
Abstract
Hepatitis B virus X protein (HBx) contributes to Hepatitis B virus (HBV)-related liver cancer. However, its impact on hepatocyte proliferation and genomic stability remains elusive. We studied the role of HBx expression on the progression of cell cycle and liver polyploidization during proliferation and liver carcinogenesis. Full-length HBx transgenic mice (FL-HBx) were developed to investigate liver ploidy as well as hepatocyte proliferation, along normal liver maturation and during cancer initiation (chemical carcinogen treatment). Investigation of postnatal liver development in FL-HBx showed an aberrant G1/S and G2/M transitions, triggered (1) a delay of the formation of hepatocytes binucleation, (2) the early synthesis of polyploidy nuclei (≥4n) and (3) DNA damage appearance. Moreover, HBV infection during hepatocytes proliferation in a humanized liver mouse model led, to modifications in polyploidy of hepatocytes. In initiation of hepatocellular carcinoma, FL-HBx protein decreased ChK1 phosphorylation, Mre11 and Rad51 expression, upregulated IL-6 expression and impaired apoptosis. This was related to DNA damage accumulation in FL-HBx mice. At day 75 after initiation of hepatocellular carcinoma, FL-HBx mice revealed significant cell cycle changes related to the increased amount of 4n nuclei and of markers of cancer progenitor cells. Finally, PLK1 upregulation and p38/ERK activation in FL-HBx mice were implicated in aberrant polyploidization favoring DNA damage propagation and hepatocyte transformation. In conclusion, our data indicate that FL-HBx protein increases DNA damage through the hijack of hepatocyte polyploidization. That leads to enhancement of hepatocellular carcinoma initiation in an inflammatory context.
Similar content being viewed by others


Introduction
Hepatitis B virus (HBV) chronic infection is related with liver damage that may progress to hepatocellular carcinoma (HCC) [1]. The host immune response to liver infection by inducing chronic inflammation is the major onset of the appearance of HCC (90% of HCCs develop in cirrhotic livers) [1]. However, the importance of a direct contribution of HBV in HCC development has been reported [1]. Indeed, HBV genome integration in the cell genome and the expression of viral proteins are possibly involved in the generation of host genetic instability [1]. The expression of the HBV X protein (HBx) is crucial for viral replication and is assumed to participate in the development of HCC. HBx protein expression has pleiotropic biological effects implicated in the modulation of signalling pathways involved in the control of the progression of cell cycle, in the responses to cellular stress and in apoptosis; underling the role of HBx in HCC [2]. It was previously stated that HBx protein activates cell cycle progression either through hastening checkpoint transition or by impairment of the miR-122 expression, and subsequently the release from G1/S arrest [3]. In contrary, HBx protein has been described to contribute to liver cell proliferation inhibition by the activation of p21 and p27 cell cycle inhibitors or by cell cycle arrest at the G1/S phase [4, 5]. Our recent data, based on partial hepatectomy, showed that HBx may delay liver regeneration in transgenic mice [6]. Several studies highlighted the contribution of HBx expression in the disruption of DNA repair process. Indeed, previous reports showed that HBx impaired DNA repair either through protein/protein interaction with partners directly implicated in this cellular machinery [7,8,9] or by alteration of the nucleic acid metabolism [10]. HBx cell DNA damage might be induced by oxidative stress involved in reactive oxygen species production, or by oxidative stress sensors regulation [11, 12]. HBx protein by associated induction of DNA damage and aberrant DNA repair machinery may account for the delay of cell cycle in G2/M phase, as previously reported [11, 12]. Otherwise, HBx may also lead to the aberrant mitotic checkpoint, possibly by increasing the number of centrosome in cells [13, 14]. In HBx expressing cells, DNA re-replication has been associated with polyploidy [15]. In fact, it was reported that in untransformed hepatocytes, HBx protein could activate polo-like kinase 1 (PLK1), a protein involved in cell cycle mitosis entry, and subsequently spreading DNA damage and inducing hepatocyte polyploidy [16].
