
Abstract
The retinoblastoma protein (RB) restricts cell cycle gene expression and entry into the cell cycle. The RB-related protein p130 forms the DREAM (DP, RB-like, E2F, and MuvB) complex and contributes to repression of cell cycle-dependent genes during quiescence. Although both RB and DREAM bind and repress an overlapping set of E2F-dependent gene promoters, it remains unclear whether they cooperate to restrict cell cycle entry. To test the specific contributions of RB and DREAM, we generated RB and p130 knockout cells in primary human fibroblasts. Knockout of both p130 and RB yielded higher levels of cell cycle gene expression in G0 and G1 cells compared to cells with knockout of RB alone, indicating a role for DREAM and RB in repression of cell cycle genes. We observed that RB had a dominant role in E2F-dependent gene repression during mid to late G1 while DREAM activity was more prominent during G0 and early G1. Cyclin D–Cyclin-Dependent Kinase 4 (CDK4)-dependent phosphorylation of p130 occurred during early G1, and led to the release of p130 and MuvB from E2F4 and decreased p130 and MuvB binding to cell cycle promoters. Specific inhibition of CDK4 activity by palbociclib blocked DREAM complex disassembly during cell cycle entry. In addition, sensitivity to CDK4 inhibition was dependent on RB and an intact DREAM complex in both normal cells as well as in palbociclib-sensitive cancer cell lines. Although RB knockout cells were partially resistant to CDK4 inhibition, RB and p130 double knockout cells were significantly more resistant to palbociclib treatment. These results indicate that DREAM cooperates with RB in repressing E2F-dependent gene expression and cell cycle entry and supports a role for DREAM as a therapeutic target in cancer.
Similar content being viewed by others


Introduction
The DREAM (DP, RB-like, E2F and MuvB) complex comprises the retinoblastoma (RB)-like protein p130 (RBL2), a repressor E2F (E2F4 or E2F5) and dimerization partner DP (DP1 or DP2), and the MuvB (synthetic multivuval class B) core containing LIN9, LIN37, LIN52, LIN54, and RBBP4 [1, 2]. The intact DREAM complex is present during the quiescent phase (G0) of the cell cycle and contributes to the repression of genes required for the entry into the cell cycle [1]. DREAM binds and represses the promoters of two sets of genes during G0: early cell cycle genes required for DNA synthesis with peak expression during late G1 and early S phase, and late cell cycle genes required for progression through mitosis with peak expression during G2 and M phase [3, 4]. During S phase, the MuvB core recruits B-MYB (MYBL2) and FOXM1 (MMB–FOXM1 complex) to activate late cell cycle gene expression [3, 5]. During quiescence, the LIN54 component of MuvB binds specifically to CHR elements found in late cell cycle gene promoters while the E2F4-DP1 heterodimer binds to E2F elements contained in early cell cycle gene promoters [6,7,8,9,10]. Together, E2F4 and MuvB enable DREAM complex binding to promoters containing E2F and CHR elements to repress early and late gene expression during G0. When cells progress from G0 to S phase, p130 is released from E2F4-DP1 and MuvB [1, 11]. Whether release of p130 from E2F4-DP1 and MuvB is required to enable increased levels of early cell cycle genes is not known.
RB binds and inhibits the activator E2Fs (E2F1, E2F2, E2F3a) that function to promote early cell cycle gene expression and entry into S phase [6]. While RB can also bind to the repressor E2F4, it is unable to bind to the MuvB core and does not form a DREAM complex [11]. Levels of activator E2Fs are reduced in G0 due to repression by the DREAM complex [1, 12]. Therefore, the DREAM complex likely has a role during G0, while RB contributes to repression later in G1 when activator E2Fs are expressed. An emerging model proposes that DREAM and RB bind and repress an overlapping set of early cell cycle genes [13]. However, the distinction between DREAM and RB control of early cell cycle gene expression during G0 and G1 remains unclear.
Cyclin–CDK complexes promote cell cycle progression by phosphorylating RB family members during G1. Growth factor-dependent expression of Cyclin D leads to CDK4 (and CDK6)-dependent phosphorylation of RB and at least partial relief of binding to the activator E2Fs and early cell cycle gene expression [14,15,16]. Subsequently, E2F1 activation leads to increased levels of Cyclin E leading to CDK2-dependent hyper-phosphorylation of RB [17,18,19]. Hyper-phosphorylated RB undergoes a conformational change and release from E2F1 with transactivation of E2F-dependent genes and entry into S phase [15, 20]. Hyper-phosphorylated p130 has reduced binding affinity to E2F4 and MuvB in vitro [11, 21, 22]. However, the specific contribution of Cyclin D–CDK4 to inactivation of p130 and DREAM remains unclear.
Whether DREAM and RB have functionally non-redundant roles in the regulation of cellular proliferation is unknown. Genetic loss of RB1 is one of the most common mutations in cancer (reviewed in [23]), while loss of DREAM complex members is infrequent (NCI Genomic Data Commons Data Portal: https://portal.gdc.cancer.gov). Inhibitors of Cyclin D–CDK4 are used clinically for treatment of cancers containing wild-type RB [24, 25]. If Cyclin D–CDK4 similarly regulates DREAM and RB, then CDK4 inhibitors may remain functional in RB-null cells with intact DREAM complex able to repress cellular proliferation. Here, we test the specific contributions of DREAM and RB in the repression of cell cycle genes during G0 and G1. We determine the contribution of Cyclin D–CDK4 activity to DREAM complex disruption and demonstrate that DREAM and RB both contribute to cell cycle arrest in normal and cancer cells.
Results
DREAM complex is disrupted in two steps during cell cycle entry from G0
To study the role of DREAM in cell cycle regulation, we first sought to determine the mechanism of DREAM disruption during cell cycle entry from G0. We used primary early passage natal human foreskin fibroblast cells (HFFs) that grow well in standard media and reliably arrest in response to contact inhibition and serum starvation [26, 27]. We characterized the proliferation kinetics of a pooled population of HFFs and observed that HFFs had significantly reduced levels of DNA synthesis as assessed by BrdU incorporation when serum starved and contact arrested compared to asynchronous cells or cultures split 24 h earlier (Figure S1A). In addition, serum starvation of contact arrested cells led to significantly reduced levels of Cyclin D1 (CCND1) transcript and protein as well as transcripts of cell cycle genes CDC6 and MCM5 compared to contact arrested cells in media with 15% serum (Figures S1B, S1C). Using these parameters, we studied the DREAM complex in cells entering into the cell cycle when released from contact arrest compared to confluent cells that were serum starved for an additional 48 h (Figure S1D). We referred to the serum starved, contact inhibited, quiescent HFFs as 0 h. DNA synthesis did not occur until 12 h after release from contact arrest with peak levels occurring at 24 h (Fig. 1a, S1E).

