
Abstract
Copy number gains, point mutations and epigenetic silencing events are increasingly observed in genes encoding elements of the Ras/Raf/MEK/ERK signaling axis in human breast cancer. The three Raf kinases A-Raf, B-Raf, and Raf-1 have an important role as gatekeepers in ERK pathway activation and are often dysregulated by somatic alterations of their genes or by the aberrant activity of receptor tyrosine kinases (RTKs) and Ras-GTPases. B-Raf represents the most potent Raf isoform and a critical effector downstream of RTKs and RAS proteins. Aberrant RTK signaling is mimicked by the polyoma middle T antigen (PyMT), which activates various oncogenic signaling pathways, incl. the RAS/ERK axis, in a similar manner as RTKs in human breast cancer. Mammary epithelial cell directed expression of PyMT in mice by the MMTV-PyMT transgene induces mammary hyperplasia progressing over adenoma to metastatic breast cancer with an almost complete penetrance. To understand the functional role of B-Raf in this model for luminal type B breast cancer, we crossed MMTV-PyMT mice with animals that either lack B-Raf expression in the mammary gland or express the signaling impaired B-RafAVKA mutant. The AVKA mutation prevents phosphorylation of T599 and S602 in the B-Raf activation loop and thereby activation of the kinase by upstream signals. We demonstrate for the first time that B-Raf expression and activation is important for tumor initiation in vivo as well as for lung metastasis. Isogenic tumor cell lines generated from conditional Braf knock-out or knock-in mice displayed a reduction in EGF-induced ERK pathway activity as well as in proliferation and invasive growth in three-dimensional matrigel cultures. Our results suggest that B-Raf, which has been hardly studied in the context of breast cancer, represents a critical effector of the PyMT oncoprotein and invite for an assessment of its functional role in human breast cancer.
Similar content being viewed by others


Introduction
Breast cancer is one of the most common cancers worldwide [1]. Although there has been marked progress in treating breast cancer patients, mortality remains high. Especially, its high heterogeneity makes it difficult to identify common strategies to treat breast cancer [2]. Recent genomic studies identified specific mutations and gene copy aberrations as potential new therapeutic targets [1, 3]. In addition to well-known genetically altered oncogenes and tumor suppressors, e.g., TP53, RB1, BRCA1, EGFR, FGFR, or MET, many lesser-known or in terms of breast cancer lesser-characterized genes were found to be mutated or amplified. Interestingly, many genes encoding members of the Ras/Raf/MEK/ERK axis are amplified in breast cancer, incl. BRAF in triple-negative tumors [1]. Moreover, a recent study reported that 62% of poor prognosis luminal type B breast cancers have inactivated either the DAB2IP or RASAL2 tumor suppressor genes, whose products suppress the activity of this axis by promoting GTP hydrolysis in RAS proteins [4,5,6]. Concomitant loss of both RasGAPs was even observed in a quarter of luminal B tumors, was correlated with their aggressiveness and promoted invasion and metastasis in breast cancer models [6].
As the Ras/Raf/MEK/ERK pathway is often deregulated in various diseases, in particular cancer, its members are already pursued as targets for therapeutic inhibition [7, 8]. These approaches, however, have been hampered by the plasticity and robustness of this pathway mediated by the various isoforms encoding the RAS, RAF MEK, and ERK proteins and their intricate regulation by protein–protein interaction events and feedback loops [7, 9]. The underestimation of pathway complexity is best illustrated by the phenomenon of paradoxical MEK/ERK activation by inhibitor bound or otherwise inactivated Raf molecules in the context of excessive Ras signaling, thereby promoting drug resistance and secondary neoplasia [10,11,12,13,14]. The Raf family comprises three Ser/Thr-kinases: A-Raf, B-Raf, and Raf-1 (aka C-Raf) [15]. B-Raf represents the major MEK activator and is necessary for placental development, as it is illustrated by the embryonic lethal phenotype of Braf knock-out mice [16,17,18,19]. B-Raf also displays the most potent transforming activity among the three Raf kinases and is activated by somatic alterations in cancer [20, 21]. The most common mutation, the V600E substitution, occurs in about 7% of tumors and imitates the conformational changes mediated by phosphorylation of T599 and S602 in the activation segment and thus generates a constitutively active oncoprotein exempted from multiple layers of positive and negative regulation [13, 22, 23]. Consequently, B-Raf has emerged as a therapeutic target and inhibitors such as vemurafenib yield unprecedented response rates in BRAFV600E mutant melanoma and hairy cell leukemia [7].
As the genes encoding B-Raf or its upstream activators such as Ras or receptor tyrosine kinases (RTK), e.g., the EGFR or FGFR, are often amplified or activated in breast cancer [1, 3, 5, 24], we asked whether B-Raf has a functional role in breast cancer initiation and progression in vivo. Therefore, we used a murine breast cancer model in which the polyoma middle T (PyMT) antigen is expressed in the mammary epithelium under the long-terminal repeat (LTR) of the mouse mammary tumor virus (MMTV-PyMT) [25]. This model is relevant to human breast cancer as the PyMT oncoprotein employs the same signaling pathways as the aforementioned RTKs, i.e., PI3K/AKT and RAS/ERK, and represents a potent oncogenic driver in its own right [2, 26,27,28,29,30]. MMTV-LTR-driven PyMT expression directs mammary epithelial cells towards a tumor phenotype mimicking the poor prognosis luminal B subtype of human breast cancer [29, 31]. PyMT induces multi-focal tumors progressing from initial hyperplasia over adenocarcinoma to invasive carcinoma with frequent lung metastases within an immunocompetent animal [32, 33]. Interestingly, a recent study demonstrated that the spontaneous metastasis of PyMT-driven mammary tumors was not accompanied by aneuploidy and copy number variations, further supporting that activation of the aforementioned signaling pathways by this viral oncoprotein is necessary and sufficient for full-blown tumor progression, including metastasis [34]. In the present study, we show that B-Raf activation represents a driver of initiation and tumor growth in vivo and that loss of B-Raf activity resulted in a drastic reduction of metastases and improved overall survival.
Results
Specific deletion of B-Raf in mammary epithelial cells impairs tumor progression and metastasis
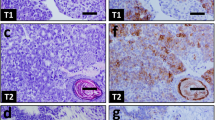
To investigate the role of B-Raf in breast cancer, we employed the MMTV-LTR-PyMT (PyMT)-driven model that is characterized by multi-focal occurrence of mammary carcinomas in multiple mammary glands and a high incidence of lung metastasis [25]. Due to the embryonic lethality caused by homozygous transmission of germ-line Braf knock-out alleles [18], we crossed conditional Braf knock-out mice (BraffloxE12) [35] with the WAP-Cre deleter strain [36]. This isogenic approach allows Cre-mediated excision of the floxed exon 12, which is critical for both B-Raf expression and function, from the Braf locus in a mammary epithelial cell-specific manner upon pregnancy (Fig. 1a). Braf recombination and the subsequent loss of its protein product in the tumor tissue were confirmed by genotyping PCR (Fig. 1b) and immunohistochemistry (Fig. 1c and Supplementary Figure 1), respectively. In homozygous WAP-Cre;PyMT;BrafΔE12/ΔE12 mice, B-Raf expression was absent in cancer cells but not in the tumor stroma. It should be noted that, even in case of a complete Cre-mediated recombination within the tumor cell compartment, all other cell types of the tumor microenvironment do not produce Cre and hence retain B-Raf expression. This could explain the faint and more prominent BraffloxE12 PCR amplicons in tumor 1 and 2, respectively (Fig. 1b). Alternatively and not mutually exclusive to this scenario, individual tumor cell subclones might have escaped Cre-mediated recombination, as it has been described for other PyMT-driven tumor models using MMTV-Cre [37, 38]. Indeed, closer inspection of primary tumor tissue identified isolated patches of B-Raf positive tumor cells (Supplementary Figure 1). This finding is in agreement with previous reports showing that the WAP-Cre deleter can generate genetic mosaics [36, 39].