Polyploidy is a characteristic event in human and mouse hepatocytes, corresponding to abnormal DNA content with atypical number of homologous chromosomes [17]. Up to 90% of adult murine [18] and around 40% of human hepatocytes are polypoid [19]. Definition of the polyploidy is based either on the cell nuclei number (cellular ploidy) or on the DNA content per nucleus (nuclear ploidy). In the liver, amount in polyploidy cells is dependent to aging. Indeed, in young mice the majority of hepatocytes are diploid while the amount of polypoid hepatocytes significantly rises in older mice [20, 21]. In previous reports, it was demonstrated that hepatocyte polypoid is produced in successive steps in a process indicated as “physiological polyploidization”. In young mice, the synthesis of binucleate cells (2 × 2n) from diploid hepatocytes is generated during full karyokinesis with inadequate cytokinesis [22, 23]. If hepatocytes undergo a new cell cycle, hepatocytes either carry out a normal cytokinesis, forming tetraploid mononuclear cells (4n), or in case of cytokinesis failure, octoploid binuclear hepatocytes are generated (2 × 4n). Mechanisms implicated in polyploidization throughout the development of the liver remains elusive, however polyploidy may contribute to genetic diversity in conferring specific metabolism to cell to undergo terminal differentiation [17, 24]. In parallel to involvement liver development, polyploidy has been also considered as a mechanism of liver adaptation to stress [25, 26]. It should be noted that in chronic hepatitis B or C infected patients, different levels of hepatocyte ploidy have been observed. This suggests in these two viral infections, that distinct molecular pathways are implicated in alteration of ploidy [19]. Interestingly, aneuploidy evidenced in patient chronically infected with HBV, may contribute to HCC predisposition [27].
Most of the data obtained on the deregulation of cell cycle by the HBx protein were realized by means of in vitro models. The aim of the present study was to investigate, in mouse models, the properties of the full-length HBx (FL-HBx), during physiological liver proliferation on cell cycle progression and polyploidization as well as during initiation of carcinogenesis.
Results
Abnormal G1/S and G2/M cell cycle transitions in the hepatocytes of FL-HBx transgenic mice
The two FL-HBx transgenic strains used in the study were previously characterized [6, 28]. For both strains a similar impact on liver regeneration after partial hepatectomy and DEN-induced HCC development was evidenced [6, 28]. As a preliminary, we confirmed the expression of FL-HBx transcript in postnatal and DEN-treated transgenic animals at timing of the experiments (supplementary results and supplementary Fig.1).
To clarify the controversial data reported in literature about HBx protein and cell proliferation, we performed an in vivo analysis of effects of FL-HBx expression on hepatocytes cell cycle regulation. For that, we chose postnatal developing liver as a model of physiological proliferation (Fig. 1). In agreement with postnatal liver development, we observed a robust but transient hepatocyte proliferation, as showed by Ki-67 and BrdU labeling (Fig. 1b). In WT mice, Ki67 expression was inversely proportional to the age of mice. In contrast, in FL-HBx transgenic mice, by days 17 and 20, a strong Ki67 staining of the nuclei was observed. However, by day 25, Ki67 staining was comparable in the two groups. Amount of hepatocytes in S-phase was achieved by BrdU incorporation assays. On day 17, the amount of BrdU labelled nuclei was higher in FL-HBx mice compared to WT mice. The greater amount of BrdU incorporation in FL-HBx transgenic mice was confirmed by BrdU accumulation assay (days 17 to 20) (Fig. 1b) indicating an increase of hepatocytes number in S-phase in these mice. Interestingly, that corroborated, on days 17 and 20, with an upregulation in cyclin A2 transcription in FL-HBx transgenic mice (Fig. 1c). Furthermore, the higher levels of the cyclin D1 and Cdk4 proteins, in transgenic mice on days 15 and 17, demonstrated the deregulation of the G1 phase (Fig. 1d and supplementary Figs 2 and 3), and of the cyclin E1 and Cdc6 mRNAs on days 17 and 20 (Fig. 1c). Thus FL-HBx shorter the G1 phase and induced an early entry of hepatocytes in S-phase.