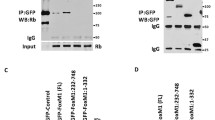
DREAM complex is disrupted in two steps during cell cycle entry from G0. a HFFs were split from contact arrest and S phase levels were measured by BrdU incorporation after 24 h (3 bio rep). b HFFs were induced to enter the cell cycle and lysates were prepared at times indicated and analyzed by immunoblot. c–f HFFs were induced to enter cell cycle and were analyzed by immunoprecipitation and immunoblot using indicated antibodies. Arrow indicates on-target band. Different exposures are included for some antibodies with blot including IgG having darkest exposure. Samples were ran in single gel except for d and f in which two gels were ran and were blotted identically. g–i Chromatin occupancy of p130, LIN9, and E2F4 on early and late cell cycle promoters in HFFs was measured by chromatin immunoprecipitation (ChIP)-qPCR using p130 (6 bio rep, g), LIN9 (6 bio rep, h), and E2F4 (3 bio rep, i) antibodies. Significance was determined Student’s t-test. P-values are indicated as * for < 0.05, ** for < 0.01, *** for < 0.001, and **** < 0.0001. See also Figure S1, S2
To determine the fate of the DREAM complex during cell cycle entry, we prepared lysates from cells harvested at intervals during the initial 12 h of cell cycle entry when cells are predominately in G1 (2N DNA, BrdU-) (Fig. 1b). Although no DNA synthesis occurred during the initial 12 h, we found substantial changes in p130 phosphorylation. Phosphorylation of p130 was initially detected at 6 h after splitting with peak levels occurring at 10 and 12 h. Total p130 levels declined at 24 h after splitting when many cells were in S phase, consistent with prior reports of p130 protein degradation during S phase (Fig. 1b) [28]. Cyclin D1, a key modulator of cell cycle progression from G0, was detected at low levels at 0 h and increased with time after release from contact arrest. The specificity of the p130-pS672 antibody was confirmed with λ phosphatase and prolonged serum starvation (Figure S1F-G). Similar kinetics of p130-pS672 phosphorylation and expression of Cyclin D1 were observed in independently prepared populations of HFFs (Figure S1H).
Phosphorylation of p130 has been reported to reduce its interaction with E2F4 [22, 29]. Since p130 phosphorylation was detected at 6 h of cell cycle entry (early G1) with peak levels at 12 h, we suspected that this change might perturb the DREAM complex. To assess the state of the DREAM complex, we performed immunoprecipitation with antibodies to p130, MuvB component LIN9, and E2F4 using lysates prepared from cells at 0, 6, 12, and 24 h (Fig. 1c). An immunoprecipitation for total p130 was blotted for p130-pS672 and showed strong signal at 12 and 24 h, similar to levels of p130-pS672 in input. Total p130 co-precipitated E2F4 at 0 h, with a slight reduction in levels at 6 h and a strong reduction at 12 and 24 h (Fig. 1c). In contrast, p130 co-precipitated MuvB components LIN54 and LIN9 at 0, 6, and 12 h, with reduced interaction found at 24 h. The interaction between LIN9 and p130 at 12 h was weak but consistently detected. Similarly, LIN9 co-precipitated p130 at 0, 6, and 12 h but co-precipitated E2F4 only at 0 and 6 h. These data indicate that p130 had reduced binding to E2F4 after 6 h but retained binding to MuvB components LIN9 and LIN54 up to at least 12 h after cell cycle entry. Consistent with this finding, p130-pS672 was able to interact with LIN9 and LIN54 at 12 h (Fig. 1d, e). In contrast, E2F4 co-precipitated LIN9 strongly at 0 h, weakly at 6 h and not at 12 h (Fig. 1f). We were unable to detect p130-pS672 interaction with E2F4 (Figure S1I). These results indicate that p130 and MuvB binding to E2F4 was restricted to the initial 6 h before p130 phosphorylation while p130 retained binding to MuvB for at least 12 h after cell cycle entry.
Given the dissociation of E2F4 from MuvB and p130 after 6 h of cell cycle entry, we tested the impact on specific binding to promoters of cell cycle-regulated genes by chromatin immunoprecipitation and quantitative PCR (ChIP-qPCR) (Fig. 1g–i). To measure the effect of cell cycle entry on chromatin occupancy, we determined fold change occupancy by normalizing percent input values to 0 h. In serum-starved quiescent cells (0 h), p130, LIN9, and E2F4 bound to the promoters of the early cell cycle genes MCM5 and RAD54L and the late cell cycle gene CCNB2. However, by 6 h of cell cycle entry, p130 and LIN9 had significantly reduced binding to these promoters that continued to decrease further at 12 and 24 h (Fig. 1g, h). The relative level of LIN9 binding to MCM5, RAD54L, and CCNB2 promoters was reduced to ~50% at 6 h (Fig. 1h). In contrast, E2F4 binding to these promoters did not change during the initial 6 h compared to 0 h but was significantly reduced by 12 h (Fig. 1i). Raw percent input values show similar kinetics and significant promoter occupancy above IgG and non-cell cycle promoter negative controls (Figure S2). The persistence of E2F4 binding to chromatin with reduced levels of p130 and LIN9 after 6 h was consistent with the release of p130 and MuvB from E2F4 observed by immunoprecipitation (Fig. 1c–f). We observed that E2F4 persisted in the nuclear fraction during the initial 12 h after cell cycle entry, while p130 nuclear levels declined after 6 h, consistent with reports that E2F4 persists in the nucleus throughout G1 (Figure S1J) [30].
Together, these data indicate that phosphorylated p130 and MuvB lose interaction with chromatin but continue to bind each other between 6 and 12 h after cell cycle entry, while E2F4 retains binding to cell cycle genes promoters during this time. At 12 h after cell cycle entry, E2F4 was lost from chromatin, and p130 and MuvB remained bound to each other. Loss of interaction of phosphorylated p130 from MuvB was observed at 24 h when many cells are in S phase.
Cyclin D–CDK4 inhibition blocks HFF cell cycle progression
Since phosphorylation of p130 occurred when p130 and MuvB dissociated from E2F4, we asked whether Cyclin D–CDK4 kinase activity contributed to the process of DREAM complex disassembly. First, we determined whether Cyclin D–CDK4 activity was required for HFFs to enter S phase after release from contact arrest using palbociclib, an inhibitor of Cyclin D–CDK4 activity [25, 31]. Addition of palbociclib when cells were split from confluence resulted in a dose-dependent decrease in the fraction of cells in S phase when measured 24 h later, with 200 nM reducing S phase levels to <5% (Fig. 2a). Furthermore, palbociclib treatment reduced S phase entry if added at any time during initial 12 h of cell cycle entry, while addition after 12 h had a reduced effect, indicating that Cyclin D–CDK4 activity was required during the initial 12 h to enable cell cycle entry (Fig. 2b). Palbociclib treatment reduced levels of phosphorylated p130 and RB in a dose-dependent manner (Fig. 2c).

Cyclin D–CDK4 promotes phosphorylation of p130 and cell cycle progression. a HFFs were split into media with palbociclib and BrdU incorporation was measured in cells harvested after 24 h (3 bio rep). Student’s t-test was calculated compared to 0 nM. b 200 nM palbociclib was added at times after split and BrdU incorporation was measured in cells harvested at 24 h post split (2 bio rep). Student’s t-test was calculated compared to 0 h. c HFFs were split into palbociclib and harvested at indicated times and analyzed by immunoblot. * indicates nonspecific band. d Parental HFFs were transduced with pLenti-E1Fa-CDK4-WT, K35M, and I12A and contact arrested before harvested for immunoblot. Arrow indicates on-target band. e CDK4 expressing HFFs were released from contact arrest into palbociclib and harvested after 24 h and analyzed by immunoblot. f CDK4 expressing HFFs were released from contact arrest into palbociclib and analyzed by RT-qPCR (3 bio rep). Student’s t-test was calculated by comparing to 0 nM. g CDK4 expressing HFFs were released from contact arrest into palbociclib and S phase was measured by BrdU incorporation after 24 h. Statistics comparing curves was completed by two-way ANOVA (3 bio rep). P-values are indicated as * for < 0.05, and ** for < 0.01, *** for < 0.001. See also Figure S3
To determine whether palbociclib-mediated inhibition of cell cycle progression was dependent on CDK4 activity in HFFs, we utilized a mutant of CDK4 (I12A) that is partially resistant to palbociclib (personal communication, Nicole Persky and Cory Johannessen, Broad Institute). We generated HFFs expressing pLenti-E1Fα-CDK4 (untagged) with wild type (WT), kinase dead (K35M), or the I12A allele using lentiviral transduction. We detected a stronger CDK4 signal at ~38 kDa in HFFs expressing pLenti-E1Fa-CDK4 compared to parental HFFs without change in CDK6 levels (Fig. 2d). Palbociclib treatment led to decreased p130 phosphorylation in CDK4-WT and CDK4-K35M cells while CDK4-I12A cells contained phosphorylated p130 even at higher doses (Fig. 2e). Consistent with this effect on p130 phosphorylation, CDK4-I12A cells had a reduction of MuvB interaction with p130 after palbociclib treatment (Figure S3A-B). CDK4-I12A cells persistently expressed early cell cycle genes CDC6 and MCM5 while CDK4-WT or CDK4-K35M cells showed a significant reduction in expression after treatment with palbociclib (Fig. 2f). CDK4-I12A cells had significantly higher S phase or relative cell density compared to WT and K35M expressing cells in the presence of palbociclib (Fig. 2g, S3C). These data demonstrate that palbociclib treatment inhibits cell cycle progression of HFFs, at least in part, by specifically inhibiting CDK4 kinase activity.