Mammary gland specific knock-out of B-Raf leads to reduction of breast tumor growth and impairs metastatic lung colonization. a Crossing scheme to obtain PyMT expressing mice allowing WAP-Cre-mediated inactivation of the Braf locus (ΔE12). Mammary specific Cre activity and Braf knock-out (KO) was induced by pregnancy and tumors were let grown until they reached the ethical endpoint. For endpoint analysis, tumors and lungs were subjected to immunohistochemistry. b Representative Braf genotyping results of tail and tumor DNA for Cre positive homozygous and heterozygous floxE12 mice. c Immunohistochemical staining for B-Raf in tumor tissue confirms recombination of Braf locus on protein level. Bars indicate same magnification of all section images. The red and yellow arrows indicate tumor and stromal cells, respectively. d Average number of tumors per mouse at week 14 revealed a 50% reduction in KO animals compared to WT (+/+: n = 12; ΔE12/+: n = 7; ΔE12/ΔE12: n = 5; one-way ANOVA with post hoc testing; *p < 0.05, **p < 0.01). e Homozygous Braf KO animals show a significant elongation of overall survival defined as time until animals reach end-stage tumor burden (+/+: n = 15; ΔE12/+: n = 9; ΔE12/ΔE12: n = 12; log-rank test; *p < 0.05; **p < 0.01). f Quantification of lung micrometastases reveals a significant protection of mice that lost B-Raf protein (+/+: n = 6; ΔE12/+: n = 5; ΔE12/ΔE12: n = 5; one-way ANOVA with post hoc testing; *p < 0.05). Note that animals that carry a floxed allele(s) but lack a Cre transgene were also included in the plus/plus population as they retain B-Raf expression. g Representative histology sections of lungs of tumor bearing mice showing metastases (red arrows)
By comparing cohorts of PyMT transgenic mice with heterozygous and homozygous deletion of Braf exon 12 in cancer cells with the Braf proficient control cohort (+/+), we detected a significantly reduced frequency of tumors per mouse formed in WAP-Cre;PyMT;BrafΔE12/ΔE12 animals (Fig. 1d). Exon 12 deletion also caused a significant gene-dosage dependent delay in the development of end-stage tumor burden (Fig. 1e), and most importantly B-Raf deficiency in mammary cancer and epithelial cells strongly impaired the development of lung micrometastases (Fig. 1f, g).
B-Raf deletion impairs MEK/ERK signaling, cancer cell growth, and cancer cell motility
To assess the consequences of B-Raf deficiency on ERK pathway activity and cellular behavior, we isolated and cultured cells from homozygous PyMT;BraffloxE12/floxE12 tumors. After spontaneous outgrowth and immortalization, tumor cells were infected with a tightly controlled doxycycline (Dox) inducible retroviral expression system [40] allowing conditional Cre expression (Fig. 2a). In two independently generated cell lines, named Py-E-24 and Py-E-18, treatment with Dox led to in vitro recombination of the Braf locus (Fig. 2a and Supplementary Figure 2a). Four days after induction of Cre expression, B-Raf levels were strongly reduced as confirmed by Western blots (Fig. 2b and Supplementary Figure 2b). EGF stimulation of one of the floxE12 lines (Py-E-24) revealed impaired MEK and ERK phosphorylation upon loss of B-Raf protein. Likewise, we observed a reduction in the ERK-mediated phosphorylation of c-Fos on S32, which has a critical role in the stabilization of this transcription factor (Fig. 2b, c) [41]. These signaling defects could be confirmed in the additional floxE12 cell line (Py-E-18) (Supplementary Figure 2b/c). We further demonstrate that loss of B-Raf expression was not associated by the compensatory upregulation of the two other Raf family members, Raf-1 and A-Raf (Supplementary Figure 2e). In this independent experiment, we demonstrated again a reduction in the levels of phosphorylated c-Fos and observed a reduction in the expression of total c-Fos, FosB, and phospho-Fra1 in the B-Raf-deficient population (Supplementary Figure 2f). The reduction of phospho-Fra1 is of particular interest as a recent paper demonstrated that this immediate early gene product represents an excellent read-out for long-term persistence effects of ERK signaling [42]. These data further underscore that B-Raf deficiency impairs the ERK-mediated immediate early gene response in PyMT-driven breast cancer cells.

B-Raf-deficient tumor cells show decreased MAPK signaling and proliferation. a Scheme to gain PyMT cell lines with floxed Braf loci and containing a doxycycline (Dox)-regulated Cre expression system as exemplified for the BraffloxE12 allele. PyMT-positive homozygous BraffloxE12 mice were killed once average sized tumors grew out. Afterwards, tumor cells were infected with the two-component doxycycline inducible Cre expression system (pSH582, pSH461MK-EV/Cre) [40]. With this isogenic set-up, cells can be compared in the same assay with or without B-Raf protein expression. Representative Braf genotyping results from genomic DNA of floxE12 tumor cells containing the dox inducible Cre vector. The same primers as in Fig. 1 were used. b The floxE12 PyMT (Py-E-24) cell line was treated ±Dox 4 days prior to the experiment. Cells were stimulated with EGF (10 nM/ml) for the indicated time points and analyzed by Western blotting. The detection of each protein of interest was performed on the same gel but on non-contiguous lanes. Hence, contiguous lanes were separated in the floxE12 and ΔE12 subpanels. c Quantification of pMEK, pERK and pc-Fos levels at 5 min after EGF stimulation (n = 3, one-way ANOVA with post hoc testing, **p < 0.01, ***p < 0.001). d Py-E-24 cells were grown in a clonogenic proliferation assay. To this end, cells were treated ±Dox 4 days prior to the experiment. Afterwards, 500 cells were seeded per 6-well, grown for 21 days and afterwards stained with GIEMSA solution (Roth, Germany). e One thousand Py-E-24 cells were seeded in a 2% 3D Matrigel matrix as described previously [55] and treated ±Dox for Cre activation. Afterwards, cells were grown for 8 days and proliferation was assessed by microscopy. f Anchorage-independent growth assay. Cells were treated with doxycycline for 2 days. Five days after removing doxycycline, cells were plated either in normal 96-well microplate or poly-HEMA-coated 96-wells microplate. Left: Representative image of cells growing in poly-HEMA-coated plates. Right: Metabolic activity assessed by XTT assay of cells treated with or without Dox. Absorbance was measured 2 days after culture on normal tissue culture control plates (Con) or poly-HEMA-treated wells. Shown are two independent biological replicates carried out in octuplicates (Exp.1 and Exp.2). g Cellular migration of PyMT cells ±Dox analyzed by xCELLigence device and quantification of migrated cells at 24 h (n = 3, one-way ANOVA with post hoc testing, **p < 0.01)
Commensurate with the mild but reproducible ERK signaling defect in EGF-stimulated PyMT-driven breast cancer cells, loss of B-Raf was also accompanied by a clear reduction of colony growth in 2D cell culture and in MatrigelTM 3D matrix model in Py-E-24 cells (Fig. 2d, e) and, albeit to a lesser extent, in Py-E-18 cells (Supplementary Figure 3a). Py-E-18 cells were also growth impaired under anchorage-independent growth conditions (Fig. 2f), suggesting that B-Raf contributes to the stress resistance of PyMT cells.
