95-activated platforms and DED filaments, the detailed molecular mechanism of c-FLIP action remains elusive. Here, we further addressed the mechanisms of c-FLIP action at the DISC using biochemical assays, quantitative mass spectrometry, and structural modeling. Our data strongly indicate that c-FLIP can bind to both FADD and procaspase-8 at the DED filament. Moreover, the constructed in silico model shows that c-FLIP proteins can lead to the formation of the DISCs comprising short DED filaments as well as serve as bridging motifs for building a cooperative DISC network, in which adjacent CD95 DISCs are connected by DED filaments. This network is based on selective interactions of FADD with both c-FLIP and procaspase-8. Hence, c-FLIP proteins at the DISC control initiation, elongation, and composition of DED filaments, playing the role of control checkpoints. These findings provide new insights into DISC and DED filament regulation and open innovative possibilities for targeting the extrinsic apoptosis pathway.
Similar content being viewed by others


Introduction
CD95/Fas is one of the best studied death receptors (DRs) that trigger the extrinsic apoptotic pathway. Stimulation of CD95 by CD95L results in the formation of a death-inducing signaling complex (DISC) which comprises CD95 and Death Effector Domain (DED)-containing proteins (denoted hereafter as DED proteins): FADD, procaspase-8/10 as well as c-FLIP [1, 2]. Procaspase-8a/b is activated in the DED filaments formed via DED interactions at the DISC [3,4,5]. The DED filament assembly is based on the so-called type I, II, and III interactions of DEDs [4]. Activation of procaspase-8 at the DED filaments leads to induction of the caspase cascade followed by the demolition of the cell.
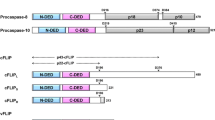
The major regulation of procaspase-8a/b activation at the DED filaments is mediated by c-FLIP proteins [6]. One long and two short c-FLIP isoforms, named Long (L), Short (S), and Raji (R), i.e., c-FLIPL, c-FLIPS, and c-FLIPR, have been reported to be present at the DISC [6, 7]. All three isoforms possess two DED domains in their N-terminal region (DED1 and DED2, 1–73, and 92–170, respectively), while c-FLIPL also has catalytically inactive caspase-like domains (p20 and p12) in its C-terminal region. c-FLIPL at the DISC can act both in a pro- as well as in an anti-apoptotic manner [8,9,10,11]. The pro-apoptotic function of c-FLIPL is mediated by the formation of catalytically active procaspase-8/c-FLIPL heterodimers in which the active center of procaspase-8 is stabilized by c-FLIPL [8, 9, 12]. Moreover, the proapoptotic role of c-FLIPL strongly depends upon its amounts at the DISC and subsequently upon the number of the formed procaspase-8/c-FLIPL heterodimers. Intermediate levels of c-FLIPL lead to the formation of a sufficient number of procaspase-8/c-FLIPL heterodimers resulting in the promotion of caspase-8 activity [13]. However, upon high concentrations of c-FLIPL, it plays only an antiapoptotic role due to the replacement of procaspase-8 from the DED filament [13]. Short c-FLIP isoforms, c-FLIPS and c-FLIPR, at the DISC act in the antiapoptotic manner. It was reported that c-FLIPS and c-FLIPR block caspase-8 activation by interrupting the procaspase-8 chains/filaments at the DISC [14]. In contrast, it was reported that short c-FLIP isoforms also incorporate into DED filaments and inhibit caspase-8 activation by forming inactive heterodimers [4, 15].
Several models of c-FLIP binding to the DISC have been reported. The initial view on c-FLIP binding to the DISC was that c-FLIP binds directly to the DED of FADD and thereby competes with procaspase-8 for binding to the DISC [16]. Subsequent studies have provided evidence for a strong requirement for both DED1 [17] as well as DED2 of c-FLIP for its binding to FADD [17, 18]. Furthermore, it has been reported that c-FLIP interacts via α2/α5 interface of its DED2 with the α1/α4 region of FADD DED, while procaspase-8 uses the other binding site for docking to FADD located at the α2/α5 region of FADD DED [19]. Recently, the hierarchical assembly model of the DISC has been reported which involves cooperative binding of c-FLIP to FADD in a procaspase-8 dependent manner, suggesting that c-FLIP requires procaspase-8 for binding to the DISC [14]. Finally, according to the recent structure obtained via cryoEM analysis of the DED filament, c-FLIP proteins incorporate into filaments and commingle with procaspase-8 [4], which suggests yet another mode of c-FLIP-DISC interaction. Taken together, there seem to be several ways of c-FLIP incorporation into DED filaments. Hence, the exact molecular mechanisms of c-FLIP action in DED filaments still require detailed analysis.
In this work we took advantage of HeLa cell lines overexpressing different c-FLIP isoforms to address the mechanisms of c-FLIP action at the DISC. Quantitative mass spectrometry analysis of CD95 DISCs assembled in these cells uncovered that overexpression of c-FLIP isoforms leads to the formation of shorter DED filaments and an increase of their amount at the DISC. Structural modeling of the DED filaments supported by quantitative mass spectrometry allowed to clarify the observed effects and suggested a model of с-FLIP control of DED filament assembly at the DISC.
Results
c-FLIP is directly recruited to FADD in the absence of caspase-8
To get more insight into the mechanisms of the DISC assembly, we used T leukemia Jurkat-caspase-8 deficient cells (Jurkat C8−/−). These cells lack caspase-8 and are resistant to CD95-induced apoptosis [20,21,22]. Jurkat C8−/− cells were stimulated with CD95L followed by immunoprecipitation (IP) of the DISC. In the DISC immunoprecipitation (DISC-IP) from Jurkat C8−/− cells all core components of the DISC were detected: CD95, FADD, procaspase-10d/a, and both c-FLIP isoforms (Fig. 1a). This was consistent with the expression of the core DISC components in Jurkat C8−/− cells (Supplementary Fig. 1) and, correspondingly, their apparent ability to form a complex at CD95 even in the absence of procaspase-8 (Fig. 1a). Strikingly, a prominent recruitment of both c-FLIP isoforms to the DISC has been observed (Fig. 1a). Furthermore, CD95L treatment of Jurkat C8−/−cells did not lead to procaspase-10 cleavage or effector caspase-3 activation (Fig. 1b, right). Thus, procaspase-10 did not activate caspase-3 and prime apoptosis in the absence of procaspase-8 in Jurkat-C8−/− cells, even though procaspase-10 is an initiator caspase and can potentially trigger apoptosis via dimerization at the DISC. The absence of apoptosis priming by procaspase-10 in Jurkat-C8−/− cells is in accordance with previous reports [23] and might be explained by low amounts of procaspase-10 at the DISC [15]. As expected, this was in contrast to Jurkat cells with caspase-8 expression, in which CD95 stimulation led to the efficient DISC formation as well as caspase-3 and PARP1 cleavage (Fig. 1a, b). Accordingly, the core components of the CD95 DISC: CD95, FADD, procaspase-8/10 and c-FLIP proteins were detected in the DISC-IP from Jurkat cells along with cleavage products of procaspase-8a/b, procaspase-10d/a and c-FLIPL indicating DISC assembly (Fig. 1a, b). The DISC assembly in Jurkat cells led to the activation of a caspase cascade in contrast to Jurkat C8−/− cells. Moreover, DISC-IPs from Jurkat C8−/− cells indicated that both c-FLIP isoforms might be directly recruited to FADD in the absence of procaspase-8 which asks for further investigation of the role of c-FLIP isoforms at the DED filament.