Deregulation of G1/S and G2/M transitions in FL-HBx mice. a Experimental design. b Time-course analysis of Ki67 expression and BrdU incorporation in WT and FL-HBx mice. Immunohistochemical staining for Ki67 and BrdU staining and their quantification. c mRNA levels for the cyclins E1, A2 (CcnE1, CcnA2) and Cdc6. d Immunoblot analysis of cyclin D1 (CcnD1), Cdk4, and cyclin B1 (CcnB1) expression, and histone H3 and Cdk1 phosphorylation levels; in the liver of WT and FL-HBx mice on the indicated days (additional mice data are presented in supplementary Fig.2 and quantification in supplementary Fig.3). e Percentage of mitotic cells in WT and FL-HBx mice. Liver sections were stained for PCNA, pHH3, and Hoechst. Cells in G2 are PCNA+/pHH3+ and mitotic cells are PCNA−/pHH3+. Bars: median. Unpaired Student’s t -tests; *p < 0.05; **p < 0.005; ***p < 0.0005
We then investigated the regulation of G2/M transition. Entry in mitosis is prompted by the activation of the CcnB1/Cdk1 complex, that under the control of dephosphorylation of the Cdk1 protein on tyrosine 15 [29]. From days 15 to 20, although comparable levels of cyclin B1 was observed in the both groups of mice, histone H3 (Ser 10) and of Cdk1 (Tyr 15) were highly phosphorylated in transgenic mice (Fig. 1d and supplementary Figs. 2 and 3) suggesting a block of hepatocytes in G2/M transition. Calculation in hepatocytes of the mitotic events confirmed the G2/M transition defect (Fig. 1e). In WT mice, an increase of the percentage (30–46%) of mitotic cells was observed between days 13 and 20. Whereas, in FL-HBx transgenic mice, the percentage (30–34%) stayed stable. Finally, on day 25, a similar numbers of mitotic cells were observed in both groups, showing a transient arrest of hepatocytes in G2/M transition in FL-HBx transgenic mice.
Our results indicate that under the physiological proliferation stimuli effect, FL-HBx induces in liver, an acceleration in S-phase entry and a delay in G2/M transition of hepatocytes (supplementary Fig. 4).
FL-HBx modifies the physiological polyploidization in a proliferating liver
We next investigated whether FL-HBx-induced cell cycle alteration might modify the polyploidization observed in normal liver development (Fig. 2). In a first attempt, we investigated the cellular hepatocytes polyploidization, in animals aged from day 13 to 3 months. On day 13, in both groups of mice, the proportion of binucleate hepatocytes was <5% (Fig. 2b). In agreement with previous reports, for control mice, from days 15 to 25, a quickly increased of binucleate hepatocytes (around 28%) was evidenced (Fig. 2b). While, in transgenic mice, on days 15 and 17, binucleate hepatocytes remained unchanged (about 6%). Then, from day 20 to 25, the number of binucleate hepatocytes increase till around 25% (Fig. 2b). At 3 months, the percentage of binucleate hepatocytes was about 27% in both groups of animals (Fig. 2c).