In addition, deletion of B-Raf significantly impaired the motility of both Py-E-24 and Py-E-18 in xCELLigence in vitro migration assays (Fig. 2g and Supplementary Figure 3b). Py-E-18 cells, however, did not show this effect in a wound healing assay (Supplementary Figure 4a) and in an invasion assay (Supplementary Figure 4b). Moreover, in an experiment using an independent PyMT-driven cell line, in which we depleted B-Raf by shRNAs, we did not observe an obvious effect on migration (Supplementary Figure 4d/e). Taken together, our data suggest that the effects of B-Raf deficiency on migration are dependent on the cellular context and experimental setting. Nevertheless, our data so far provide in vitro and in vivo evidence for B-Raf as an enhancer of cancer progression and metastasis in the MMTV-PyMT mouse model for luminal type B breast cancer.
An intact B-Raf activation segment is required to promote breast cancer progression and metastasis
Raf kinases act not only via their enzymatic function but also as scaffolds and allosteric activators within signaling complexes [15]. To assess whether B-Raf activity is required to promote tumor initiation and progression in the MMTV-PyMT model, we crossed the PyMT allele with knock-in mice [23] constitutively expressing the signaling impaired B-RafAVKA protein (Supplementary Figure 5). B-RafAVKA lacks the two critical phosphorylation sites, T599 and S602, in its activation segment, whose phosphorylation is promoted by active Ras and is required to induce the active conformation of the Raf kinase domain [43,44,45]. As a result, the B-RafAVKA protein displays strongly impaired kinase activity and reduces the signaling output of the ERK pathway in vivo [23]. In contrast to Braf- deficient mice, however, heterozygous and homozygous BrafAVKA animals are viable, allowing their analysis in genetically engineered murine tumor models. In regard to the present study, homozygous BrafAVKA females display no overt phenotype in mammary gland development and lactation [23]. To assess whether B-RafAVKA modulates breast cancer development, cohorts of PyMT;BrafWT and PyMT;BrafAVKA mice were monitored weekly by palpation to determine tumor onset and growth. First tumors became detectable from week 9 onwards. However, tumor penetrance was 100% in PyMT;BrafWT animals at week 15, whereas homozygous PyMT;BrafAVKA mice showed a longer latency with the latest tumor onset at week 18 (Fig. 3a). This delayed tumor onset was accompanied by a significant reduction in tumor number per animal in PyMT;BrafAVKA mice compared to PyMT;BrafWT mice (Fig. 3b). This protection was not observed in heterozygous mice, which displayed no significant change in tumor latency or number compared to BrafWT. These findings suggest a role of B-Raf activation segment phosphorylation during tumor initiation. To investigate the early stages of breast tumorigenesis, we prepared mammary gland whole mounts of 7-week-old PyMT mice in which no palpaple tumors are present (Fig. 3c). Staining of the ductal trees confirmed hyperplasia in all glands. However, there were differences in quantity and localization of the hyperplasia between different samples. Some glands displayed local oligo-focal hyperplasia only at the ductal roots, while others showed multi-focal outgrowths distributed over the whole ductal tree. In addition, we defined an intermediate phenotype (Fig. 3c). When we correlated the distribution of the different growth phenotypes with the Braf genotype, we observed that glands with multi-focal precursor lesions were absent in PyMT;BrafAVKA/AVKA animals, while the multi-focal growth pattern prevailed in the control glands (Fig. 3c). Since multi-focal carcinogenesis is typical for PyMT mice, the altered growth pattern in PyMT;BrafAVKA/AVKA glands appears to be in line with the delayed tumor onset and reduced incidence of mammary cancers observed for this genotype. Because PyMT acts upstream of RAS [28], and consequently also of activation segment phosphorylation, our findings provide evidence that full B-Raf activation is required for tumor initiation.

Impairment of B-Raf activity slows down tumor growth and invasiveness. a Kaplan–Meier graph showing results from weekly tumor palpation. Compared to wildtype (WT) animals, BrafAVKA mice present a significant longer latency until palpable tumors become detectable (+/+: n = 16; A/+: n = 27; A/A: n = 16; log-rank test; *p < 0.05). b The average number of palpable tumors is significantly reduced in mice with impaired B-Raf activity (+/+: n = 15; A/+: n = 20; A/A: n = 12; one-way ANOVA with post hoc testing; **p < 0.01). c Exemplary whole mounts of mammary glands of 7-week-old mice show three different stages of hyperplasia. Basic or primary hyperplasia is restricted to one local area, whereas extended hyperplasia affects the whole ductal tree. Additionally, there is an intermediate phenotype. Quantification showed that AVKA mice show mainly primary restricted hyperplasia. WT and heterozygous mice additionally display extended hyperplasia (+/+: n = 8; A/+: n = 7; A/A: n = 5). Chi-square test was used for statistical analysis (*p < 0.05). d Composition of different tumor size per mouse at week 19 depending on the genotype. Homozygous BrafAVKA mice represent mainly very small and medium sized tumors, whereas wildtype and heterozygous mice display a greater proportion of very large tumors (+/+: n = 14; A/+: n = 19; A/A: n = 11; chi-square test; **p < 0.01). e Representative images of mammary gland tumors of mice with different Braf genotypes. Mice were killed at week 19, tumors and lungs were harvested and fixed in 4% PFA. Tumors of BrafAVKA mice are smaller on average and present less blood supply. White bar represents 0.45 cm. f Kaplan–Meier showing a significant increase in overall survival of AVKA mice compared to WT (+/+: n = 7; A/A: n = 8; log-rank test; ***p < 0.001). g Quantification revealed in homozygous BrafAVKA mice a drastic reduction in number of micrometastases compared to WT and heterozygous animals (+/+: n = 5; A/+: n = 5; A/A: n = 6; one-way ANOVA with post hoc testing; *p < 0.05; **p < 0.01). h Representative histology sections of lungs of tumor-bearing mice and quantification. Ki-67 (#M7249, DAKO, Germany) was used to visualize proliferating tumor cells in the lung tissue
Additionally, we observed a reduction in average tumor size at the day of sacrifice in homozygous PyMT;BrafAVKA/AVKA mice compared to PyMT;BrafWT/WT and heterozygous animals (Fig. 3d, e). Grouping of tumor sizes into three different categories, small (<0.5 cm), intermediate (0.5–1 cm) and large (>1 cm), revealed that PyMT;BrafAVKA/AVKA mice mainly developed small and intermediate tumors. In contrast, PyMT;BrafWT/WT and heterozygous mice show a significantly higher number of large tumors (Fig. 3d). In addition, a summative tumor burden of 3 cm, which defines the end-stage of the tumor model, was reached significantly later in PyMT;BrafAVKA/AVKA mice (Fig. 3f). Further, we quantified the presence of micrometastases in the lungs of PyMT;BrafAVKA/AVKA, PyMT;BrafWT/WT and heterozygous mice. Strikingly, we observed a strong and significant reduction of number of metastasis in PyMT;BrafAVKA/AVKA mice (Fig. 3g, h). Taken together, the homozygous BrafAVKA/AVKA mutation causes smaller tumors and less metastasis in the MMTV-PyMT breast cancer model.