c-FLIP is recruited to the CD95 DISC in Jurkat-caspase-8 deficient cells. a Jurkat (wt) and Jurkat-caspase-8 deficient (C8−/−) cells were stimulated with 650 ng/mL CD95L for 20 min. CD95 DISC-IPs were performed using anti-APO-1 (anti-CD95) antibodies and analyzed by Western Blot along with corresponding total cell lysates (Inputs). Actin was used as a loading control. BC-control IP with ‘beads-only’, without the addition of antibodies. One representative experiment out of five is shown. b Jurkat and Jurkat-caspase-8 deficient (C8−/−) cells were stimulated with 100 ng/mL CD95L from one to 3 h. Total cell lysates were analyzed by Western Blot and probed for the indicated proteins. Actin was used as a loading control. Abbreviations: l.e.-long exposure; s.e.-short exposure; casp-8/10/3-procaspase-8/10/3. One representative experiment out of two is shown
The generated c-FLIP-overexpressing HeLa cells form CD95 DISC but show different pattern of caspase-8 activity
To further study the role of c-FLIP proteins we have generated HeLa cell lines stably overexpressing both c-FLIP isoforms, c-FLIPL and c-FLIPS, named HeLa-CD95-FL and HeLa-CD95-FS, respectively (Supplementary Fig. 2). Both cell lines were characterized by a high overexpression of c-FLIP proteins (Supplementary Fig. 3a, b). In particular, the expression of c-FLIPS in HeLa-CD95-FS cells was thousand times higher than in parental HeLa-CD95 cells, while c-FLIPL was 630 times higher expressed in HeLa-CD95-FL cells compared with parental HeLa-CD95 cells (Supplementary Fig. 3a, b). Importantly, the surface expression of CD95 was not changed in c-FLIP-overexpressing cell lines compared with the parental HeLa-CD95 cells (Fig. 2a).

Overexpression of long and short c-FLIP isoforms does not fully suppress caspase activation. a HeLa-CD95, HeLa-CD95-FL, and HeLa-CD95-FS cells were stained with FITC-labeled anti-CD95 antibodies followed by imaging flow cytometry analysis. Shown is one representative result out of two for HeLa-CD95 and HeLa-CD95-FL cells. b HeLa-CD95 cells, HeLa-CD95-FL cells, and HeLa-CD95-FS cells were stimulated with 125 ng/mL CD95L for indicated time intervals. Cell viability was measured using the Cell Titer-Glo®-Luminescent Cell Viability Assay estimating the ATP content. Mean and standard deviation of three independent experiments are shown; *p < 0,05. (c + d) HeLa-CD95, HeLa-CD95-FL, and HeLa-CD95-FS cells were stimulated with CD95L (250 ng/mL) for indicated time intervals. Total cellular lysates were analyzed by Western Blot using the indicated antibodies. Actin and GAPDH were used as loading controls. One representative experiment out of three independent experiments is shown
Upon high expression, both c-FLIPL and c-FLIPS down-modulate CD95-mediated caspase activation and apoptosis [6, 24]. In line with this, the time-dependent analysis of cell viability loss of HeLa-CD95, HeLa-CD95-FL, and HeLa-CD95-FS cells upon CD95L stimulation has shown that HeLa-CD95 cells are more sensitive towards CD95L treatment compared with HeLa-CD95-FL and HeLa-CD95-FS cells (Fig. 2b). Importantly, as shown in the Fig. 2a, the surface expression of CD95 was similar in HeLa-CD95, HeLa-CD95-FL, and HeLa-CD95-FS cells indicating that differences in response are attributed to c-FLIP isoforms.
Furthermore, the expression of the intracellular core DISC components: FADD, procaspase-8/10 was also not altered upon c-FLIPL/S overexpression in HeLa-CD95-FL and HeLa-CD95-FS cells (Fig. 2c, d). However, caspase cleavage and activation were largely influenced by c-FLIPL/S overexpression in HeLa-CD95-FL and HeLa-CD95-FS compared with HeLa-CD95 cells (Fig. 2c, d). Importantly, two c-FLIP isoforms had different effects on caspase cascade. In CD95L-treated HeLa-CD95-FL cells the processing of procaspases-8, -10, -3 and PARP1 was largely diminished compared with CD95L-treated HeLa-CD95 cells (Fig. 2c). In particular, the cleavage products p43/p41 and p18 of procaspase-8a/b as well as p19/p17 of caspase-3 were detected in HeLa-CD95–FL cells with much lower intensity compared with HeLa-CD95 cells (Fig. 2c). Overexpression of c-FLIPS almost completely blocked CD95L-induced processing of caspases and their substrates. This inhibition was much stronger than in c-FLIPL-overexpressing cells. Only minor cleavage events of PARP1 and procaspase-8a/b were detected in HeLa-CD95-FS cells upon CD95L stimulation (Fig. 2d). Importantly, expression of the core DISC components: procaspase-8, procaspase-10, and FADD was not changed in HeLa-CD95-FL and HeLa-CD95-FS compared with HeLa-CD95 cells, which supports the observation that differences in apoptotic response of these cells are solely based on the properties of c-FLIPL versus c-FLIPS (Fig. 2c, d).
In accordance with cell viability and caspase activation data, apoptosis induction was decreased in HeLa-CD95-FL compared with HeLa-CD95 cells, which was monitored by Annexin V (A)/SytoxOrange (S) staining in combination with imaging flow cytometry analysis (Fig. 3a, b). Furthermore, HeLa-CD95–FS cells also demonstrated a low sensitivity towards CD95L-induced apoptosis at later time points (Fig. 3c).