Cyclin D–CDK4 promotes DREAM complex disruption by phosphorylation of p130
Since palbociclib sensitivity was limited to the initial 12 h of cell cycle entry and led to reduced cell cycle gene expression, we reasoned that sustained palbociclib treatment could maintain an intact DREAM complex after cell cycle entry. To determine whether continued Cyclin D–CDK4 activity was required to maintain p130 phosphorylation, cells were released from contact arrest and treated with palbociclib for 2-h intervals just prior to harvest (Fig. 3a). Cells harvested at 6 h of cell cycle entry (lane 3; 6−) contained phosphorylated p130 but when cells were treated with palbociclib from 6 to 8 h, (lane 4; 8+), levels of p130-pS672 detection were reduced. Similarly, reduced phosphorylation levels were observed at 10 and 12 hours post release (Fig. 3a). Phosphorylated RB (pT821) was also reduced at 12+ h compared to untreated cells at 10 h. These data indicate that p130 phosphorylation is rapidly turned over during G1 and continuous CDK4 activity is required for maintenance of p130 phosphorylation.

Cyclin D–CDK4 promotes DREAM complex disruption by phosphorylation of p130. a HFFs were split into drug-free media and 200 nM palbociclib was added to media 2 h before harvesting. Lysates were prepared and analyzed by immunoblot. b mRNA abundance was measured by RT-qPCR from cells treated as in a (3 bio rep). Statistics calculated using Student’s t-test. P-values as indicated * <0.05. c HFFs were split and harvested after 0, 6, or 12 h. Cells were untreated, or treated with 200 nM palbociclib for 12 h or the last 6 h before harvest at 12 h. Lysates were subjected to immunoprecipitation using p130, LIN9, or IgG antibodies and analyzed by immunoblot. * indicates heavy chain. See also Figure S4
To determine whether the transient reduction of p130 and RB phosphorylation with the 2 h treatment of palbociclib affected cell cycle gene levels, we performed RT-qPCR (Fig. 3b). We observed a significant reduction in CCNE2 (Cyclin E2), MCM5, and CDC6 mRNA levels when cells were treated with palbociclib from 6 to 8 and from 8 to 10 h, with reduced effect of treatment from 10 to 12 h. Reduced levels of MCM5 protein was also observed when comparing lysates from 8 h untreated to 10 h with 200 nM (lane 5, lane 6) as measured by immunoblot (Fig. 3a). Since Cyclin D–CDK4 activity was required to promote p130 phosphorylation and expression of DREAM-repressed cell cycle genes, we suspected that palbociclib could maintain an intact DREAM complex during cell cycle entry (Fig. 3c). We observed that treatment with palbociclib between 6 and 12 h after release or during the entire 12 h after release led to increased levels of p130, E2F4, and LIN9 co-immunoprecipitation at 12 h compared to untreated samples as measured by IP and immunoblot. These data indicate that CDK4 inhibition maintains the intact DREAM complex during cell cycle entry.
Since CDK2 also phosphorylates p130, we reasoned CDK2 could also promote DREAM complex disruption. However, we were unable to perform similar experiments since no potent and selective CDK2 inhibitor is currently available [32]. For example, CVT-313, a commonly used CDK2 inhibitor, had low potency and required at least 6–8 h of treatment to affect changes in p130 and RB phosphorylation (Figure S4A-C). Furthermore, CVT-313 treatment in cells lacking p130 and RB resulted in significantly reduced levels of genes unrelated to the cell cycle, suggesting off-target effects on transcription by CVT-313 (Figure S4D). Although we cannot rule out an additional role for CDK2, the effects of palbociclib indicate an important role of Cyclin D–CDK4 in DREAM complex disruption.
DREAM and RB cooperate to repress early and late cell cycle gene expression in G0 and G1
To distinguish the contribution of p130 (RBL2) and RB (RB1) in cell cycle gene expression and sensitivity to palbociclib, we generated RB1 and p130 knockout HFFs using CRISPR-Cas9 mediated gene editing [33]. HFFs were transduced with pLenti-CRISPR V2-puromycin against RB1 or control (sgControl-P) and selected with puromycin (Figure S5A). Knockout of p130 was accomplished using pLenti-CRISPR V2-neomycin followed by selection in G418 (Figure S5B). RB and p130 knockout HFFs were generated with two rounds of transduction and selection, creating Control (sgControl-P, sgControl-N), sgP130 (sgControl-P, sgP130-N), sgRB1 (sgRB1-P, sgControl-N), and sgRB1+sgP130 (sgRB1-P, sgP130-N) cell lines. Knockout of p130 and RB was confirmed by immunoblot of lysates of cells under serum starvation and contact arrest conditions (Fig. 4a).

DREAM and RB cooperate to repress early and late cell cycle gene expression in G0 and G1. a Knockout of RB, p130 was measured by immunoblot from serum starved and contact arrested HFFs expressing sgRNA against control (sgCTL), RB (sgRB1), p130 (sgP130), or RB and p130 (sgRB1+sgP130). b Relative mRNA abundance of indicated transcript was measured by RT-qPCR in contact arrested, serum-starved HFFs (4 bio rep). c S phase levels were measured by BrdU incorporation in HFFs in contact arrest and serum starvation (3 bio rep) or 24 h split conditions (6 bio rep). d HFFs were harvested after splitting and mRNA abundance of indicated transcript was measured by RT-qPCR (4 bio rep). e HFFs were contact arrested and serum starved and lysates were subjected to immunoprecipitation using p107, LIN9, and IgG antibodies and analyzed by immunoblot. Different exposures of same membrane shown for LIN9. Statistics calculated using Student’s t-test. P-values as indicated * < 0.05, ** < 0.01. See also Figs. S5–7
To assess the efficiency of RB1 and p130 knockout in CRISPR-edited HFFs, we performed next-generation sequencing on an amplicon generated by nested PCR centered on the PAM site of the corresponding sgRNA at the p130 (RBL2) and RB (RB1) genetic loci. gDNA was isolated from HFFs four passages after antibiotic selection was complete. We found diverse editing of the RB1 gene in all reads with the four highest represented reads shown in Figure S6A. A frameshift mutation in the RBL2 locus was observed in 100% of reads from the sgP130 and the sgRB1+sgP130 cells resulting in a fragment containing the N-terminal 90 residues (Figure S6A-C). Since HFFs were unable to tolerate single cell cloning, we maintained the knockout cells as a population. We cannot exclude the possibility of a small fraction of knockout cells retaining a WT p130 or RB1 allele in our population.