At this point, however, it is not resolved whether the reduced tumor burden is determined by impaired tumor initiation or if there is an additional defect in the growth of established tumors. Furthermore, in the present experiment, the BrafAVKA allele was constitutively expressed in all cells of the mouse. To achieve a cancer cell-specific expression of B-RafAVKA, we crossed BraffloxAVKA animals, which maintain expression of wildtype B-Raf prior to Cre-mediated recombination [23], with MMTV-PyMT mice and the WAP-Cre deleter strain [36]. Upon pregnancy, the floxed minigene ensuring the expression of wildtype B-Raf is excised from the BraffloxAVKA locus leading to mammary epithelium specific expression of B-RafAVKA (Fig. 4a and Supplementary Figure 5). Importantly, the numbers of tumors per mouse (Fig. 4b) as well as of pulmonary micrometastases (Fig. 4c, d) were significantly reduced upon B-RafAVKA expression in mammary epithelial cells. Strikingly, there was no difference in occurrence of end-stage tumors in dependence of the B-Raf status (Fig. 4e), indicating that cancer cell specific BrafAVKA mutation primarily impairs tumor and metastasis initiation.

WAP-Cre BraffloxAVKA show impaired tumor growth and a reduction in lung micrometastasis. a Crossing scheme to obtain PyMT-expressing mice allowing WAP-Cre-mediated knock-in of BrafAVKA into the Braf locus (AVKA). WAP-Cre and PyMT were transferred via the male parent to avoid pregnancy-induced Cre activity or tumor growth in the breeding pairs. Braf locus heterozygous animals were used to generate littermates with all Braf genotypes as proper controls. Mammary-specific Cre activity and Braf knock-out (KO) was induced by pregnancy and tumors were let grown until they reached the ethical endpoint. Mice were killed, tumors and lungs were harvested and fixed in 4% PFA. b Average tumor number per mouse showed significant protection for homozygous mice (+/+: n = 7; A/+: n = 6; A/A: n = 5; one-way ANOVA with post hoc testing; *p < 0.05). c Quantification of lung micrometastasis reveals a drastic reduction in metastasis in homozygous AVKA mice. Heterozygous mice show intermediate phenotype with partial protection. d Representative histology sections of lungs of tumor bearing mice. e Kaplan–Meier showing the overall survival of WAP-Cre BraffloxAVKA mice (+/+: n = 9; A/+: n = 11; A/A: n = 7; log-rank test)
Defective phosphorylation of the B-Raf activation segment impairs MEK/ERK signaling and invasive cell growth but not cancer cell motility
To assess the molecular mechanisms for the observed in vivo effects, we isolated and cultured cells from homozygous PyMT;BraffloxAVKA tumors. After spontaneous outgrowth and immortalization, tumor cells were infected with the aforementioned doxycycline (Dox) inducible Cre expression system (Fig. 5a), allowing the switching from B-RafWT to B-RafAVKA expression in vitro. Successful recombination of the BraffloxAVKA alleles was confirmed by genotyping PCR and subsequent sequencing of the PCR-product (Fig. 5b). Next, we asked whether this independently generated isogenic PyMT-driven tumor cell model would recapitulate the attenuated growth behavior observed in the spontaneously arising tumors in the PyMT;BrafAVKA/AVKA (Fig. 3a) and the PyMT;BraffloxAVKA/floxAVKA;WAP-Cre models (Fig. 4). Therefore, we treated Py-A-45 cells either with Dox in vitro (B-RafAVKA) or left them untreated (B-RafWT) and implanted them subsequently orthotopically into the mammary fat pads of immunocompromised Rag2−/−;γc−/− mice (Table 1). In agreement with the PyMT; BrafAVKA/AVKA and the PyMT; BraffloxAVKA/floxAVKA; WAP-Cre models, we also observed an effect of B-RafAVKA expression on the growth of most individual tumors as 66.66% the B-RafWT expressing tumors caused the termination of the experiment, while the B-RafAVKA expressing tumors triggered the killing in only 37.50% of the animals. Moreover, six of the B-RafAVKA inoculates did not grow at all, while all B-RafWT-expressing tumors engrafted successfully. This suggests that B-RafAVKA expressing tumors are intrinsically impaired in their proliferative capacity in vivo and to manifest tumors.

B-Raf impairment decreases MAPK signaling and invasiveness in tumor cells. a Scheme to generate PyMT cell lines with floxed Braf loci and containing a doxycycline (Dox)-regulated Cre expression system as exemplified for the BraffloxAVKA allele. PyMT-positive homozygous BraffloxAVKA mice were eliminated once average sized tumors grew out. Tumor cells were spontaneously immortalized as described in Materials and methods and infected with the two-component doxycycline inducible Cre expression system (pSH582, pSH461MK-EV/Cre) [40]. b Sequencing results of isolated genomic DNA before and after Dox-dependent induction of Cre recombinase in Py-A-45 cells proofing successful exchange from WT sequence into AVKA sequence. c Py-A-45 cells ±Dox stimulated with EGF for indicated time points. Dox-treated cells presented knock-in of B-RafAVKA protein and a reduction of pMEK levels at peak level at 5 min after stimulation. d Quantification of pMEK levels at 5 min EGF stimulation (n = 3, one-way ANOVA with post hoc testing, *p < 0.05). e Cellular migration of PyMT cells ±Dox analyzed by xCELLigence device and quantification of migrated cells at 24 h (n = 3). f Py-A-45 cells were seeded in 3D Matrigel matrix and treated ±Dox for Cre activation. Expression of B-RafAVKA reduces cellular proliferation and invasiveness
EGF stimulation of Dox-treated and untreated cells revealed a slightly reduced MEK phosphorylation in the cells expressing B-Raf with the phosphosite-deficient activation segment (Fig. 5c, d). Like in B-Raf-deficient PyMT cells (Supplementary Figure 2e), induction of B-RafAVKA expression was not associated by the compensatory upregulation of Raf-1 or A-Raf (Supplementary Figure 6a/b).