c-FLIP isoforms differentially modulate CD95L-induced apoptosis at the DISC. HeLa-CD95 (a), HeLa-CD95-FL (b) and HeLa-CD95-FS (c) cells were stimulated with 125 ng/mL CD95L for indicated time intervals. Cell death was measured by Annexin-V-FITC (A) and SytoxOrange (S) staining using imaging flow cytometry. Mean and standard deviation are shown (n = 3). Three representative pictures for each cell condition generated by AMNIS® are presented. HeLa-CD95 cells (d), HeLa-CD95-FL cells (e) and HeLa-CD95-FS (f) cells were stimulated with 125 ng/mL (d) or 250 ng/mL (e + f) CD95L from 1 to 3 h. CD95 DISC-IPs were performed using anti-APO-1 (anti-CD95) antibodies and analyzed by Western Blot analysis for the indicated proteins. Actin was used as a loading control. Total cellular lysates of the stimulated conditions are shown (Input). Abbreviations: l.e.-long exposure; s.e.-short exposure; casp-8/10/3-procaspase-8/10/3; BC-control IP with ‘beads-only’, without the addition of antibodies. One representative experiment out of three is shown
Despite their low sensitivity to CD95L stimulation, in both c-FLIP overexpressing cell lines caspase processing was observed implicating that c-FLIP isoforms do not fully block caspase-8 activation (Fig. 2c, d). Moreover, c-FLIPL blocked caspase-8 activation to a lesser extent compared with c-FLIPS. The latter might be attributed to the fact that, in contrast to c-FLIPS, c-FLIPL has a long C-terminus that is crucially involved in promoting the caspase-8 activity via the formation of the procaspase-8/c-FLIPL heterodimers and the stabilization of the active center of caspase-8 [12, 13]. Subsequently, we have suggested that even upon high overexpression of c-FLIPL there might be still some active caspase-8 generated at the DISC of HeLa-CD95-FL cells. This would lead to the differential effects of c-FLIPL versus c-FLIPS on caspase-8 activation at the DISC though both isoforms are highly expressed in these cells.
To test this hypothesis, next we analyzed the CD95 DISC assembly and caspase-8 activation in these cells comparing them to the HeLa-CD95 cells. Upon 125 ng/mL CD95L stimulation, in HeLa-CD95 cells efficient CD95 DISC assembly was observed, resulting in procaspase-8/10 activation and processing (Fig. 3d). Because HeLa-CD95–FL and HeLa-CD95-FS cells have shown a low sensitivity upon 125 ng/mL stimulation (Fig. 3b, c), we have increased the concentration of CD95L to 250 ng/mL CD95L, as this concentration induced caspase activation in these cell lines (Fig. 2c, d). Indeed, the CD95 DISCs were efficiently formed under these conditions in HeLa-CD95–FL and HeLa-CD95-FS cells (Fig. 3e, f). However, the pattern of procaspase-8/10 processing was quite different between these two cell lines (Fig. 3e, f). Namely, the analysis of the DISC in HeLa-CD95-FL cells showed that procaspase-8a/b and procaspase-10d/a undergo almost complete processing to their cleavage products p43/p41 and p18 as well as p47/p43 and p20, respectively (Fig. 3e). In contrast, in HeLa-CD95-FS cells only a small degree of procaspase-8a/b and procaspase-10d/a processing was observed (Fig. 3f). In particular, only low amounts of procaspase-8-p43/p41 were detected in the DISC-IP. This goes along with the suggested different action of c-FLIPL and c-FLIPS, respectively, which also fits to the previous reports [9, 13].
The analysis of caspase-8 activity at the DISC-IPs from HeLa-CD95, HeLa-CD95–FL, and HeLa-CD95-FS cells was consistent with the data on the composition of the DISCs by Western Blot. These results further supported the hypothesis on different actions of c-FLIPL versus c-FLIPS (Fig. 4a–c). Indeed, the DISCs of HeLa-CD95-FL cells were characterized by a relatively fast increase in caspase-8 activity, supporting the formation of the heterodimers c-FLIPL/caspase-8 and their subsequent high catalytic activity (Fig. 4b). Comparing absolute amounts of caspase-8 activity, the highest caspase-8 activity was observed at the CD95 DISC from HeLa-CD95, intermediate levels of caspase-8 activity were detected at the DISC from HeLa-CD95-FL, and the lowest activity was found at the DISC of HeLa-CD95-FS cells (Fig. 4d). Notably, in the caspase-8 activity (Fig. 4) and DISC-IP experiments (Fig. 3d-f) a two times lower concentration of CD95L for stimulation of HeLa-CD95 cells compared with HeLa-CD95-FL and HeLa-CD95-FS cells was used. This still resulted in a higher degree of caspase-8 activation in HeLa-CD95 cells compared with HeLa-CD95 cells overexpressing c-FLIP isoforms. These data further support that, despite a high c-FLIP overexpression, CD95 DISC formation takes place in HeLa-CD95-FL/-FS cells, leading to caspase-8 activation. However, the total amount of caspase-8 activity is not that high at the DISCs of HeLa-CD95-FL/-FS cells, with the lowest value at the DISC of HeLa-CD95-FS cells and a higher value at the DISC of HeLa-CD95-FL cells. Hence, this allows to suggest that the quantity of active caspase-8 at the DISCs of HeLa-CD95-FL/-FS cells is apparently not high enough to overcome the threshold for apoptosis induction in these cells [25]. This explains the observed inhibitory effects upon high overexpression of both isoforms on CD95-induced cell death despite the efficient DISC formation and a detection of caspase activity in these cell lines.

c-FLIP isoforms differentially modulate caspase-8 activity at the DISC. HeLa-CD95 cells (a), HeLa-CD95-FL cells (b), and HeLa-CD95-FS cells (c) were stimulated with 125 ng/mL (a) or 250 ng/mL (b + c) CD95L from 1 to 3 h. Caspase-8 activity at CD95 DISC-IPs was analyzed by Caspase-Glo®-8-Assay. The caspase activity of nontreated cells was taken as one relative unit [RU]. (d) The absolute amount of caspase-8 activity at the CD95 DISC-IPs from a–c is presented
Absolute quantitative mass spectrometry indicates the formation of short DED filaments and an increase in DISC formation upon c-FLIP overexpression
To get further insight into the mechanisms of c-FLIP action in DED filaments and to get an explanation for the observed caspase-8 activity despite high c-FLIPL/S overexpression we have performed a quantitative AQUA mass spectrometry analysis of the CD95 DISCs. In particular, CD95 DISCs were immunoprecipitated from HeLa-CD95, HeLa-CD95-FL, and HeLa-CD95-FS cells and their stoichiometry was analyzed using AQUA peptides designed by us previously [5, 15]. The AQUA peptides have been designed against the DED parts of caspase-8 and c-FLIP, which remain at the DISC and are not cleaved off in the course of the proteolysis [5, 26].
In the DISC-IPs of HeLa-CD95 cells, high procaspase-8 amounts compared with FADD and c-FLIP were observed (Fig. 5, Supplementary Fig. 4), which is consistent with previous reports [5]. Furthermore, c-FLIP was present in the DED filaments of HeLa-CD95 cells in sub-stoichiometric amounts, which also fits well to the previous observations [3, 5]. The ratios between c-FLIP and caspase-8 at the DISC did not markedly change within observed time intervals (Fig. 5d).