Disruption of hepatocyte polyploidization and maturation in FL-HBx mice during liver development. a Liver sections stained for β-catenin and Hoechst. b Time-course analysis after birth of binucleate and mononucleate (2n, ≥4n) hepatocytes, in FL-HBx and control mice. c Percentages of binucleate and mononucleates (2n, 4n, ≥8n) hepatocytes in 3 month-old WT and FL-HBx mice. d Liver sections from 17-day-old WT and FL-HBx mice stained for β-catenin, glutamine synthetase, and Hoechst. e C/EBP-α, PepCK, and AFP transcript levels in livers of 17 day-old WT and FL-HBx mice. Bars: median. Unpaired Student’s t -tests; *p < 0.05; **p < 0.005; ****p < 0.00005
Nuclear ploidy was then analyzed. Along the liver development, nuclei with 2n content were predominantly evidenced (98–75%) in hepatocytes (Fig. 2b, c). The population of hepatocyte with polyploidy nuclei (≥4n) was augmented at days 15 and 17 in FL-HBx transgenic mice (4.2 ± 1.4% vs. 2.5 ± 1.3% at day 15 and 5.9 ± 4.3% vs. 2.2 ± 0.9% at day 17) (Fig. 2b). However, the comparable ratio of polyploidy nuclei (≥4n) was determined in both groups of mice by days 20 (2.5 ± 0.8% for WT and 2.8 ± 1.0% for FL-HBx) and 25 (WT: 9.1 ± 2.5% for WT and 7.1 ± 2.5% for FL-HBx) (Fig. 2b). At 3 months, in absence of liver proliferative stimuli, polyploidy became similar in the two groups of animals (Fig. 2c).
We then investigated whether hepatocytes polyploidy deregulation may disturb the terminal differentiation of the liver. At day 17, both groups of mice showed in the perivenous zone an analogous levels of glutamine synthetase (Fig. 2d); but, for transgenic mice, a lower levels of the transcription of two markers of hepatocyte differentiation (C/EBP-α and PepCK) and a higher levels of AFP transcripts, a characteristic feature of immature hepatocytes, were observed (Fig. 2e). However, at 3 months of age, no differences in transcription were observed (data not shown). Thus, our data are reliable with a transitory impairment of hepatocytes physiological polyploidization and maturation, in FL-HBx transgenic mice during postnatal liver proliferation.
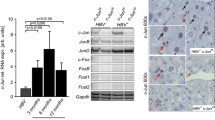
Finally, to address the consequences of chronic HBV infection on hepatocytes polyploidy, we used a mouse model with humanized liver and thus susceptible to HBV infection, the uPA+/+/Scid/RAG/GammaC (Supplementary Fig. 5). To avoid experimental bias, the same batch of human hepatocytes was used for mice transplantation (Supplementary Fig. 5a). 3 months after infection, liver and serum samples were obtained. Human albumin levels and HBV copies were evaluated to determine the transplantation rates and the viremia, respectively (Supplementary Fig. 5b). Co-immunostaining with Human cytokeratin 18 and human albumin were used to visualize human hepatocyte clusters (Supplementary Fig. 4c). Interestingly, in agreement with the data on FL-HBx transgenic mice, a lower number of binucleate human hepatocytes was observed in HBV-infected animals [HBV(−): 13.3 ± 1.3%, HBV(+): 9.2 ± 1.2%] (Supplementary Fig. 5d, left panel). In HBV-infected mice, the human hepatocytes population was enriched in polyploid cell [nuclei ≥4 N; HBV(−): 65.0 ± 6.2%, HBV(+): 70.5 ± 7.8%] consequent to the increase of ≥8n nuclei [HBV(−): 5.40 ± 2.0%; HBV(+): 9.5 ± 1.3%]. Furthermore, a significantly greater percentage of ≥8n nuclei in comparison with ≥4n nuclei was observed in human hepatocytes of HBV-infected mice [HBV(-): 8.2 ± 2.6% vs. HBV(+): 13.6 ± 1.7%] (Supplementary Fig. 4d, right panel). These results indicate an association between HBV infection and induction of abnormal polyploidization in hepatocytes.
Altogether, our data illustrate that under the physiological liver proliferation, FL-HBx hijacks transiently the normal polyploidization of hepatocytes and that correlates with a defect in hepatocytes maturation. In addition, we observed that HBV infection modifies hepatocytes polyploidization similarly to that observed in FL-HBx transgenic mice.