To test the effect of RB and p130 loss on cell cycle gene expression during G0 and G1, transcript levels of early and late cell cycle genes were measured by RT-qPCR under serum starved and contact inhibited conditions (Fig. 4b). Loss of p130 (sgP130) did not have a significant effect on levels of cell cycle gene expression compared to control cells under quiescent conditions (Fig. 4b). In contrast, loss of RB alone (sgRB1) led to a significant increase in levels of E2F1, MYBL2, and BUB1 in quiescent conditions. Furthermore, when RB and p130 were lost in combination (sgRB1+sgP130), we observed significantly higher levels of early and late cell cycle genes compared to cells with RB knockout alone. Increased cell cycle gene expression in sgRB1+sgP130 cells was not explained by increased cell cycle kinetics as no difference in rates of BrdU incorporation was measured in contact arrested or 24 h released cells (Fig. 4c). Furthermore, no change in growth rate in low serum conditions was observed when p130 or RB were lost compared to the other HFF derivatives, consistent with reports on the effect of loss of RB in other human cell lines (Figure S5C-D) [34]. sgRB1+sgP130 cells enter S phase with slightly quicker kinetics (21 versus 24 h) but at 24 h all four cell lines have similar levels of BrdU incorporation (Figure S5E). These data indicate that DREAM and RB cooperate to repress the expression of early and late cell cycle genes during G0 under serum starvation and contact arrest conditions.
We next tested the effect of DREAM and RB on expression of early cell cycle genes during cell cycle entry (Fig. 4d). CCNE2 transcript levels were unaffected in quiescent cells (0 h) with p130 loss but were significantly increased in cells with RB loss, similar to reports using mouse cell lines [34]. CCNE2 levels further increased when p130 and RB were both lost, indicating a cooperative effect of DREAM and RB during quiescence. We tested the effect on gene expression at 8 and 10 h after release from quiescence. Again, we observed no effect of p130 loss alone compared to control cells while RB1 knockout cells had significantly increased levels at 8 and 10 h after release compared to control cells. Furthermore, combined knockout of p130 and RB led to significantly increased levels of CCNE2 at 8 and 10 h after release. The cooperative effect of DREAM and RB to repress CCNE2 at 0, 8, and 10 h was also observed for CDC6 and MCM5. No significant differences in cell cycle gene expression were found in cells harvested at 24 h even when RB and p130 was lost, consistent with our finding that sgRB1+sgP130 cells have similar S phase levels to control cells at 24 h (Figure S5F). These data indicate that DREAM and RB cooperate to repress cell cycle gene expression during progression from G0 into G1 and that loss of RB and p130 is not sufficient to alter the proliferation rate of primary cells.
p107 does not form DREAM complex or repress early cell cycle genes during G1
The RB-related protein p107 is a cell cycle-regulated gene and, as expected, higher levels of p107 were observed in sgP130 and sgRB1 cells (Fig. 4a). We asked whether p107 could substitute for p130 and form a DREAM-like complex in the knockout cells. Lysates were prepared from confluent cells and blotted (Input) or immunoprecipitated with p107 and LIN9 antibodies (Fig. 4e). Antibodies to p107 co-precipitated E2F4 in sgRB1, sgP130, and sgRB1+sgP130 cells (Fig. 4e). In addition, p107 co-precipitated LIN9 in sgP130 and sgRB1+sgP130 cells, but not in control or sgRB1 HFFs. The interaction between LIN9 and E2F4 in cells lacking p130 was substantially reduced compared to control or sgRB1 cells, indicating that p107 was unable to form a strong link between MuvB and E2F4 and unable to form a complete DREAM-like complex.
To test whether p107 contributed to repression of cell cycle gene expression in sgP130, sgRB1, or sgRB1+sgP130 HFFs, we knocked down p107 with siRNA (siRBL1). siRBL1 transfection led to knockdown of p107 and not p130 in each cell line compared to siControl when measured by protein and mRNA levels (Figure S7A, S7B, S7F). Knockdown of p107 did not consistently lead to de-repression of early cell cycle genes MCM5, CDC6, and E2F1 (Figure S7C-E). Since Cyclin D–CDK4 activity drives the separation of MuvB and E2F4, we tested whether inhibition of CDK4 activity could promote DREAM complex assembly with p107 when p130 is lost. Inhibition of Cyclin D–CDK4/6 did not lead to increased levels of E2F4-MuvB interaction in sgRB1+sgP130 cells (Figure S7G). These data indicate that although p107 can bind E2F4 or MuvB when p130 was absent, knockdown of p107 did not significantly or consistently repress early cell cycle gene expression during G1.
RB and DREAM regulate an overlapping set of early cell cycle genes
Combined loss of p130 and RB led to higher levels of several cell cycle genes when compared to loss of RB alone in quiescent cells and during early G1 cell cycle entry. This raised the question of whether all RB regulated genes were also regulated by the DREAM complex or whether there are genes uniquely regulated by RB and not by DREAM. To address this question, we utilized a published meta-analysis of TP53 and cell cycle-regulated genome-wide expression and ChIP-seq studies to generate high-confidence target gene maps for DREAM, p53, RB-E2F, and MMB–FOXM1 [35]. The meta-analysis employed stringent cutoffs to ensure high-confidence targets gene lists.
To determine whether any cell cycle-regulated genes were exclusively repressed by RB-E2F (referenced as RB-only genes) and not repressed by DREAM, we compared the list of DREAM and RB-E2F targets (Fig. 5a). We identified a strong overlap of DREAM and RB target genes, with 419 of 506 (82.81%) RB-E2F genes also bound by DREAM in various ChIP-seq studies. In addition, there were 552 DREAM-only genes not bound by RB, but these were primarily G2/M or late cell cycle genes without E2F elements and containing CHR elements in their promoters. There were 86 genes classified as bound by RB and not by DREAM. To test whether these 86 genes could also be regulated by DREAM, we re-evaluated the data that led to their exclusion (Fig. 5b). We excluded genes lacking an E2F or CHR element (unlikely to bind specifically to RB or DREAM), those containing a CHR element (likely to bind to MuvB), and genes with evidence for p130, B-MYB, or FOXM1 binding in at least 1 ChIP-seq dataset. This re-analysis reduced the list of 86 to 18 potential RB-only genes. Next, we ranked the 18 potential RB-only genes by their p53 expression score since genes with lower p53 expression scores were more likely to be cell cycle-regulated (by p21 inhibition of CDK activity) and annotated whether they had been identified in E2F4, RB, and E2F1 ChIP-seq datasets. We identified CCNE2, CHAF1B, DONSON, and MCMBP to be the top four ranked genes most likely to be a true RB-only gene (Supplemental Table 1).