Quantification of procaspase-8, c-FLIP, and FADD at the CD95 DISC by AQUA mass spectrometry. HeLa-CD95 cell, HeLa-CD95-FL cells, and HeLa-CD95-FS cells were stimulated with 125 ng/mL (HeLa-CD95) or 250 ng/mL (FL + FS) CD95L for 1, 2, and 3 h. CD95-DISC-IPs were performed with anti-APO-1 (anti-CD95) antibodies and were separated by 1D SDS PAGE with subsequent quantification by AQUA peptide-based mass spectrometry. Experimental data are shown for 1 h (a), 2 h (b), 3 h (c) of CD95L stimulation and all three measured time intervals merged together (d). Absolute amounts of peptides at the DISC is plotted with the box plot (median, 25/75% confidence intervals) on the left side of the figure. Calculated ratios of caspase-8:c-FLIP:FADD in the DISC (mean values and standard deviations) are shown with the bars on the right side of the figure. Calculated means and standard deviations are shown on the bottom of each panel. Values that are beyond plot range are indicated with arrows. Statistical significance of mean value differences (p < 0.05) for HeLa-CD95-FL is indicated; # corresponds to p < 0.05 calculated by two-tailed Mann–Whitney test; ##: corresponds to p < 0.05 by both two-tailed Mann–Whitney U-test and Student’s t-test. Box plots were plotted using the Python matplotlib and seaborn libraries
Strikingly, overexpression of c-FLIP isoforms completely changed the ratios of DED proteins in the filaments. In the DISC-IPs from c-FLIPs-overexpressing cells, FADD, c-FLIP, and procaspase-8 proteins composed about 20, 54, and 26% of the total DED protein amount, respectively (Fig. 5a–c, Supplementary Fig. 4). This indicates the formation of shorter DED filaments compared with HeLa-CD95 cells with the similar amounts of procaspase-8 and FADD. Strikingly, the amount of c-FLIPS was only two times higher than that of procaspase-8 and FADD in these DISCs. In the DISC-IPs from c-FLIPL–overexpressing cells, FADD, c-FLIPL and procaspase-8 composed 35, 38, and 27% of the total amount in the DISC. This suggests similar amounts of procaspase-8 and c-FLIPL per one FADD molecule in the DISC (Fig. 5a–c, Supplementary Fig. 4). Strikingly, the composition of the DED filament was not changing up to 3 h of stimulation for all three cell lines (Fig. 5d), supporting our previous reports on the formation of the prodomain platform [27].
Another feature of the stoichiometry of the DISCs from c-FLIP-overexpressing cells was that the overexpression of c-FLIP isoforms seems to increase the DISC quantities (Fig. 5, Supplementary Fig. 4). It must be noted that even though we used a lower CD95L concentration of 125 ng/mL for HeLa-CD95 versus 250 ng/mL used for HeLa-CD95-FL and HeLa-CD95-FS cells, the absolute amount of the DED proteins: procaspase-8, FADD and c-FLIP at the DISC-IPs was approximately ten times higher in HeLa-CD95-FL and HeLa-CD95-FS cells compared with HeLa-CD95 cells. This indicated an increase in the DISC quantities upon c-FLIP overexpression (Fig. 5, Supplementary Fig. 4).
Taken together, the observed ratio of procaspase-8/c-FLIPL was approximately 1:1 in HeLa-CD95-FL, while the procaspase-8/c-FLIPs ratio was 1:2 in HeLa-CD95-FS cells. To get more insights into these intriguing results we expanded our analysis to computational structural modeling.
Structural modeling suggests how c-FLIPS/c-FLIPL controls the composition stoichiometry of DED filaments
To get further insights into the role of c-FLIP at the DED filament we applied in silico structural modeling techniques to construct a model of the DISC. The structural model comprising CD95/FADD death domains (DDs) and FADD DEDs was obtained via the estimation of optimal mutual spatial alignment of DDs and DEDs (see details in Supplementary modeling procedures). The region linking FADD DDs and DEDs was considered to be flexible and thereby mediate conformational changes of FADD induced by DD oligomerization. The flexibility of this region is supported by its low sequence conservation (Fig. 6a). The constructed DISC model has indicated that DDs of CD95 and FADD did not compose any steric hindrance for the formation of DED filaments at the DEDs of FADD (Fig. 6c). Furthermore, this model suggested that growth of the DED filament is possible in three directions from FADD DEDs, initiated by binding of DED proteins to FADD DEDs via type I or II or III interactions (Fig. 6b, c).

Structural models of CD95/FADD platforms. a Multiple sequence alignment of FADD proteins for different organisms. The region linking FADD DD and DED is indicated. The alignment is colored according to T-Coffee color scheme. b The scheme of the DED and types of interfaces involved in the DED filament assembly. Types and direction of interactions between DEDs in the filament are indicated with arrows. c Representative schemes (left) and corresponding structural models of CD95/FADD platforms (right) triggering DED filament assembly. DD and DED are shown as hexagons. CD95 DDs are shown in green, FADD DDs are shown in purple and FADD DEDs are shown in brown. FADD region (aa 84–92) linking DD and DED is shown in red. Three chains a, b, c of DEDs bound by type I interaction composing DED filament are denoted and highlighted with dashed lines. Structural models of c-FLIP – FADD (d) and procaspase-8 – FADD (e) heterodimers bound via type I/II/III interactions. The heatmap of binding energies estimated with Rosetta modeling package is shown in the center. Interaction interfaces of c-FLIP (d) or procaspase-8 (e) DEDs and corresponding estimated binding energies with FADD are indicated. Interaction interfaces with predicted selectivity to FADD are highlighted with gray boxes. DED1 and DED2 of c-FLIP are shown in orange and yellow colors, respectively. DED1 and DED2 of procaspase-8 are shown in dark blue and light blue colors, respectively. Reference structure of caspase-8 DED filament (PDB ID 5l08) used to predict heterodimer complexes is shown in transparent gray colors
Type I, II, or III interactions of DEDs mediate DED filament assembly and are based on interactions of Ia/Ib, IIa/IIb, and IIIa/IIIb interfaces (Fig. 6b). Quantitative mass spectrometry analysis has indicated that the amount of c-FLIP at the DED filament upon c-FLIPS overexpression is approximately two times higher than that of FADD and caspase-8 (Fig. 5). These data in combination with the recruitment of c-FLIP to FADD in caspase-8-deficient cells (Fig. 1) strongly indicate that there are several ways of c-FLIP incorporation into DED filaments. Hence, next we aimed to estimate in silico the binding efficiency of c-FLIP and procaspase-8 DEDs to different FADD DED interfaces, e.g., Ia/Ib, IIa/IIb, and IIIa/IIIb, and thereby to select the most efficient interactions between DEDs of FADD, procaspase-8, and c-FLIP (Fig. 6d, e). The rationale behind this analysis was to identify all putative binding sites of c-FLIP at the DED filament in silico. To this point, we estimated interaction energies of all FADD DED interfaces with different c-FLIP and procaspase-8 DED interfaces using the Rosetta modeling package [28] (see details in Supplementary modeling procedures). Briefly, the homology model of c-FLIP was generated (Supplementary Fig. 5) followed by predictions of all structural combinations of c-FLIP/FADD and procaspase-8/FADD heterodimers (Fig. 6d, e). We assumed that the DEDs in these heterodimers are bound by type I, II, or III interactions. These complexes were further refined with the Rosetta high-resolution protein-protein docking refinement protocol resulting in obtaining an ensemble of 900 conformations for each heterodimer. The top 5% scoring conformations were used to calculate the average interaction energies (Fig. 6d, e).