The ability of those cells to migrate was not different from the control (Fig. 5e), but there was a striking reduction in 3D colony growth and invasive strand formation in the B-RafAVKA-expressing cells (Fig. 5f).
In summary, the analysis of B-RafAVKA in the context of the MMTV-PyMT breast cancer model provides evidence that the phosphorylation of T599 and S602 in the B-Raf activation segment primarily affects tumor initiation and the invasive seeding capacity of cancer cells. In light of the data for the constitutive BrafAVKA knock-in (Fig. 3), there might be also a contribution to tumor progression, which could also be influenced by B-Raf functions in cells of the tumor stroma. Comparing the selective, WAP-Cre-mediated deletion of B-Raf (Fig. 1) and expression of B-RafAVKA (Fig. 4) reveals that the main difference is the significantly delayed occurrence of end-stage tumors in WAP-Cre;PyMT;BrafΔE12/ΔE12 mice, while there was no effect on this parameter in WAP-Cre;PyMT;BrafAVKA/AVKA animals. In contrast, the number of tumors developing per mouse and the number of metastases were reduced to a similar extent in both models. This strengthens the evidence for a role in B-Raf activation segment phosphorylation in early tumor stages and metastatic seeding, while other capabilities of B-Raf, such as its scaffold function in signaling complexes, might determine its role in advanced primary cancer growth.
Discussion
Despite all the advances in the breast cancer field, new therapeutic targets are urgently needed for drug resistant tumors [46]. Ras/ERK signaling is often deregulated in breast cancer and elevated pERK levels have been described to drive tumor progression [6, 47, 48]. However, RAS or BRAF mutations themselves are rare in breast cancer and therefore their prognostic importance is not understood yet [1, 3, 49,50,51]. Thus, alternative mechanisms such as the aforementioned BRAF amplifications in basal-like cancers or the loss of RasGAPs, which is observed in about 62% of luminal type B tumors [6], could contribute to increased ERK activity. Moreover, B-Raf represents a main effector of various RTKs and their effector proteins such as Gab2 and Shp2, which are often overexpressed or aberrantly activated in breast cancer [2, 52, 53]. However, the role of B-Raf in an in vivo breast cancer model has not been characterized so far, although MDA-MB-231, one of the most frequently used human breast cancer cell lines, expresses the B-RafG464V oncoprotein [54] and active Raf proteins strongly impact on the acinar morphogenesis of MCF-10A cells [55,56,57]. Here, we show that B-Raf is strongly involved in the initiation and lung metastasis of PyMT-driven breast cancers in vivo. This is of particular interest given that PyMT recruits various pro-oncogenic effectors and that activation of the PI3K pathway in particular has been traditionally implicated in luminal type breast cancers and models thereof [26, 28]. However, our observation that B-Raf deficiency impairs lung metastasis in the MMTV-PyMT model ties in with recent observations that MCF-10A cells become invasive by the concomitant loss of the RasGAPs DAB2IP and RASAL2 and that this new trait required the ERK and PI3K effector pathways of RAS [4,5,6]. Furthermore, recently performed sequencing approaches in murine triple-negative breast cancer models driven by the loss of Trp53, either singly or in combination with Brca1 alterations, identified several oncogenic B-Raf and Raf-1 fusion proteins as spontaneously arising drivers [58]. Taken together, these data further illustrate the importance of the ERK axis for various molecular subtypes of breast cancer.
A striking finding of our study is that absence of either B-Raf protein or activity impacts the lung metastasis potential of PyMT-driven tumor cells. Considering the various steps of the metastatic cascade [59], one could envisage several mechanisms by which B-Raf promotes lung metastasis. Although our data show that the absence of B-Raf protein or activity impairs the migratory/invasive behavior in some but not all PyMT cell lines, we do not posit that B-Raf is a critical contributor to tumor cell trafficking in this context. Likewise, our tail vein injection experiments failed to show a contribution of B-Raf to lung colonization (Supplementary Figure 7). Our data showing the influence of B-Raf on the proliferative potential of PyMT-driven breast cancer cells in vitro, also under anchorage-independent conditions, and the requirement for signaling competent B-Raf to promote the growth of spontaneous tumors in the primary models and of orthotopically implanted tumor cells rather suggest that B-Raf accelerates the growth of primary tumors. Our data further suggest that B-Raf contributes to the generation of “metastatic seedlings” that are fit enough to survive the perils of metastasis [59].
Genetic approaches have shown that Raf kinases, in particular, the Raf-1 isoform, also exert crucial non-catalytic functions by serving as scaffold proteins [15, 60]. Moreover, the embryonic lethal phenotype of BraffloxE12 mice significantly differs from that of BrafD594A animals [17, 61], expressing catalytically inactive B-Raf, or that of the viable BrafAVKA mice, expressing activation impaired B-Raf [23]. This Braf allele series suggests that B-Raf also exerts non-catalytic functions, as it is also supported by the paradoxical ability of tumor-associated kinase-dead B-Raf molecules to activate the ERK pathway [10, 13, 62]. These data raise the question whether B-Raf activity is also required for PyMT-driven carcinogenesis. By using our unique in vivo model for expression of a catalytically impaired B-Raf protein [23], we clearly demonstrate that it is not only the B-Raf protein but also its kinase activity that is crucially involved in the progression of mammary gland tumors. We could also show that the impairment of B-Raf expression or activity leads to a reduction in ERK pathway output. We further observed decreased proliferation of PyMT cells and the loss of their migratory potential and invasive protrusions in 3D cultures. The latter two features, however, were strongly influenced by the context of the individual PyMT cell line, which can be explained by the natural heterogeneity displayed by PyMT-driven tumors. Thus, in addition to our previous finding that the AVKA mutation impairs oncogenic signaling by non-V600E BRAF mutants [23], we demonstrate here for the first time that blocking activation segment phosphorylation can be used to dampen signaling by upstream acting oncoproteins like PyMT in vivo. Interestingly, deletion of the murine Gab2 and Shp2/Ptpn11 alleles, which act upstream of RAS and RAF, also prolongs tumor-free survival in MMTV-NeuNT and MMTV-PyMT mice, respectively, and therefore phenocopies the effects we have observed with loss or impairment of B-Raf signaling [38, 53, 63]. In summary, our study highlights the importance to further investigate Raf kinases, in particular B-Raf, in breast cancer and points towards a novel druggable vulnerability of breast cancer cells to impairment of the Raf-MAPK pathway. This might be of particular relevance as MEK inhibitors are currently trialed in breast cancer (NCT01467310, NCT01964924, and NCT02685657) and as newer Raf inhibitors avoiding paradoxical ERK activation are currently being developed with the hope that they could also be employed in cells, which display aberrant B-Raf activity in the absence of BRAF mutations [64, 65]. Such inhibitors, either alone or in combination with MEK and ERK inhibitors, might be particularly relevant for tumors with aberrant RAS activity, e.g., caused by loss of RasGAPs or RTK mutation/amplification, as no RAS inhibitors are clinically available so far [66].