DREAM and RB repress an overlapping set of cell cycle genes. a Venn diagram of DREAM and RB-E2F targets identified in Fischer et al. [35]. b Schematic of exclusion “RB-E2F” genes from final potential RB-only list. c Chromatin occupancy of p130 (8 bio rep), E2F4 (6 bio rep), and LIN9 (8 bio rep) on indicated promoters was measured by ChIP-qPCR using chromatin isolated from contact arrested and serum-starved HFFs. d Relative mRNA levels of indicated transcripts in contact arrested and serum-starved HFFs was measured by RT-qPCR (6 bio rep). Statistics calculated using Student’s t-test. P-values as indicated * < 0.05, ** < 0.01, *** < 0.001. See also Table S1
To test whether these candidate genes were regulated by RB-only and not DREAM, we prepared chromatin from contact arrested HFFs that were serum starved for 48 h. We observed significant enrichment of LIN9 and E2F4 at the CCNE2 promoter and significant enrichment of p130, E2F4, and LIN9 at the DONSON promoter (Fig. 5c). These data indicate that DREAM can bind specifically to these candidate RB-only genes, despite missing the stringent cutoff set in the published meta-analysis. We also measured contribution of p130 and RB to expression of CHAF1B (Fig. 5d), DONSON (Fig. 5d), MCMBP (Fig. 5d), and CCNE2 mRNA (Fig. 4e), and observed an increase in gene expression when RB and p130 were lost in combination when compared to loss of RB alone. Together, the data demonstrated cooperation of DREAM and RB to repress the expression of the top four candidate RB-only genes, indicating that these genes were falsely identified as RB-only genes and were also repressed by DREAM.
p130 and RB contribute to sensitivity to palbociclib
Since palbociclib treatment inhibits CDK4 activity and maintains the DREAM complex and RB inhibition during cell cycle entry, we tested whether DREAM or RB were required for sensitivity to palbociclib. We measured S phase levels of cells split into palbociclib and found control and sgP130 cells were similarly sensitive to increasing concentrations of palbociclib while sgRB1 HFFs had reduced sensitivity compared to control cells at 200 nM (Fig. 6a, S8). Combined loss of p130 and RB led to a significant increase in resistance to palbociclib compared to RB loss alone. Increase in resistance of sgRB1+sgP130 cells to palbociclib was also observed using crystal violet as a measure of relative cell density (Fig. 6b).

p130 and RB contribute to sensitivity to palbociclib. a HFFs were split from confluency into palbociclib and BrdU incorporation was measured in cells harvested after 24 h (3 bio rep). Student’s t-test calculated compared to control cells. Two-way ANOVA used to compare curves. b Relative cell density was measured by crystal violent for cells seeded into palbociclib and harvested after 4 days (3 bio rep). Significance calculated as in a (c, d) mRNA abundance was measured by RT-qPCR at 8 or 10 h after splitting. 10+ cells were treated with 200 nM palbociclib for 2 h prior to harvest (5 bio rep). e Same as c and d, but lysates were analyzed by immunoblot. P-values are as indicated * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001, NS not significant. See also Figure S8
To test the impact of palbociclib on cell cycle gene expression in cells with RB and p130 loss, we treated control, sgRB1, sgP130, or sgRB1+sgP130 HFF cells with palbociclib for two hours prior to harvesting during cell cycle entry (Fig. 6c, d, compare 10+ to 8 and 10−). Significantly reduced levels of CDC6 and MCM5 gene expression was observed in control cells and p130 knockout cells containing an intact RB. In contrast, palbociclib treatment did not reduce MCM5 or CDC6 levels in sgRB1 cells despite reduction of p130 phosphorylation (Fig. 6c–e). These data indicate that although palbociclib can block disruption of the DREAM complex during cell cycle entry during the initial 6 h of cell cycle entry (Fig. 3c), reduced levels of cell cycle genes after 8 h by palbociclib was only dependent on RB status. Thus, DREAM is active and can repress gene expression during G0 and early G1, but after disruption of p130 from E2F4 by Cyclin D–CDK4 activity, DREAM was unable to reduce early cell cycle gene levels.
DREAM contributes to palbociclib sensitivity in cancer cell lines
Next, we tested whether DREAM contributed to palbociclib sensitivity in palbociclib-sensitive cancer cell lines. A549 is a non-small cell lung cancer cell line with wild type DREAM, RB, and p53 pathways [36]. To test whether palbociclib treatment could regulate the DREAM complex in A549 cells during cell cycle entry, lysates were collected before and after 24 h release into the cell cycle in the presence of 500 nM palbociclib (Fig. 7a). The DREAM complex was intact as determined by LIN9 co-precipitation with p130 and E2F4 in cells treated with palbociclib (Fig. 7a).

DREAM and RB contribute to sensitivity to palbociclib in cancer cell lines. a Parental A549 cells were harvested after 0 or 24 h of cell cycle entry with palbociclib (500 nM) and immunoprecipitated with LIN9 and IgG antibodies. b Knockout of RB, p130 was measured by immunoblot from A549 cells expressing sgRNA against control (sgCTL), RB (sgRB1), p130 (sgP130), or RB and p130 (sgRB1+sgP130). Cells were split from confluency and harvested after 24 h. c A549 cells were split into palbociclib and S phase was measured after 24 h by BrdU incorporation and PI staining (4 bio rep). Statistical significance was calculated by two-way ANOVA test. d A549 cells were split palbociclib (500 nM) and transcript levels were measured by RT-qPCR after 24 h (3 bio rep). Significance calculated by Student’s t-test. e T47D cells were split into palbociclib and harvested after 24 h and analyzed by immunoprecipitation and immunoblot using indicated antibodies. f Knockout of RB, p130 measured by immunoblot in T47D cells expressing sgRNA against control (sgCTL), RB (sgRB1), p130 (sgP130) or RB, and p130 (sgRB1+sgP130). * indicates nonspecific band. g T47D cells were split into palbociclib and BrdU incorporation was measured after 24 h (3 bio rep). h Model of cooperative effect of DREAM and RB control of cell cycle gene expression during G0 and G1. P-values are indicated as * for < 0.05, and ** for < 0.01, *** for < 0.001, and **** for < 0.0001. See also Figures S9–12
To determine whether DREAM and RB status contributes to palbociclib sensitivity in the A549 cells, we generated control, sgP130, sgRB1, and sgRB1+sgP130 derivative cell lines and assessed for knockout of RB and p130 by blotting lysates prepared from cells obtained 24 h after release from contact arrest (Fig. 7b). Loss of p130 or RB individually resulted in a significant increase in S phase levels after palbociclib treatment (Fig. 7c, S9). Notably, loss of RB and p130 together resulted in further significant increase in S phase levels compared cells with loss of p130 or RB alone as measured by ANOVA or Student’s t-test comparison for each concentration (Fig. 7c, S10A-E). A549 sgRB1 cells had ~70% of the S phase levels compared to sgRB1+sgP130 cells (Figure S10F). Level of resistance to palbociclib was also assessed by crystal violet staining after 4 days treatment (Figure S10G). Again, double knockout of RB and p130 led to higher rates of proliferation while treated with palbociclib compared to cells with loss of RB1 alone. Resistance to palbociclib was not an indirect effect of increased growth kinetics since all knockout cells grew at relatively the same rate (Figure S11). Reduced levels of the early cell cycle gene CDC6 after palbociclib treatment was dependent on p130 and RB status with sgRB1+sgP130 cells demonstrating significantly higher expression than all others (Fig. 7d). CCNE1 levels were not affected by loss of p130 in palbociclib treated cells, which may be due to other cell-specific increase of CCNE1 levels. These data indicate that intact DREAM and RB status contributes to sensitivity to palbociclib treatment in A549 cells.
To test whether p130 contributed to palbociclib sensitivity in other cancer cell lines, we used the palbociclib-sensitive T47D breast cancer cell line that is ER-positive with wild-type RB1 and DREAM complex [37, 38]. Palbociclib treatment induced DREAM complex formation in T47D cells as measured by LIN9 co-precipitation of p130 and E2F4 (Fig. 7e). To test the contribution of DREAM and RB to palbociclib-dependent cell cycle arrest in T47D cells, we generated p130 and RB knockouts (Fig. 7f). We tested the effect of p130 and RB knockout on T47D cell sensitivity to palbociclib by measuring S phase levels by BrdU incorporation after 24 h (Fig. 7g, S12). We observed that control and sgP130 cells were strongly sensitive to palbociclib at low doses while loss of RB alone resulted in a significant increase in S phase levels. Similar to HFFs and A549s, combined loss of p130 and RB (sgRB1+sgP130) led to significant increased S phase levels compared to sgRB1-only palbociclib treated T47D cells. We observed that sgRB1 cells had ~70% of the S phase levels compared to sgRB1+sgP130 cells, indicating that loss of p130 in combination with RB contributes to ~30% increase in S phase levels with palbociclib treatment (Figure S10H). These data demonstrate DREAM and RB contribute to CDK4 inhibition-induced cell cycle arrest in two different cancer cell lines.