According to this analysis, the c-FLIP DED2 Ib interface had a higher binding energy to FADD Ia interface (α1/α4 region) in comparison to the binding energy of the corresponding procaspase-8 DED2 Ib interface (Fig. 6d, e). Moreover, the c-FLIP DED1 Ia interface had a lower binding energy to the FADD Ib interface (α2/α5 region) compared with the corresponding DED1 Ia interface of procaspase-8 (Fig. 6d, e). Moreover, the c-FLIP DED2 IIIa interface demonstrated stronger interactions with the FADD IIIb interface compared with the IIIa interface of procaspase-8 (Fig. 6d, e).
Based on these findings we have proceeded with predictions of the configuration of the DED platform and the modes of binding of c-FLIPs to the DED filament. The core of this model is initiated by the formation of a ‘FADD platform’ composed of three FADD DEDs, considering the well-established line of evidence for a trimerized structure of CD95 [29]. The FADD DED oligomer gives the possibility for initiation of three DED chains formed via type I interactions (chains “a”, “b”, “c”, Fig. 7), described by Fu et al. [4]. In accordance with previous reports, it was considered that adjacent chains are bound to each other via type II and III interactions [4]. It was assumed that each FADD DED can interact with procaspase-8 DED1 in the “forward” and c-FLIP DED2 in the “backward” directions (Fig. 7a). According to the estimation of binding energies, the FADD platform can accommodate procaspase-8 DED1 and c-FLIP DED2 bound to Ib/IIIa and Ia/IIIb interfaces of FADD DED, correspondingly. This model topology is supported not only by the analysis of interaction energies in silico, but also by our findings of c-FLIP recruitment to the DISC in Jurkat-C8−/− cells, as well as by previous reports highlighting DED1-caspase-8/FADD and DED2-c-FLIP/FADD type I interactions [18, 19]. The analysis of the structural model together with energy estimations suggests that binding of at least two more DED proteins is necessary to stabilize the DED filament by providing interactions between DED chains. This implies that the formation of the DISC comprising short DED filaments would require binding of DED proteins to chains “a” and “c” in “forward” and “backward” directions of FADD platform, correspondingly, that would connect chains via type II/III interactions (Fig. 7a).

Model of the cooperative DED filaments assembly. a Scheme of FADD platform triggering DISC assembly. Three chains a, b and c bound by type I interaction in the DED filament are denoted and highlighted with dashed lines. Interaction sites of chains a and c in the DED filament are indicated by duplicating them in the transparent color on the top and bottom of diagram. This implicates the shift on −6 and + 6 positions in the DED chain, correspondingly, in accordance with the DED filament symmetry. b The schematic model of single DISC assembly in HeLa-CD95-FS (left), HeLa-CD95-FL (middle), and HeLa-CD95 (right) cells. c Structural (top) and schematic model (bottom) of the cooperative DISC assembly in HeLa-CD95-FL cells. The configuration of FADD platform is highlighted. c-FLIP DED1 and DED2 are shown in orange and yellow colors, correspondingly. Caspase-8 DED1 and DED2 are shown in dark blue and light blue colors, correspondingly. FADD DED is shown in brown. C-terminal domains of c-FLIP and caspase-8 are shown as yellow and blue rectangles, correspondingly. CD95 DD and FADD DD are shown in green and purple colors, respectively
Using the structural model of the FADD platform and quantitative mass spectrometry data we suggest several configurations of the DISC (Fig. 7b). In HeLa-CD95 cells the DISC would comprise long DED filaments largely composed of procaspase-8. Overexpression of c-FLIP would lead to the formation of a DISC comprising short DED filaments. In HeLa-CD95-FS cells c-FLIPS molecules would be prevailing approximately two times in comparison with FADD and caspase-8. In HeLa-CD95-FL cells equal amounts of c-FLIPL and procaspase-8 are expected in the DED filaments (Fig. 7b). The molecular architecture of the latter is likely mediated by C-terminal domains of c-FLIPL.
Furthermore, an additional stabilization of the DED filament has been predicted leading to the formation of ‘cooperative’ DISC networks (Fig. 7c). Assembly of the cooperative DISC networks is enabled by association of the adjacent DISCs via DED filaments. This is only possible due to FADD DED having distinct binding sites for c-FLIP DED2 and procaspase-8 DED1 serving as a connection between filaments. We expect that simultaneous formation of multiple type I/II/III interactions between adjacent DISC complexes would lead to a high affinity interaction compensating potentially energetically unfavorable interactions that limit the growth of ‘c-FLIP-only’ or ‘c-FLIP-procaspase-8-only’ DED filaments. The formation of cooperative networks would provide more opportunities for the homo- and heterodimerization of procaspase-8 and c-FLIPL compared with the short filaments and subsequently more efficient activation of procaspase-8. The formation of a cooperative DISC network can be also one of the factors leading to enhancement of the numbers of CD95 DISCs upon c-FLIP overexpression detected by quantitative mass spectrometry, as the longer filaments are expected to have enhanced stability [16, 27].
Taken together, our structural modeling has delineated two modes of c-FLIP binding to the DED filament and has shown how c-FLIP isoforms play a role in the control of DED filament assembly by defining the length of DED filaments and their composition.
Discussion
In this study further insights into DISC architecture and the role of c-FLIP proteins in DED filaments function were deciphered. This was only possible via application of state-of-the-art quantitative mass spectrometry analysis supported by structural modeling. Contemporary structural modeling is a powerful tool that allows getting new insights into molecular mechanisms of signaling pathways. Using this technology, we have developed an in silico structural model of the CD95 DISC. This model allowed to suggest molecular mechanisms of DED filament assembly as well as to explain multiple modes of c-FLIP interactions with the DED filament.
One of the important aspects of our model was the prediction of distinct binding sites on FADD DED for both procaspase-8 and c-FLIP. The association between DEDs of these proteins is mediated by all three types of DED interactions (type I, II, and III). Using our model we demonstrate that FADD DED can trigger filament growth in a ‘forward’ direction by binding to Ia/IIIb interfaces of procaspase-8 DED1 domains or it can trigger filament growth in an opposite ‘backward’ direction by binding to Ib/IIIa interfaces of the c-FLIP DED2 domain. Furthermore, our findings shed light on the existing controversies on c-FLIP binding to the DISC. As mentioned above, from one side, c-FLIP is reported to interact via the α2/α5 interface of its DED2 (Ib interface) with the α1/α4 region of FADD DED (Ia interface), while procaspase-8 uses the other binding site for docking to FADD located at the α2/α5 region of FADD DED (Ib interface) [19]. From another side, a cooperative binding of c-FLIP to FADD in a procaspase-8 dependent manner has been suggested [14]. Our model combines both hypotheses showing that both mechanisms play an important role in DED filament assembly.