PLK1/p38/ERK activation induces aberrant liver polyploidization thereby promoting DNA damage propagation in FL-HBx transgenic mice
To determine the mechanism responsible of hepatocytes cell cycle and polyploidization deregulation, we examined whether the G2/M arrest might be associated with FL-HBx-induced DNA damage. Thus, DNA damage induction was analyzed by evaluating the level of phosphorylation of H2AX protein. A higher H2AX phosphorylation was evidenced in FL-HBx transgenic mice livers, on days 15–20 (Fig. 3a and supplementary Figs 2 and 3). This was confirmed by an increase of H2AX phosphorylated nucleic at day 17 (Fig. 3b). On day 20, the higher levels of phosphorylation of H2AX were associated with a lower expression of DNA repair protein, Rad51 (Fig. 3a and supplementary Figs. 2 and 3). Furthermore, in FL-HBx transgenic mice, at days 17 and 20, an increase of Rad51 transcription, associated with the activation of the DNA repair machinery was observed (Fig. 3b); indicating that the FL-HBx protein regulates negatively the translation of Rad51 protein.

Pathways regulating polyploidization and hepatocyte phenotype in postnatal FL-HBx mice. a Immunoblot analysis of Mre11 and Rad51 levels and histone H2AX, p38 and ERK1/2 phosphorylation in FL-HBx and control mice (additional mice data are presented in supplementary Fig. 2 and quantification in supplementary Fig.3). b Liver sections were stained for β-catenin, pH2AX, and Hoechst. Percentage of pH2AX positive nuclei in WT and FL-HBx mice at day 17. RT-qPCR analysis of c Rad51 and d Plk1, mRNA levels in livers of WT and FL-HBx mice on the indicated days. Bars: median. Unpaired Student’s t -tests; *p < 0.05; **p < 0.005
The activation of p38 in hepatocytes participates to growth arrest in developing liver caused by low-level oxidative stress [30, 31]. We checked if the abnormal cell cycle and polyploidization evidenced in transgenic mice might be linked with p38-MAPK activation. Interestingly, in time-course of our analysis (days 15–20), p38 was highly activated in FL-HBx transgenic mice on day 15 (Fig. 3a and supplementary Figs. 2 and 3). In addition, higher phosphorylation of ERK1/2 on days 15 and 17 was also detected in FL-HBx transgenic (Fig. 3a and supplementary Figs. 2 and 3). This stronger activation of MAPKs p38/ERK1/2 in FL-HBx transgenic mice correlates, as previously reported in vitro, with a higher transcription of PLK1 (days 17 and 20) [16, 32] (Fig. 3d).
In order to validate the interplay between p38/ERK1/2 MAPKs and PLK1, and their role in hepatocytes polyploidization, we performed the inhibition of p38 with SB203508 in WT and FL-HBx transgenic mice (Fig. 4 and supplementary Fig. 6). Considering cell cycle regulation, inhibition of p38 induced a significant decrease of PLK1 transcription which was associated with a moderated diminution of Ki67 expression in both groups of mice (Fig. 4b, Supplementary Fig. 6a). For the S phase, a significant reduction in BrdU incorporation was only observed in SB203580-treated control mice (Supplementary Fig. 6b). However, in FL-HBx transgenic mice, SB203580 treatment induced less phosphorylation of ERK1/2 and an increase in Mre11 expression which correlate with recovery at least partial of the number of mitotic events (Fig. 4c and supplementary Fig. 6c). This was associated, in FL-HBx transgenic mice with a partial rescue of binucleation and smaller numbers of polypoid nuclei (≥4n) hepatocytes. By contrast, in control mice, this was associated with an increase of hepatocytes binucleation and a lesser percentage of polypoid nuclei (Fig. 4d). Finally, in both groups of mice, the modulation of p38 pathway was associated with an increase of the transcription of C/EBP-α, a marker of hepatocyte maturation (Supplementary Fig.5e).

Inactivation of p38 restores normal hepatocytes polyploidization and maturation. a Protocol of the p38 inhibition by SB203508 in WT and FL-HBx transgenic mice. b PLK1 mRNA levels; c ERK phosphorylation and Mre11 levels in the livers of WT and FL-HBx mice treated or not. Histograms represent western blots quantification using β-actin as control. d Percentage of binucleate hepatocytes and 2n and ≥4n nuclei; and e C/EBP-α mRNA levels; in WT and FL-HBx mice treated or not. Bars: median. Unpaired Student’s t or Mann–Whitney U-tests; *p < 0.05; **p < 0.005
Taken together, our data reveal that FL-HBx protein by upregulating PLK1 through p38 and ERK1/2 pathways, induces the attenuation of cell cycle checkpoint control leading to disruption of hepatocyte polyploidization and DNA damage propagation (supplementary Fig.4).