Discussion
Our model is that the DREAM complex containing p130, E2F4, DP, and MuvB represses the expression of early cell cycle genes during G0 and early G1 (Fig. 7h). Midway through G1, Cyclin D expression leads to activation of CDK4 leading to phosphorylation of p130 and the first step of DREAM complex disruption with release of p130 and MuvB from E2F4 and reduced binding of p130 and MuvB to cell cycle-regulated promoters. During late G1, it is likely that Cyclin E-CDK2 activity contributes to release of p130 from the MuvB core when maximum early cell cycle gene expression occurs to push cells through the Restriction Point and into S phase [39].
We observed that loss of RB alone, but not p130, resulted in a significant increase in levels of cell cycle genes during quiescence and in G1. However, p130 loss combined with RB1 loss consistently led to a significant increase in cell cycle gene levels compared to RB1 loss alone. These data indicate RB has a dominant role in the repression of cell cycle genes while DREAM contributes to repression in the absence of RB. Our results are consistent with a recent report that found LIN37 knockout (a MuvB component) in combination with RB resulted in increased levels of gene expression in G0 [40]. However, our approach of knocking out p130 uniquely affected the DREAM complex while knockout of LIN37 could also impair activation of the MMB–FOXM1 complex in S/G2. In addition, we found no evidence for cell cycle-regulated genes that were exclusively regulated by RB and not by DREAM. Thus, DREAM and RB cooperate to repress cell cycle gene expression in G1.
It is unlikely that RB directly compensates for p130 loss by forming a DREAM complex since RB does not have the capability to bind to the MuvB complex [1]. Furthermore, p130 likely does not act to repress activator E2Fs in the absence of RB since p130 has a strong specificity for repressor E2F4-5 and does not bind E2F1-3 [41, 42]. Our model is that RB is able to control expression of early cell cycle genes through its direct repression of transcriptional activation by activator E2Fs, so the de-repression by loss of DREAM does not result in increased gene expression. However, in a RB-null cell, DREAM can weakly maintain repression of some cell cycle gene that can be overwhelmed by de-repressed activator E2Fs, resulting in increased cell cycle gene expression and S phase entry. The activity of DREAM in RB-null cells seems to be restricted to the initial 6 h of cell cycle entry as two-hour palbociclib treatment at 10 h in RB-null cells did not reduce cell cycle gene expression. In a RB and p130 null cell, lack of DREAM repression of cell cycle gene expression and lack of RB repression of activator E2Fs results in a further increase in cell cycle gene expression and near complete loss of repression of cell cycle genes. This dominant effect of RB activity was also observed in a mouse model of small cell lung cancer in which loss of p130 in combination with RB and p53 results in more aggressive tumors than RB and p53 loss alone [43, 44].
The specific role of the RB-like protein p107 is less studied in the context of a normal cell cycle. We found levels of p107 were increased in HFFs lacking p130 or RB, consistent with previous reports [45,46,47]. Although p107 could strongly interact with either E2F4 or LIN9, we were unable to detect significant levels of LIN9 binding to E2F4 in the absence of p130. Furthermore, reduced levels of p107 by siRNA did not result in a consistent change in cell cycle gene expression in the p130 and RB knockout cells. Our data indicate that p130 and RB have significant roles in repression of early cell cycle gene expression during G1 and p107 does not appear to provide any additional activity.
We identified Cyclin D–CDK4 as necessary for DREAM complex disruption during cell cycle entry from G0. Inhibition of CDK4 activity with palbociclib prevented the disruption of DREAM complex during cell cycle entry. While Cyclin D–CDK4 phosphorylation disrupted the p130-E2F4 interaction, it was not sufficient to disrupt p130 binding to MuvB. It was recently reported that continuous treatment of T98G cells with palbociclib for 16 h after release from quiescence maintained the interaction of p130 with LIN37 (MuvB component) [11]. This result likely reflected maintenance of p130 interaction with E2F4 as well as MuvB. Phosphorylation of RB at S608, the analogous site to p130-S672, results in a conformational change in RB that inhibits E2F binding [20]. If a similar structural mechanism exists in p130, this could explain the mechanism of p130-MuvB release from E2F4 during cell cycle entry.
Our findings reveal a potential role for DREAM as a tumor suppressor. We found that DREAM contributes to palbociclib sensitivity in normal and cancer cells which is novel and important since CDK4 inhibitors such as palbociclib are currently used clinically to treat a variety of tumor types alone and in combination with other drugs that reduce Cyclin D protein levels [24, 38, 48]. Despite the low frequency of p130 mutations in cancer, functional inactivation of RB by overexpression of Cyclin D1 or reduced levels of the CDK4 inhibitor, p16 (CDKN2A), are likely to also inactivate DREAM [49, 50]. Similarly, viral oncogenes such as E7 in human papillomavirus and Large T antigen in Merkel cell polyomavirus induced cancers are likely to inactivate both RB and DREAM [51, 52]. Importantly, the DREAM complex contributed to reduced cellular proliferation in a gastrointestinal stromal tumor cell line treated with imatinib and in ovarian cancer [53, 54]. In some tumor types such as small cell lung cancer (SCLC), genetic loss of p130 (RBL2 gene) has been reported [55]. In a mouse model of SCLC with RB and p53 loss, loss of p130 accelerates the rate of tumor formation [43, 44, 55]. Therefore, DREAM is likely to complement RB’s role in regulating proliferation of cancer cells.
Materials and methods
Human foreskin fibroblasts
Fibroblasts were isolated from de-identified natal foreskin specimens and grown in DMEM (Cellgro) containing 15% fetal bovine serum supplemented with 1% Pen Strep (GIBCO), and 1% Glutamax (GIBCO), and incubated at 37 °C with 5% CO2. Cells were treated with Palbociclib (PD-0332991) (SelleckChem # S1116) dissolved in sterile water or with CVT-313 (Santa Cruz Biotechnology, sc-221445) dissolved in DMSO by direct addition to media.
Cell lines
A549 and T47D cells were obtained from ATCC.
Lentiviral sgRNA expression vectors
The pLenti-CRISPR V2 sgRNA expression vectors that express Cas9 and either a puromycin-resistance gene (Addgene #52961) or a G418 resistance gene were used [56]. sgRNA sequences were designed using the Broad Institute sgRNA designer and cross-referenced for the fewest off-target effects using the MIT CRISPR design site. Oligonucleotides were synthesized containing 5′ and 3′ BsmBI sites to facilitate ligation into vector. The sgRNA oligos (including BsmBI sites) are reported in Supplemental Table 2.
Amplicon sequencing of RB1, RBL2 loci
Genomic DNA was isolated using Qiagen DNeasy Blood & Tissue kit and subjected to nested PCR using Pfu Ultra DNA polymerase. Genomic DNA was used a template to create larger fragment using primers centered on sgRNA target. The larger fragment was purified and used as template in subsequent PCR reaction. Smaller fragments were purified and sequenced by Next Generation Sequencing at Massachusetts General Hospital Center for Computational & Integrative Biology. PCR primers are listed in Supplemental Table 2.
RNAi knockdown of RBL1
Overall, 1.0 × 106 cells were seeded in 60 mm dishes and transfected with 5 pmol of siControl (Non-Targeting siRNA #1, D-001210-01-05) or siRBL1 (SMARTpool, M-003298-02-0005) from GE-Dharmacon using Lipofectamine RNAiMAX Transfection Reagent (Life Technologies, 13778030). Cells were harvested 48 h after transfection.