The molecular mechanisms of priming the DED filament assembly at the DED of FADD and sterical constraints of this process are highly important for understanding apoptosis initiation. In this regard, it is considered that the binding of procaspase-8 DED1 to FADD is necessary for initiation of filament growth. At the same time, it has not been clear whether the binding site on FADD targeted by c-FLIP DED2 is sterically accessible and if there are any structural constraints prohibiting this interaction. Using our structural model, we show that the FADD DD/DED platform poses no sterical hindrance for the binding of c-FLIP DED2, providing a basis for ‘multidirectional’ filament growth and the assembly of ‘cooperative’ DED filaments. Our modeling approach showed that the flexibility of the loop linking DD and DED of FADD might be a key structural feature that enables a conformational change, which in turn might trigger DED filament initiation. In the future studies, introducing mutations into this loop of FADD or its targeting with rationally designed molecular probes might provide new insights into apoptosis regulation.
The suggested structural model of the DED filament answers several questions. The first question is how caspase-8 can be activated upon high c-FLIP overexpression. The analysis of our 3D structural filament model shows that the formation of procaspase-8 dimers takes place even in short DED filaments. This might be supported by the interactions between adjacent DED2 of procaspase-8 from adjacent DED chains via type II interfaces (Fig. 7b). This architecture is only possible due to the independent specific binding sites of FADD for c-FLIP and procaspase-8 in the FADD DED platform. The FADD specific binding site for procaspase-8 allows a correct spatial localization of procaspase-8 DEDs, which leads to the proximity-induced dimerization of C-terminal domains. c-FLIP DEDs in turn control the exact topology of the DISC by limiting the amount of procaspase-8 and at the same time stabilizing the DISC structure via the binding to FADD DED. Moreover, c-FLIP-induced formation of a long DED filament, e.g., cooperative DISC networks, would further stabilize the DISC and increase the probability for procaspase-8 homo- and heterodimerization (Fig. 7c). Those predictions are in full agreement with the obtained experimental data on caspase-8 activity, that show that even upon high c-FLIPL/S overexpression procaspase-8 is activated in the DISC (Fig. 4).
Another question is how c-FLIP overexpression leads to an increased number of DISCs as obtained in our mass spectrometry experiments. It seems plausible that c-FLIP-dependent formation of cooperative DISC networks should enhance the stability of the filaments, thereby resulting in increased DISC amounts upon c-FLIP overexpression. The phenomenon of length-dependent stabilization of a filamentous structure is well known and has been widely studied for other systems [27]. Another reported observation of DISC behavior is the internalization and degradation of the DISC complex [30, 31]. It has been shown that internalization of the DISC is dependent on caspase-8 activity. Hence, a high level of c-FLIP proteins naturally decreases caspase-8 activity at the DISC and thereby DISC internalization. These factors certainly contribute to the higher amounts of the DISCs obtained in this study upon overexpression of c-FLIP proteins.
The cooperative assembly of DED filaments might represent an important mechanism for efficient filament growth. Indeed, a high local concentration of short DED filaments might generate a platform to trigger filament growth. Adjacent filaments of FADD platforms that are formed independently from each other can associate into one entity due to their close proximity. The latter might be facilitated by crowding of CD95 receptors on the membrane surface. Further insights into the cooperative assembly of DED filaments might be obtained by understanding their stability and the efficiency of recruitment of the individual components. Within these lines, the important information which is needed to draw further conclusions on the dynamics of the elongation of long DED filaments is the values of association constants between individual DED proteins. For example, quantitative mass spectrometry data indicate that the strength of c-FLIP/c-FLIP interactions within the DED filament is rather low, otherwise the assembly of long c-FLIP chains upon overexpression of both c-FLIP isoforms would be observed. The molecular mechanisms limiting the growth of c-FLIP DED filaments are yet to be elucidated. At the same time, we hypothesize that c-FLIPL-induced cooperative networks can be formed to higher extend, in particular, due to the higher level of caspase-8 per DISC complex, putative caspase-8/caspase-8 interactions between adjacent DISC complexes and the interactions of the C-terminal domains of procaspase-8 and c-FLIPL (Fig. 7c). Moreover, in our model we did not consider yet another component of the DED filament, procaspase-10, due to its low abundance at the DISC and reported roles as ‘not-priming’ initiator caspase in extrinsic apoptosis with pro-survival function [15, 23, 32]. Nevertheless, the role of this caspase in DED filament dynamics has to be considered in the future studies.
Finally, it has to be noted that the high overexpression of c-FLIP isoforms that was used in this study allowed dissecting the role of c-FLIP in the DED filaments. This approach has proven to be a very powerful tool in combination with quantitative mass spectrometry and structural modeling. Future studies using the technologies of quantitative or systems biology have to show whether the composition of the DISCs in these cells lines is close to that of primary cancer cells with high overexpression of c-FLIP proteins and can be used for targeting the extrinsic apoptosis pathway.
Our data show that c-FLIP isoforms play the role of control checkpoints in the dynamics of DED filaments by controlling their growth and stoichiometry. In particular, c-FLIP isoforms can largely modulate the DED filament composition by the assembly of short DED filaments or by promoting the formation of long cooperative networks of DED filaments. Taken together, using a powerful combination of state-of-the-art computational and experimental approaches we uncovered how c-FLIP proteins control the architecture of DED filaments and procaspase-8 activation at the CD95 DISC. This effective combination can also be used for the analysis of signaling of other cell death inducing complexes and opens new horizons for the development of therapeutic applications.
Material and methods
Cell lines
Human cervical cancer HeLa-CD95 cells (HeLa cells overexpressing CD95) [33], HeLa-CD95-FS cells (CD95/c-FLIPs-overexpressing cells), and HeLa-CD95-FL cells (CD95/c-FLIPL-overexpressing cells) were maintained in DMEM/Ham’s F-12 (Merck Millipore, Germany) supplemented with 10% heat-inactivated fetal calf serum, 1% Penicillin-Streptomycin and 0.0001% Puromycin in 5% CO2. T cell leukemia Jurkat (ACC 282) and Jurkat-caspase-8 deficient cells were maintained in RPMI 1640 (Thermo Fisher Scientific Inc., USA) supplemented with 10% heat-inactivated fetal calf serum and 1% Penicillin-Streptomycin in 5% CO2.