Increased in DNA damage and inflammation correlate with abnormal polyploidization and HCC initiation in FL-HBx transgenic mice
In a previous repot, we have demonstrated that the occurrence of DEN-induced HCC was promoted by FL-HBx protein [28], suggesting that early HCC development might be enhanced by FL-HBx. For this assessment, we explored the consequence of FL-HBx expression on initiation and promotion of HCC, in particular on hepatocyte DNA damage, proliferation, and polyploidization.
Four hours after DEN treatment, increased in H2AX phosphorylation was demonstrated by a higher DNA damage induction in FL-HBx transgenic mice (Fig. 5b). This was confirmed by immunofluorescence on liver slices, where a significant increase of pH2AX positive nuclei was observed in FL-HBx mice (Fig. 5c). The increase level of H2AX phosphorylation in FL-HBx transgenic mice does not only correspond to perturbation of the S-phase. Indeed, in FL-HBx mice, DNA damage accumulation was sustained with a decrease expression of Rad51 and Mre11 DNA repair genes and inactivation of cell cycle checkpoint 1(ChK1) (Fig. 5b). Furthermore, in FL-HBx transgenic mice, four hours after DEN treatment, a decrease of caspase-3 cleavage was detected (Fig. 5b). In FL-HBx transgenic mice, these observations correlate with a higher expression of genes implicated in stress sensor activity (Gadd45-α), anti-apoptosis (Bcl2) and inflammation (IL-6, TGFβ; and a trend for TNF-α) (Fig. 5d). Thus, during the initial stage of HCC, the FL-HBx protein, in addition to deregulate pathways implicated in DNA repair or apoptosis, potentiates DEN-induced inflammation thereby favoring the survival of initiated hepatocytes.

Potentiation of DEN-induced HCC initiation/promotion in FL-HBx mice 4 h after DEN treatment. a Experimental design of HCC induction. b Immunoblot analysis of H2AX, ChK1 phosphorylation, Mre11, Rad51, and caspase 3 levels in the livers of WT and FL-HBx mice. c Liver sections stained for β-catenin, pH2AX and Hoechst. Bars represent 250 µm. The graph shows the number of pH2AX-positive nuclei per mm2 in WT and FL-HBx mice. d Gadd45-α, Bcl2, IL-6, TNF-α, and TGF-β mRNA levels, normalized against WT mice receiving PBS, in the livers of WT and FL-HBx mice. Histograms represent immunoblots quantification using β-actin as control. Bars: median. Unpaired Student’s t-tests; *p < 0.05; **p < 0.005
We next evaluated in both groups of mice, the associated profile of hepatocyte proliferation and polyploidization during the initiation/promotion of HCC. After 48 h of treatment, BrdU incorporation was not modified (data not shown). In contrast to WT treated mice, where an increase of Ki67-positive nuclei was observed (1137 ± 406.3 for PBS vs. 1728 ± 455.1 for DEN), in FL-HBx treatment mice, not an additional increase of Ki67-positive nuclei was determined (Fig. 6a). At the transcription level, this was associated with a significant increase of Plk1 and decrease of Rad51 in FL-HBx treated mice (Fig. 6b), this correlates with p38 overactivation (Fig. 6c).