Materials and methods
Mouse strains
The generation and genotyping of BraffloxAVKA and BrafAVKA mice was described previously [23]. BraffloxE12 mice were kindly provided by Dr. Manuela Baccarini (Vienna) and genotyped as described previously [18]. All mouse lines were backcrossed onto a C57BL/6N background by at least 8 generations. MMTV-LTR-PyMT mice were maintained on a C57BL/6N background in-house. WAP-Cre mice [36] were kindly provided by Dr. Rolf Kemler (Max-Planck-Institute for Immunobiology and Epigenetics, Freiburg) and maintained on a C57BL/6N background. The WAP-Cre and MMTV-PyMT transgenes were paternally transferred to avoid pregnancy-induced Cre activity or tumor growth in breeding females. Braf locus heterozygous animals were used to generate littermates with all three Braf genotypes.
Animal procedures
Mice were bred and maintained in the animal facilities of the University Medical Center Freiburg according to institutional guidelines. Animals were kept in specific pathogen-free conditions, fed with standard diet and given access to food and water ad libitum. Temperature in the animal facility was 21–23 °C with a 45–60% humidity. A day/night rhythm of 12 h light and 12 h darkness was maintained. Experiments were carried out in accordance with the German law for animal protection. All studies were approved by the government commission for animal protection and the local ethics committee (G-14/54). Investigators were blinded for genotypes previous to data analysis. For transplantation experiments, Rag2−/−;γc−/− mice (BALB/C background) lacking lymphocytes and NK cells were purchased from the local stock of the animal facility at Freiburg University Medical Center and housed in the specific pathogen-free barrier facility of the University Medical Center Freiburg in accordance with the institutional guidelines and approved by the local animal ethics committee (G-15/23).
Tail vein transplantation
PyMT cells (1 × 105) transduced with the pBabe luciferase vector were intravenously injected into the tail vein of six to twelve weeks old Rag2−/−;γc−/− mice. Bioluminescence imaging (BLI) was performed as described recently [40].
Orthotopic transplantation
As described previously [67], immortalized PyMT tumor cells were re-suspended in 25 µl phosphate buffered saline (PBS) (Gibco, Carlsbad, CA, USA) mixed with an equal volume of phenol-red free matrigel (Corning Lifesciences, Corning, NY, USA), and transplanted into inguinal mammary glands of female Rag2−/−;γc−/− mice via 5 mm lateral incisions. Tumor growth was monitored by palpation twice a week until the largest tumor diameter reached 1.5 cm.
Whole mount mammary gland analysis
Glands were isolated and fixed in 4% paraformaldehyde solution. Afterwards cells were washed in phosphate buffered saline, stained with carmine red solution (2% carmine red, 5% aluminum potassium sulfate) overnight and fat was removed using xylene.
Immunohistochemistry
Immunohistochemistry (IHC) was performed as described previously [68]. In brief, tissue was dehydrated via a graded ethanol series, embedded into paraffin and sectioned at 5 µm. The sections were processed and stained using standard methods with the primary antibodies against B-Raf H-145 (1:200, Santa Cruz SC-9002) and Ki-67 (1:200, Dako M7248). Diaminobenzidine (DAB) solution was incubated for 10 s before the sections were counterstained with Mayer’s Hematoxylin.
Tissue culture, cell line isolation, and retroviral transduction
Tumor tissue was digested in DMEM 5% Collagenase A (Roche, Germany) and plated in in vitro cell culture dishes. Cells were then cultivated in standard DMEM growth medium (4.5 g/l glucose, 10% fetal calf serum (FCS), 2 mM l-glutamine, 10 mM HEPES, 200 U/ml penicillin and 200 μg/ml streptomycin) until spontaneously immortalized tumor cells grew out. Anchorage-independent growth was assessed in poly-Hema-coated plates as described previously [13]. PyB6 313 cells were isolated from primary tumors of 24 week-old female PyMT mice as previously described [69] and were spontaneously immortalized by keeping freshly isolated cells in culture for 6 weeks. During this period, cells were split whenever 80% confluency was surpassed. These cells were isolated from a TgN(MMTVPyVT)634-Mul/J mouse backcrossed to a C57BL6/N background for 10 generations. For retroviral transduction, Plat-E packaging cells were transfected as described previously [13]. Three days after transfection, Plat-E supernatant was filtered with a 0.22 μm Millipore filter, diluted 1:4 in DMEM-10% FBS, and complemented with polybrene to a final concentration of 8 μg/ml to reduce cell surface charges. 24 h before retroviral infection, cells were plated in 10 cm dishes and selection was started 72 h after infection (10 µg/ml Blasticidin S Hydrochlorid, Roth).
XTT assay
XTT assay (11465015001 Roche) was performed according to the instruction by the manufacturer. Briefly, 20,000 cells were seeded in 96-wells microplates with or without poly-HEMA coating. One day after seeding, the medium was removed and 100 μl fresh medium was added to each well. Subsequently, 50 μl of XTT labeling mixture (50 μl XTT labeling reagent and 1 µl electron coupling reagent) was added to each well. The microplates were incubated at 37 °C in a 5% CO2 atmosphere for 24 h. Absorbance was measured with an Infinite M200 plate reader (Tecan) at 490 nm and normalized to reference readings at 650 nm.
Cell migration
To monitor real time migration of primary tumor cells the xCELLigence migration assay from ACEA Biosciences was performed using CIM-Plates 16. As a chemo-attractant within the lower chamber, growth medium containing 10% FCS was used. Primary tumor cells were are added to the upper chamber and allowed to migrate for up to 24 h. The xCELLigence device was set to measure the resistance at the membrane located between the two chambers every 15 min, resulting in a graph representing the migration of analyzed cells.
Western blotting
Cells were lysed in RIPA buffer (50 mM Tris/HCl, pH 7.4; 1% Triton X-100; 137 mM NaCl; 1% glycerin; 1 mM sodium orthovanadate; 0.5% sodium deoxycholate, 0.1% sodium dodecylsulfate, 0.5 mM EDTA; 0.01 µg/µl leupeptin, 0.1 µg/µl aprotinin, 1 mM AEBSF). Lysates were analyzed by Western blotting using 10% SDS-PAGE gels as described previously [23] using the following antibodies. Anti-Raf-B (F-7 or H-145), anti-Fos (K-25), anti-Tubulin (B-5-1-2), anti-Raf-1 (C-12), anti-A-Raf (C-20) and anti-pan 14-3-3 (H8) were all purchased from Santa Cruz Biotechnology, USA. Anti-phospho-c-Fos (pS32; D82C12), anti-FosB (5G4), anti-phospho-Fra1 (S265; D22B1), anti-HSP90 (#4874), anti-phospho-MEK1/2 (pS217/221), anti-phospho-MEK1/2 (pS221), anti-MEK1/2, anti-p42/p44 MAPK, and anti-phospho-MAPK (pT202/pY204); (ERK1/2 were all purchased from Cell Signaling Technologies, USA). Anti-GAPDH antibodies were purchased from Abcam (ab9484). Blotted proteins were visualized with a Fusion Solo chemiluminescence reader and quantified using FusionCapt software (Vilber Lourmat, Germany).