Antibodies and reagents
The following antibodies were used for Western Blot analysis: polyclonal anti-caspase-3 antibody (#9662), polyclonal anti-PARP antibody (#9542) and monoclonal anti-RIPK1 XP antibody (#3493) from Cell Signaling Technology, Massachusetts, USA; polyclonal anti-mCherry antibody (ab183628) from abcam, Cambridge, UK; polyclonal anti-actin antibody (A2103) from Sigma-Aldrich, Germany; polyclonal anti-CD95 antibody (sc-715), polyclonal anti-GAPDH antibody (sc-48166) from Santa Cruz, USA; monoclonal anti-caspase-10 antibody (M059–3), monoclonal anti-FADD antibody (clone 1C4), monoclonal anti-caspase-8 antibody (clone C15) and monoclonal c-FLIP antibody (clone NF6) were a kind gift of Prof. P.H. Krammer, (DKFZ, Heidelberg). Horseradish peroxidase-conjugated goat anti-mouse IgG1, -2a, -2b, goat anti-rabbit and rabbit anti-goat were from Santa Cruz (California, USA). The monoclonal anti-APO-1 antibodies (mouse-IgG3) were used for immunoprecipitations (IPs). All chemicals were of analytical grade and purchased from Merck (Darmstadt, Germany) or Sigma (Germany). The anti-APO-1 antibodies were a kind gift of Prof. P. H. Krammer (DKFZ, Heidelberg). Recombinant LZ-CD95L was produced as described [33].
Analysis of total cellular lysates by Western Blot
The Western Blot analysis of total cellular lysates was performed in accordance to our previous reports [34].
CD95 DISC-IP
The CD95 DISC-IP (Immunoprecipitation) from 5 × 106 HeLa-CD95 cells or 10 × 106 Jurkat cells were done as described before [33, 35]. The anti-APO-1 (anti-CD95) antibodies were a kind gift of Prof. P. H. Krammer (DKFZ, Heidelberg). DISC-IPs were washed with PBS for four times, which followed by Western Blot analysis or caspase-8 activity assays.
Caspase-8 activity assay
Protein A beads samples with CD95 DISC-IPs were re-suspended in 95 µL of CHAPS-Buffer (50 mM HEPES pH = 7.2; 50 mM NaCl; 10 mM EDTA; 5% Glycerin; 10 mM DTT; 0.1% CHAPS) and transferred into a 96-well plate. Caspase-8 activity was measured according to manufacturer’s instructions (Caspase-Glo® 8 Assay, Promega, Germany). The caspase-8 activity of untreated cells was taken as one relative unit (RU). Every condition was performed in duplicate. The luminescence intensity was analyzed by the microplate reader Infinite M200pro (Tecan, Switzerland).
Cell viability quantification by ATP assay
A total of 1.2 × 104 HeLa-CD95/HeLa-CD95-FL/HeLa-CD95-FS cell were seeded in a 96 well plate a day before experiments. After corresponding treatment, 50 µL of the CellTiter-Glo® solution was added to each well. Measurements were performed according to manufacturer’s instructions (CellTiter-Glo® Luminescent Cell Viability Assay, Promega, Germany). The luminescence intensity was analyzed by a microplate reader Infinite M200pro (Tecan, Switzerland). The viability of untreated cells was taken as 100%. Every condition was performed in duplicate.
Cell death analysis by imaging flow cytometry
A total of 7.5 × 105 cells were plated in six wells and treated with or without the indicated concentration of CD95L in a time-dependent manner. To analyze early and late apoptosis cells were stained by a combination of SytoxOrange and Annexin-V-FITC. Quantification of different cell population was performed by imaging flow cytometry using AMNIS® FlowSight® with subsequent analysis as described previously [36].
CD95 surface analysis by imaging flow cytometry
A total of 1 × 106 cells were stained with FITC anti-human CD95 (Fas) antibody (BioLegend, USA) for 15 min according to manufacturer’s instructions. The surface receptor staining was quantified by imaging flow cytometry using AMNIS® FlowSight®. Unstained 1 × 106 cells were measured as a control.
AQUA mass spectrometry
Immunoprecipitates (protein A sepharose beads washed after IPs) were re-suspended in 50 mM NH4HCO3 and incubated in 1 mM DTT at 56 °C for 45 min, which was followed by the subsequent S-Methyl methanethiosulphonate (MMTS) treatment (5 mM MMTS, 30 min). Tryptic digestion was performed by addition of 0.5 µg Trypsin (Trypsin Gold, Promega) and incubation at 37 °C for 24 h. AQUA peptides for DED of procaspase-8 (NLYDIGEQLDSEDLASL[K 13C6; 15N2]; FLSLDYIPQ[R 13C6, 15N4]), FADD (IDSIED[R 13C6, 15N4]) and c-FLIP (LSVGDLAELLY[R 13C6, 15N4]; DVAIDVVPPNV[R 13C6, 15N4]) [5, 26] were spiked into the tryptic digestion solution in an absolute amount of 100 fmol of each peptide. After digestion, the supernatant was collected and dried in a vacuum centrifuge. The peptides were re-dissolved in 5 µl 0.1 % trifluoroacetic acid (TFA) and purified on ZIP-TIP, C18-nanocolumns (Millipore, Billerica, USA). Peptides were eluted in 7 µl 70% (v/v) acetonitrile (ACN) and subsequently dried in a vacuum centrifuge. Samples were dissolved in 10 µl 2% ACN/0.1% TFA and separated on a 75 μm I.D., 25 cm PepMap C18-column (Dionex, Sunnyvale, USA) applying a gradient from 2 to 45% ACN in 0.1% formic acid over 120 min at 300 nl/min using an Ultimate 3000 Nano-HPLC (Thermo Scientific, San Jose, USA). Mass spectrometry was performed on a hybrid dual-pressure linear ion trap/orbitrap mass spectrometer (LTQ Orbitrap Velos Pro, Thermo Scientific, San Jose, USA) in exclusive orbitrap full MS mode (FTMS; resolution 60,000; m/z range 400–2000). Absolute protein quantification was achieved using Skyline analysis platform [37] for MS-peak integration on extracted ion chromatograms of selected peptide masses. The consideration of the monoisotopic precursor mass and at least two C13-isotopic variants ([M + 1] and [M + 2]) has been chosen for more accurate and confident quantification. The peak qualities of the quantified peptides were controlled by the “isotope dot product” (idotp) set to >0.95. Idotp provides a measure to assess precursor isotope distribution and its correlation between expected and observed pattern with optimal matching resulting in an idotp value of “1” [27].
Statistical analysis
Graphpad Prism 8 software was used for generating and performing one-way ANOVA-tests analyzing the significance of viability assays (paired, normal distribution, multiple comparison test: Turkey test). Scipy python package was used for two-tailed Mann–Whitney and t-Student tests. Uncertainty quantification of caspase-8:c-FLIP:FADD ratios generated by mass spectrometry analysis values was calculated with linear error propagation theory as implemented in Python Uncertainties Package (http://pythonhosted.org/uncertainties/).
Structural modeling
Structural modeling procedures are presented in detail in the Supplementary modeling procedures.
References
Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. 2007;7:532–42.
Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012;19:36–41.