Protein analysis
Whole-cell lysates were prepared in EBC buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.5% NP-40, 1: 10,000 β-mercaptoethanol, 0.5 mM EDTA) with phosphatase inhibitor cocktail set I (Calbiochem) and protease inhibitor cocktail set I (Calbiochem). Protein extracts were boiled in SDS sample buffer (Boston BioProducts) before SDS-polyacrylamide gel electrophoresis (Criterion TGX precast gels, Bio-Rad). Resolved proteins were transferred to nitrocellulose membranes (Bio-Rad) with 75V for 1 h. Membranes were blocked with 5% milk in Tris Buffered Saline with 0.5% Tween-20 before incubation with primary antibodies overnight at 4 °C. Antibody detection was performed with horseradish peroxidase-conjugated secondary antibodies (Rockland) developed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and imaged with G:BOX Chemi detection system (Syngene). To perform immunoprecipitations, whole-cell lysates were incubated with antibody and magnetic Protein A/G beads (Pierce) overnight with end-over-end agitation at 4 °C. Beads were separated by magnet and washed with EBC lysis buffer five times before proteins were eluted by boiling in SDS sample buffer (Boston BioProducts) for 5 min. To perform cellular fractionation, cells were resuspended in cytoplasmic extraction buffer (10 mM Tris-HCl pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT). The nuclear pellet was then lysed with nuclear extraction buffer (20mM Tris-HCl, 25% glycerol, 1.5 mM MgCl2, 240 mM NaCl, 0.2 mM EDTA, 0.5 mM DTT). Supernatants from both steps were treated with benzonase (1:1000, Sigma Aldrich) before analysis by immunoblot.
Antibodies
The following commercial antibodies were used for IP and immunoblot analysis: anti-β-actin (D6A8, Cell Signaling), anti-Cyclin D1 (2922S, Cell Signaling), anti-Cyclin E (HE12, Santa Cruz), anti-E2F4 (C-20, Santa Cruz), IgG (2729, Cell Signaling), anti-LIN9 (BL2981, Bethyl), anti-MCM5 (ab17967, Abcam), anti-p107 (C-18, Santa Cruz), anti-p130 (C-20, Santa Cruz), anti-p130-pS672 (ab76255, Abcam), anti-RB (554136, BD Pharmingen), anti-RB-pT821 (ab32015, Abcam), and anti-Vinculin (H-10, Santa Cruz). The following antibodies were used for ChIP: anti-E2F4 (C-20, Santa Cruz), IgG (2729, Cell Signaling), anti-LIN9 (BL2981, Bethyl), and anti-p130 (C-20, Santa Cruz).
Validation of phospho-antibodies with phosphatase
Cells were lysed using 10× cell pellet volume of EBC lysis buffer with protease inhibitor (Calbiochem). Lysate was incubated with/without phosphatase inhibitor (Calbiochem) with lambda phosphatase (New England BioLabs) at 30 °C for 1 h and analyzed by immunoblot.
Cell cycle analysis
Cells were incubated with 3 μg/mL BrdU (Sigma Aldrich) for 1 h before harvesting via trypsin digestion and fixation with cold 70% ethanol. Cells were permeabilized with 2N HCl and blocked Super Block Buffer (Thermo Scientific) before incubation with anti-BrdU-FITC (Fischer Scientific) overnight. Cells were washed with PBS-T and incubated with 1 mg/mL Propidium Iodide (Sigma) with 0.5 mg/mL RNase (Sigma) before analysis by flow cytometry (BD FACS Canto II).
Crystal violet staining
Overall, 5×104 cells were seeded into 6 well plates and harvested by staining with 5% Crystal Violet stain in PBS (Sigma Aldrich) at 25 °C for 20 min and destained with ddH2O. Crystal violet was solubilized with methanol and absorbance was measured with spectrophotometer at 570 nm.
Chromatin immunoprecipitation-qPCR
Digested chromatin was prepared using SimpleChIP enzymatic Chromatin IP Kit (Cell Signaling Technologies) and incubated overnight with antibodies and magnetic ChIP Grade Protein A/G beads (Cell Signaling). Eluted chromatin was purified using QIAquick PCR Purification Kit (Qiagen) and analyzed by qPCR. Fold change occupancy was calculated by normalizing %input values at 0 h. ChIP-qPCR primers are listed in Supplemental Table 2.
RNA analysis by RT-qPCR
Total RNA was isolated using Qiagen RNeasy plus mini kit. cDNA was synthesized with a High-Capacity Reverse Transcription kit (Thermo Fischer). qPCR was performed using Brilliant III SYBR Master Mix using Aria Mx3000 or AriaMx real time PCR machines (Agilent Genomics). Raw Ct values were normalized to geometric mean of ACTB and B2M transcripts before normalization to experimental control indicated. RT-qPCR primers are listed in Supplemental Table 2.
References
Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, et al. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell. 2007;26:539–51.
Pilkinton M, Sandoval R, Colamonici OR. Mammalian Mip/LIN-9 interacts with either thep107, p130/E2F4 repressor complex or B-Myb in a cell cycle-phase-dependent context distinct from the Drosophila dREAM complex. Oncogene. 2007;26:7535–43.
Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012;26:474–89.
Schmit F, Korenjak M, Mannefeld M, Schmitt K, Franke C, von Eyss B, et al. LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle Georget Tex. 2007;6:1903–13.
Osterloh L, von Eyss B, Schmit F, Rein L, Hübner D, Samans B, et al. The human synMuv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. EMBO J. 2007;26:144–57.
Araki K, Nakajima Y, Eto K, Ikeda M-A. Distinct recruitment of E2F family members to specific E2F-binding sites mediates activation and repression of the E2F1 promoter. Oncogene. 2003;22:7632–41.
Marceau AH, Felthousen JG, Goetsch PD, Iness AN, Lee H-W, Tripathi SM, et al. Structural basis for LIN54 recognition of CHR elements in cell cycle-regulated promoters. Nat Commun. 2016;7:12301.
Muller GA, Quaas M, Schumann M, Krause E, Padi M, Fischer M, et al. The CHR promoter element controls cell cycle-dependent gene transcription and binds the DREAM and MMB complexes. Nucleic Acids Res. 2012;40:1561–78.
Muller GA, Wintsche A, Stangner K, Prohaska SJ, Stadler PF, Engeland K. The CHR site: definition and genome-wide identification of a cell cycle transcriptional element. Nucleic Acids Res. 2014;42:10331–50.
Schmit F, Cremer S, Gaubatz S. LIN54 is an essential core subunit of the DREAM/LINC complex that binds to the cdc2 promoter in a sequence-specific manner: LIN54 is an essential core subunit of the DREAM/LINC complex. FEBS J. 2009;276:5703–16.
Guiley KZ, Liban TJ, Felthousen JG, Ramanan P, Litovchick L, Rubin SM. Structural mechanisms of DREAM complex assembly and regulation. Genes Dev. 2015;29:961–74.
Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–16.
Fischer M, Müller GA. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit Rev Biochem Mol Biol 2017;52:638–62.
Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297–306.
Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife. 2014;3:e02872.
Hitomi M, Stacey DW. Cyclin D1 production in cycling cells depends on ras in a cell-cycle-specific manner. Curr Biol. 1999;9:1075–84.
Brown JR, Nigh E, Lee RJ, Ye H, Thompson MA, Saudou F, et al. Fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol. 1998;18:5609–19.
Lew DJ, Dulić V, Reed SI. Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell. 1991;66:1197–206.
Ohtsubo M, Roberts JM. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science. 1993;259:1908–12.