Plasmids
The two-component doxycycline inducible Cre expression system was generated by subloning a Kozak optimized Cre cDNA into pSH461MK-EV [40]. This vector was co-infected with pSH582 encoding Dox-regulated transcriptional silencer and transactivator proteins [40]. For generation of knockdown plasmids, the following oligonucleotides encoding the specific shRNAs in a miR-30 backbone were ordered from biomers.net (Ulm, Germany): shBraf.627, 97-mer 5′-TGCTGTTGACAGTGAGCGCCCCGCAAGATGTGGTGTTACATAGTGAAGCCACAGATGTATGTAACACCACATCTTGCGGGTTGCCTACTGCCTCGGA-3′ and shLuc.1309, 97-mer 5′-TGCTGTTGACAGTGAGCGCCCGCCTGAAGTCTCTGATTAATAGTGAAGCCACAGATGTATTAATCAGAGACTTCAGGCGGTTGCCTACTGCCTCGGA-3′. As described previously [70], these 97-mers serve as PCR templates for addition of XhoI and EcoRI restriction sites. With added restriction sites, the oligomers were cloned into the dual color pTREBAV retroviral vector. This plasmid is derived from pTRMPV [71] and allows doxycycline inducible shRNA expression. The non-silencing control plasmid pTREBAV-BpuEI_shLuc.1309 was a kind gift from Cornelius Miething (University Medical Centre Freiburg, Freiburg, Germany).
Statistical analysis
Quantitative data are presented as mean ± SE. Pairwise comparisons were performed by Student t test (two-tailed), respectively. When the data did not conform to a normal distribution, the Mann–Whitney U test was utilized. Differences in mouse survival (Kaplan–Meier survival curves) were analyzed by log-rank test. Unless noted otherwise, bar diagrams report the mean of three independent experiments. A p-value of ≤0.05 was considered statistically significant (****p < 0.0001; ***p < 0.001; **p < 0.01; and *p < 0.05).
References
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Ha JR, Siegel PM, Ursini-Siegel J. The tyrosine kinome dictates breast cancer heterogeneity and therapeutic responsiveness. J Cell Biochem. 2016;117:1971–90.
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4.
McLaughlin SK, Olsen SN, Dake B, De Raedt T, Lim E, Bronson RT, et al. The RasGAP gene, RASAL2, is a tumor and metastasis suppressor. Cancer Cell. 2013;24:365–78.
Sears R, Gray JW. Epigenomic inactivation of RasGAPs activates RAS signaling in a subset of luminal B breast cancers. Cancer Discov. 2017;7:131–3.
Olsen SN, Wronski A, Castano Z, Dake B, Malone C, De Raedt T, et al. Loss of RasGAP tumor suppressors underlies the aggressive nature of luminal B breast cancers. Cancer Discov. 2017;7:202–17.
Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–42.
Little AS, Smith PD, Cook SJ. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene. 2013;32:1207–15.
Röring M, Brummer T. Aberrant B-raf signaling in human cancer—10 years from bench to bedside. Crit Rev Oncog. 2012;17:97–121.
Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21.
Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5.
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30.
Röring M, Herr R, Fiala GJ, Heilmann K, Braun S, Eisenhardt AE, et al. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J. 2012;31:2629–47.
Yaktapour N, Meiss F, Mastroianni J, Zenz T, Andrlova H, Mathew NR, et al. BRAF inhibitor-associated ERK activation drives development of chronic lymphocytic leukemia. J Clin Invest. 2014;124:5074–84.
Desideri E, Cavallo AL, Baccarini M. Alike but different: RAF paralogs and their signaling outputs. Cell. 2015;161:967–70.
Wojnowski L, Stancato LF, Larner AC, Rapp UR, Zimmer A. Overlapping and specific functions of Braf and Craf-1 proto-oncogenes during mouse embryogenesis. Mech Dev. 2000;91:97–104.
Galabova-Kovacs G, Catalanotti F, Matzen D, Reyes GX, Zezula J, Herbst R, et al. Essential role of B-Raf in oligodendrocyte maturation and myelination during postnatal central nervous system development. J Cell Biol. 2008;180:947–55.
Galabova-Kovacs G, Matzen D, Piazzolla D, Meissl K, Plyushch T, Chen AP, et al. Essential role of B-Raf in ERK activation during extraembryonic development. Proc Natl Acad Sci USA. 2006;103:1325–30.
Zhong J, Li X, McNamee C, Chen AP, Baccarini M, Snider WD. Raf kinase signaling functions in sensory neuron differentiation and axon growth in vivo. Nat Neurosci. 2007;10:598–607.
Pritchard CA, Samuels ML, Bosch E, McMahon M. Conditionally oncogenic forms of the A-Raf and B-Raf protein kinases display different biological and biochemical properties in NIH 3T3 cells. Mol Cell Biol. 1995;15:6430–42.
Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14:455–67.
Köhler M, Brummer T. B-Raf activation loop phosphorylation revisited. Cell Cycle. 2016;15:1171–3.
Köhler M, Röring M, Schorch B, Heilmann K, Stickel N, Fiala GJ, et al. Activation loop phosphorylation regulates B-Raf in vivo and transformation by B-Raf mutants. EMBO J. 2016;35:143–61.
Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4:232–45.
Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–61.
Ali NA, Wu J, Hochgrafe F, Chan H, Nair R, Ye S, et al. Profiling the tyrosine phosphoproteome of different mouse mammary tumour models reveals distinct, model-specific signalling networks and conserved oncogenic pathways. Breast Cancer Res. 2014;16:437.
Bengsch F, Buck A, Gunther SC, Seiz JR, Tacke M, Pfeifer D, et al. Cell type-dependent pathogenic functions of overexpressed human cathepsin B in murine breast cancer progression. Oncogene. 2014;33:4474–84.
Fluck MM, Schaffhausen BS. Lessons in signaling and tumorigenesis from polyomavirus middle T antigen. Microbiol Mol Biol Rev. 2009;73:542–63.
Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76.
Dilworth SM. Polyoma virus middle T antigen and its role in identifying cancer-related molecules. Nat Rev Cancer. 2002;2:951–6.
Smith BA, Shelton DN, Kieffer C, Milash B, Usary J, Perou CM, et al. Targeting the PyMT oncogene to diverse mammary cell populations enhances tumor heterogeneity and generates rare breast cancer subtypes. Genes Cancer. 2012;3:550–63.
Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–26.
Fantozzi A, Christofori G. Mouse models of breast cancer metastasis. Breast Cancer Res. 2006;8:212.
Ben-David U, Ha G, Khadka P, Jin X, Wong B, Franke L, et al. The landscape of chromosomal aberrations in breast cancer mouse models reveals driver-specific routes to tumorigenesis. Nat Commun. 2016;7:12160.
Chen AP, Ohno M, Giese KP, Kuhn R, Chen RL, Silva AJ. Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. J Neurosci Res. 2006;83:28–38.
Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323–30.
Marcotte R, Smith HW, Sanguin-Gendreau V, McDonough RV, Muller WJ. Mammary epithelial-specific disruption of c-Src impairs cell cycle progression and tumorigenesis. Proc Natl Acad Sci USA. 2012;109:2808–13.