Dickens LS, Boyd RS, Jukes-Jones R, Hughes MA, Robinson GL, Fairall L, et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell. 2012;47:291–305.
Fu TM, Li Y, Lu A, Li Z, Vajjhala PR, Cruz AC, et al. Cryo-EM structure of caspase-8 tandem DED filament reveals assembly and regulation mechanisms of the death-inducing signaling complex. Mol Cell. 2016;64:236–50.
Schleich K, Warnken U, Fricker N, Ozturk S, Richter P, Kammerer K, et al. Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol Cell. 2012;47:306–19.
Ozturk S, Schleich K, Lavrik IN. Cellular FLICE-like inhibitory proteins (c-FLIPs): fine-tuners of life and death decisions. Exp Cell Res. 2012;318:1324–31.
Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN. c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem. 2005;280:14507–13.
Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, et al. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–14.
Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW, et al. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. 2002;277:45162–71.
Peter ME. The flip side of FLIP. Biochemical J. 2004;382:e1–3.
Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol. 2001;1:50–58.
Yu JW, Jeffrey PD, Shi Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc Natl Acad Sci USA. 2009;106:8169–74.
Fricker N, Beaudouin J, Richter P, Eils R, Krammer PH, Lavrik IN. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J Cell Biol. 2010;190:377–89.
Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, et al. Co-operative and hierarchical binding of c-FLIP and caspase-8: a unified model defines how c-flip isoforms differentially control cell fate. Mol Cell. 2016;61:834–49.
Schleich K, Buchbinder JH, Pietkiewicz S, Kahne T, Warnken U, Ozturk S, et al. Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2016;23:681–94.
Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274:1541–8.
Hwang EY, Jeong MS, Park SY, Jang SB. Evidence of complex formation between FADD and c-FLIP death effector domains for the death inducing signaling complex. BMB Rep. 2014;47:488–93.
Ueffing N, Keil E, Freund C, Kuhne R, Schulze-Osthoff K, Schmitz I. Mutational analyses of c-FLIPR, the only murine short FLIP isoform, reveal requirements for DISC recruitment. Cell Death Differ. 2008;15:773–82.
Majkut J, Sgobba M, Holohan C, Crawford N, Logan AE, Kerr E, et al. Differential affinity of FLIP and procaspase 8 for FADD’s DED binding surfaces regulates DISC assembly. Nat Commun. 2014;5:3350.
Golks A, Brenner D, Schmitz I, Watzl C, Krueger A, Krammer PH, et al. The role of CAP3 in CD95 signaling: new insights into the mechanism of procaspase-8 activation. Cell Death Differ. 2006;13:489–98.
Hoffmann JC, Pappa A, Krammer PH, Lavrik IN. A new C-terminal cleavage product of procaspase-8, p30, defines an alternative pathway of procaspase-8 activation. Mol Cell Biol. 2009;29:4431–40.
Juo P, Kuo CJ, Yuan J, Blenis J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol. 1998;8:1001–8.
Sprick MR, Rieser E, Stahl H, Grosse-Wilde A, Weigand MA, Walczak H. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J. 2002;21:4520–30.
Lavrik IN. Systems biology of death receptor networks: live and let die. Cell Death Dis. 2014;5:e1259.
Bentele M, Lavrik I, Ulrich M, Stosser S, Heermann DW, Kalthoff H, et al. Mathematical modeling reveals threshold mechanism in CD95-induced apoptosis. J Cell Biol. 2004;166:839–51.
Warnken U, Schleich K, Schnolzer M, Lavrik I. Quantification of high-molecular weight protein platforms by AQUA mass spectrometry as exemplified for the CD95 death-inducing signaling complex (DISC). Cells. 2013;2:476–95.
Schilling B, Rardin MJ, MacLean BX, Zawadzka AM, Frewen BE, Cusack MP, et al. Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation. Mol Cell Proteom. 2012;11:202–14.
Chaudhury S, Berrondo M, Weitzner BD, Muthu P, Bergman H, Gray JJ. Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS ONE. 2011;6:e22477.
Lavrik I, Golks A, Krammer PH. Death receptor signaling. J Cell Sci. 2005;118:265–7.
Algeciras-Schimnich A, Shen L, Barnhart BC, Murmann AE, Burkhardt JK, Peter ME. Molecular ordering of the initial signaling events of CD95. Mol Cell Biol. 2002;22:207–20.
Algeciras-Schimnich A, Peter ME. Actin dependent CD95 internalization is specific for Type I cells. FEBS Lett. 2003;546:185–8.
Horn S, Hughes MA, Schilling R, Sticht C, Tenev T, Ploesser M, et al. Caspase-10 negatively regulates caspase-8-mediated cell death, switching the response to CD95L in favor of NF-kappaB activation and cell survival. Cell Rep. 2017;19:785–97.
Neumann L, Pforr C, Beaudouin J, Pappa A, Fricker N, Krammer PH, et al. Dynamics within the CD95 death-inducing signaling complex decide life and death of cells. Mol Syst Biol. 2010;6:352.
Schmidt JH, Pietkiewicz S, Naumann M, Lavrik IN. Quantification of CD95-induced apoptosis and NF-kappaB activation at the single cell level. J Immunol Methods. 2015;423:12–17.
Pietkiewicz S, Eils R, Krammer PH, Giese N, Lavrik IN. Combinatorial treatment of CD95L and gemcitabine in pancreatic cancer cells induces apoptotic and RIP1-mediated necroptotic cell death network. Exp Cell Res. 2015;339:1–9.
Pietkiewicz S, Schmidt JH, Lavrik IN. Quantification of apoptosis and necroptosis at the single cell level by a combination of Imaging Flow Cytometry with classical Annexin V/propidium iodide staining. J Immunol Methods. 2015;423:99–103.
MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–8.
Acknowledgements
We acknowledge Volkswagen Foundation (VW 90315), Wilhelm Sander-Stiftung (2017.008.01), Center of dynamic systems (CDS), funded by the EU-program ERDF (European Regional Development Fund) and DFG (LA 2386) for supporting our work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Hillert, L.K., Ivanisenko, N.V., Espe, J. et al. Long and short isoforms of c-FLIP act as control checkpoints of DED filament assembly. Oncogene 39, 1756–1772 (2020). https://doi.org/10.1038/s41388-019-1100-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-019-1100-3
- Springer Nature Limited
This article is cited by
-
cFLIP in the molecular regulation of astroglia-driven neuroinflammation in experimental glaucoma
Journal of Neuroinflammation (2024)
-
MIND bomb 2 prevents RIPK1 kinase activity-dependent and -independent apoptosis through ubiquitylation of cFLIPL
Communications Biology (2021)
-
Cryo-EM structural analysis of FADD:Caspase-8 complexes defines the catalytic dimer architecture for co-ordinated control of cell fate
Nature Communications (2021)
-
Pharmacological targeting of c-FLIPL and Bcl-2 family members promotes apoptosis in CD95L-resistant cells
Scientific Reports (2020)