Burke JR, Liban TJ, Restrepo T, Lee H-W, Rubin SM. Multiple mechanisms for E2F binding inhibition by phosphorylation of the retinoblastoma protein C-terminal domain. J Mol Biol. 2014;426:245–55.
Canhoto AJ, Chestukhin A, Litovchick L, DeCaprio JA. Phosphorylation of the retinoblastoma-related protein p130 in growth-arrested cells. Oncogene. 2000;19:5116–22.
Farkas T, Hansen K, Holm K, Lukas J, Bartek J. Distinct phosphorylation events regulate p130- and p107-mediated repression of E2F-4. J Biol Chem. 2002;277:26741–52.
Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–82.
Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6:353–67.
Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48:2388–406.
Davis PK, Ho A, Dowdy SF. Biological methods for cell-cycle synchronization of mammalian cells. Biotechniques. 2001;30:1322–6. 1328, 1330–11
Tobey RA, Valdez JG, Crissman HA. Synchronization of human diploid fibroblasts at multiple stages of the cell cycle. Exp Cell Res. 1988;179:400–16.
Bhattacharya S, Garriga J, Calbó J, Yong T, Haines DS, Graña X. SKP2 associates with p130 and accelerates p130 ubiquitylation and degradation in human cells. Oncogene. 2003;22:2443–51.
Hansen K, Farkas T, Lukas J, Holm K, Rönnstrand L, Bartek J. Phosphorylation-dependent and -independent functions of p130 cooperate to evoke a sustained G1 block. EMBO J. 2001;20:422–32.
Gaubatz S, Lees JA, Lindeman GJ, Livingston DM. E2F4 is exported from the nucleus in a CRM1-dependent manner. Mol Cell Biol. 2001;21:1384–92.
Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–38.
Jorda R, Hendrychová D, Voller J, Řezníčková E, Gucký T, Kryštof V. How selective are pharmacological inhibitors of cell-cycle-regulating cyclin-dependent kinases? J Med Chem. 2018;61:9105–20.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308.
Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–87.
Fischer M, Grossmann P, Padi M, DeCaprio JA. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016;44:6070–86.
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7.
Aka JA, Adjo Aka J, Lin S-X. Comparison of functional proteomic analyses of human breast cancer cell lines T47D and MCF7. PLoS ONE. 2012;7:e31532.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35.
Odajima J, Saini S, Jung P, Ndassa-Colday Y, Ficaro S, Geng Y, et al. Proteomic landscape of tissue-specific cyclin E functions in vivo. PLoS Genet. 2016;12:e1006429.
Mages CF, Wintsche A, Bernhart SH, Müller GA. The DREAM complex through its subunit Lin37 cooperates with Rb to initiate quiescence. eLife. 2017;6. https://doi.org/10.7554/eLife.26876.
Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20.
Liban TJ, Thwaites MJ, Dick FA, Rubin SM. Structural conservation and E2F binding specificity within the retinoblastoma pocket protein family. J Mol Biol. 2016;428:3960–71.
Ho VM, Schaffer BE, Karnezis AN, Park KS, Sage J. The retinoblastoma gene Rb and its family member p130 suppress lung adenocarcinoma induced by oncogenic K-Ras. Oncogene. 2009;28:1393–9.
Schaffer BE, Park K-S, Yiu G, Conklin JF, Lin C, Burkhart DL, et al. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res. 2010;70:3877–83.
Lara MF, García-Escudero R, Ruiz S, Santos M, Moral M, Martínez-Cruz AB, et al. Gene profiling approaches help to define the specific functions of retinoblastoma family in epidermis. Mol Carcinog. 2008;47:209–21.
Mayhew CN, Bosco EE, Fox SR, Okaya T, Tarapore P, Schwemberger SJ, et al. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res. 2005;65:4568–77.
Ruiz S, Santos M, Segrelles C, Leis H, Jorcano JL, Berns A, et al. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Dev Camb Engl. 2004;131:2737–48.
Corona SP, Generali D. Abemaciclib: a CDK4/6 inhibitor for the treatment of HR+/HER2– advanced breast cancer. Drug Des Devel Ther. 2018;12:321–30.
Ortiz AB, Garcia D, Vicente Y, Palka M, Bellas C, Martin P. Prognostic significance of cyclin D1 protein expression and gene amplification in invasive breast carcinoma. PLoS ONE. 2017;12:e0188068.
Tang B, Li Y, Qi G, Yuan S, Wang Z, Yu S, et al. Clinicopathological significance of CDKN2A promoter hypermethylation frequency with pancreatic cancer. Sci Rep. 2015;5:13563.
Hesbacher S, Pfitzer L, Wiedorfer K, Angermeyer S, Borst A, Haferkamp S, et al. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget. 2016;7:32956–68.
Nor Rashid N, Yong ZL, Yusof R, Watson RJ. HPV 16E7 and 48E7 proteins use different mechanisms to target p130 to overcome cell cycle block. Virol J. 2016;13. https://doi.org/10.1186/s12985-015-0460-8.
Boichuk S, Parry JA, Makielski KR, Litovchick L, Baron JL, Zewe JP, et al. The DREAM complex mediates GIST cell quiescence and is a novel therapeutic target to enhance imatinib-induced apoptosis. Cancer Res. 2013;73:5120–9.
MacDonald J, Ramos-Valdes Y, Perampalam P, Litovchick L, DiMattia GE, Dick FA. A systematic analysis of negative growth control implicates the DREAM complex in cancer cell dormancy. Mol Cancer Res MCR. 2017;15:371–81.
George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53.
Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4.
Acknowledgements
We thank Viktor Huang and Christian Berrios for providing de-identified neo-natal foreskin specimens. We thank William G. Kaelin Jr. (DFCI) for reagents, and Nicole Persky and Cory Johannessen (Broad Institute) for personal communication regarding CDK4 mutant alleles. We thank Donglim Esther Park for RT-qPCR primers for JAG1, MDM4, and CK1α. We thank the Dana-Farber Flow Cytometry Core. We thank members of the DeCaprio laboratory for critical reading of this manuscript.
Funding
This work was supported by US Public Health Service grants R01CA63113, R01CA173023, and P01CA050661 to JAD; F31CA220800 to AES; K08CA222657 to MGO; and a Harvard Landry Biology Consortium Fellowship to AES.
Author information
Authors and Affiliations
Contributions
Conceptualization, AES and JAD; Methodology, AES and JAD; Investigation, AES, MGO, and HEN; Resources, AES, MGO, and HEN; Writing—original draft, AES; Writing—review and editing, AES, MGO, and JAD; Supervision, JAD; Funding acquisition, AES and JAD.
Corresponding author
Ethics declarations
Conflict of interest
JAD has received honoraria for participation in an advisory board from Merck & Co., Inc. JAD has received research funding from Constellation Pharmaceuticals, Inc. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Schade, A.E., Oser, M.G., Nicholson, H.E. et al. Cyclin D–CDK4 relieves cooperative repression of proliferation and cell cycle gene expression by DREAM and RB. Oncogene 38, 4962–4976 (2019). https://doi.org/10.1038/s41388-019-0767-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-019-0767-9
- Springer Nature Limited
This article is cited by
-
HDAC activity is dispensable for repression of cell-cycle genes by DREAM and E2F:RB complexes
Nature Communications (2024)
-
miR‐449a: A Promising Biomarker and Therapeutic Target in Cancer and Other Diseases
Cell Biochemistry and Biophysics (2024)
-
Cell cycle control in cancer
Nature Reviews Molecular Cell Biology (2022)
-
The MuvB complex binds and stabilizes nucleosomes downstream of the transcription start site of cell-cycle dependent genes
Nature Communications (2022)
-
Structure and function of MuvB complexes
Oncogene (2022)