Lan L, Holland JD, Qi J, Grosskopf S, Rademann J, Vogel R, et al. Shp2 signaling suppresses senescence in PyMT-induced mammary gland cancer in mice. EMBO J. 2015;34:1493–508.
Wagner KU, Boulanger CA, Henry MD, Sgagias M, Hennighausen L, Smith GH. An adjunct mammary epithelial cell population in parous females: its role in functional adaptation and tissue renewal. Development. 2002;129:1377–86.
Haug S, Schnerch D, Halbach S, Mastroianni J, Dumit VI, Follo M, et al. Metadherin exon 11 skipping variant enhances metastatic spread of ovarian cancer. Int J Cancer. 2015;136:2328–40.
Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–64.
Gillies TE, Pargett M, Minguet M, Davies AE, Albeck JG. Linear integration of ERK activity predominates over persistence detection in Fra-1 regulation. Cell Syst. 2017;5:549–63 e545.
Zhang BH, Guan KL. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. Embo J. 2000;19:5429–39.
Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJ, et al. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154:1036–46.
Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, et al. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol. 2015;22:37–43.
O’Toole SA, Beith JM, Millar EK, West R, McLean A, Cazet A, et al. Therapeutic targets in triple negative breast cancer. J Clin Pathol. 2013;66:530–42.
Adeyinka A, Nui Y, Cherlet T, Snell L, Watson PH, Murphy LC. Activated mitogen-activated protein kinase expression during human breast tumorigenesis and breast cancer progression. Clin Cancer Res. 2002;8:1747–53.
Whyte J, Bergin O, Bianchi A, McNally S, Martin F. Key signalling nodes in mammary gland development and cancer. Mitogen-activated protein kinase signalling in experimental models of breast cancer progression and in mammary gland development. Breast Cancer Res. 2009;11:209.
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–67.
Santarpia L, Qi Y, Stemke-Hale K, Wang B, Young EJ, Booser DJ, et al. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res Treat. 2012;134:333–43.
Tilch E, Seidens T, Cocciardi S, Reid LE, Byrne D, Simpson PT, et al. Mutations in EGFR, BRAF and RAS are rare in triple-negative and basal-like breast cancers from Caucasian women. Breast Cancer Res Treat. 2014;143:385–92.
Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, et al. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat Med. 2012;18:529–37.
Bentires-Alj M, Gil SG, Chan R, Wang ZC, Wang Y, Imanaka N, et al. A role for the scaffolding adapter GAB2 in breast cancer. Nat Med. 2006;12:114–21.
Jacob LS, Vanharanta S, Obenauf AC, Pirun M, Viale A, Socci ND, et al. Metastatic competence can emerge with selection of preexisting oncogenic alleles without a need of new mutations. Cancer Res. 2015;75:3713–9.
Herr R, Wohrle FU, Danke C, Berens C, Brummer T. A novel MCF-10A line allowing conditional oncogene expression in 3D culture. Cell Commun Signal. 2011;9:17.
Pearson GW, Hunter T. Real-time imaging reveals that noninvasive mammary epithelial acini can contain motile cells. J Cell Biol. 2007;179:1555–67.
Beliveau A, Mott JD, Lo A, Chen EI, Koller AA, Yaswen P, et al. Raf-induced MMP9 disrupts tissue architecture of human breast cells in three-dimensional culture and is necessary for tumor growth in vivo. Genes Dev. 2010;24:2800–11.
Liu H, Murphy CJ, Karreth FA, Emdal KB, White FM, Elemento O, et al. Identifying and targeting sporadic oncogenic genetic aberrations in mouse models of triple-negative breast cancer. Cancer Discov. 2018;8:354–69.
Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017;168:670–91.
Varga A, Ehrenreiter K, Aschenbrenner B, Kocieniewski P, Kochanczyk M, Lipniacki T, et al. RAF1/BRAF dimerization integrates the signal from RAS to ERK and ROKalpha. Sci Signal. 2017;10:eaai8482.
Kamata T, Hussain J, Giblett S, Hayward R, Marais R, Pritchard C. BRAF inactivation drives aneuploidy by deregulating CRAF. Cancer Res. 2010;70:8475–86.
Nieto P, Ambrogio C, Esteban-Burgos L, Gomez-Lopez G, Blasco MT, Yao Z, et al. A Braf kinase-inactive mutant induces lung adenocarcinoma. Nature. 2017;548:239–43.
Calleja V, Alcor D, Laguerre M, Park J, Vojnovic B, Hemmings BA, et al. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007;5:e95.
Girotti MR, Lopes F, Preece N, Niculescu-Duvaz D, Zambon A, Davies L, et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell. 2015;27:85–96.
Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–6.
Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13:828–51.
Hillebrand LE, Bengsch F, Hochrein J, Hulsdunker J, Bender J, Follo M, et al. Proteolysis—a characteristic of tumor-initiating cells in murine metastatic breast cancer. Oncotarget. 2016;7:58244–60.
Sevenich L, Schurigt U, Sachse K, Gajda M, Werner F, Muller S, et al. Synergistic antitumor effects of combined cathepsin B and cathepsin Z deficiencies on breast cancer progression and metastasis in mice. Proc Natl Acad Sci USA. 2010;107:2497–502.
Sevenich L, Werner F, Gajda M, Schurigt U, Sieber C, Muller S, et al. Transgenic expression of human cathepsin B promotes progression and metastasis of polyoma-middle-T-induced breast cancer in mice. Oncogene. 2011;30:54–64.
Dow LE, Premsrirut PK, Zuber J, Fellmann C, McJunkin K, Miething C, et al. A pipeline for the generation of shRNA transgenic mice. Nat Protoc. 2012;7:374–93.
Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol. 2011;29:79–83.
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG) via the Collaborative Research Centre 850 (projects B4; B7, and C6), the Excellence Initiative of the German Federal and State Governments (EXC 294 BIOSS), the German Cancer Consortium (DKTK, L627) and the Spemann Graduate School for Biology and Medicine (GSC-4; SGBM) via fellowships to MK, SH, NK, CK. and NR. TB was also supported by the Emmy-Noether and Heisenberg programs of the DFG. We would also like to thank Chris Ormandy for helpful discussions, Ricarda Herr for advice, and Nicole Klemm and Ulrike Reif for expert technical assistance.
Author contributions
MK, TR, and TB designed and conceived experiments, conducted data interpretation and wrote the manuscript. MK, SE, SH, ML, UB, NR, SC, CS, FMU, NK, KR, and SB performed experimental work and data interpretation. CP and RZ contributed to experimental design and data interpretation. TR and TB supervised research and project planning.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Köhler, M., Ehrenfeld, S., Halbach, S. et al. B-Raf deficiency impairs tumor initiation and progression in a murine breast cancer model. Oncogene 38, 1324–1339 (2019). https://doi.org/10.1038/s41388-018-0663-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0663-8
- Springer Nature Limited
This article is cited by
-
Cathepsin D deficiency in mammary epithelium transiently stalls breast cancer by interference with mTORC1 signaling
Nature Communications (2020)