Impact of upregulation of PLK1 through p38/ERK on polyploidy and HCC progenitor markers in FL-HBx transgenic mice. a Ki67-positive nuclei and b PLK1 and Rad51 mRNA levels; 48 h post-treatment in WT and FL-HBx mice. c p38 phosphorylation 48 h and 75 days post-treatment. Histograms represent immunoblots quantification using β-actin as control. d Cyclin A2 (CcnA2) and PLK1, and e percentage of binucleate hepatocytes and polyploid nuclei (2n, 4n, ≥8n), in the livers of WT and FL-HBx mice 75 days post-injection. f Transcription levels of Ly6D, GpC3, and AFP in the livers of WT and FL-HBx mice 75 days post-injection. Bars: median. Unpaired Student’s t or two-way ANOVA with post hoc Tukey tests; *p < 0.05; **p < 0.005; ***p < 0.0005 ****p < 0.00005
Seventy-five days after DEN treatment, we observed the persistence of these deregulations. Indeed, in treated FL-HBx transgenic mice, p38 was highly phosphorylated and cyclin A2 and PLK1 transcripts were increased (Fig. 6d). We then examined liver polyploidy. Although, 75 days after DEN injection, the percentage of binucleate hepatocytes was similar in the treated groups (26 ± 4.1% vs. 24 ± 3.4% for WT and FL-HBx mice, respectively), a significant increase of 4n hepatocytes was only evidenced in FL-HBx transgenic mice (16 ± 2.6% vs. 21 ± 3.7% for WT and FL-HBx mice, respectively) (Fig. 6e).
It was previously reported the presence of HCC progenitor cells, three or 5 months after DEN treatment mice [33]. Then, 75 days post-treatment, HCC progenitor cell markers were investigated (Fig. 6f). In DEN-treated groups, Ly6D and GpC3 mRNA levels were upregulated. In addition, a significantly higher expression of Ly6D was only observed in FL-HBx transgenic treated mice. A doubling of the levels of AFP transcripts was only noticed in treated FL-HBx transgenic mice.
Taken together, our data show that by enhancing cell cycle deregulation through PLK1/p38 axis, FL-HBx disrupts the polyploidization of hepatocytes thereby favoring DNA damage propagation. These events were then potentiated by the HBx-enhanced inflammation resulting in an increase of HCC initiation.
Discussion
We found that during physiological and pathological hepatocytes proliferation, FL-HBx by controlling the p38/PLK1 axis induces cell cycle deregulation, disrupts hepatocytes polyploidization and thus favors DNA damage propagation.
During physiological liver cell proliferation, we showed aberrant G1/S and G2/M cell-cycle transitions in FL-HBx transgenic mice. We observed an unscheduled entry into S-phase which may be caused by Cdc6 upregulation. This is consistent with reports showing, in vitro, that HBx protein promotes Cdc6 and Cdt1 proteins overproduction leading to DNA re-replication followed by polyploidization [15, 34]. These replication errors may explain the delay of G2/M progression in FL-HBx transgenic mice. In parallel, DNA damage propagation might be the result of mitosis escape and progression into cell cycle of hepatocytes with damaged DNA. Consistently, it was reported that, in HBx expressing cells, that the delay in the G2/M progression might be related to the ATM–Chk2 checkpoint activation [11]. We showed that, in FL-HBx transgenic mice, cell cycle deregulation was responsible for the generation of polyploid mononuclear hepatocytes (≥4N) directly from diploid hepatocytes (2N) to the detriment of binucleated hepatocytes. This profile of polyploidization has been reported as pathological liver development due to its association with genetic instability [35]. The transient effect observed on polyploidy deregulation during postnatal liver development was probably concurrent with the decrease in hepatocyte liver proliferation [17].
We demonstrated that, by controlling the p38/PLK1 axis, FL-HBx protein expression was responsible of the aberrant G1/S and G2/M transitions. p38 is not only involved in mitosis entry, but also in oxidative stress responses, DNA repair regulation and apoptosis [30,31,32]. In FL-HBx transgenic mice, we found that p38/ERK MAPKs pathways were strongly activated. The activation of p38/ERK MAPKs is probably involved in the lower levels of expression of DNA repair proteins, and thus participates to the increased DNA damage propagation. Furthermore, in our study, p38 MAPK activation correlates with polyploidy deregulation. Indeed, during liver development, p38 by controlling the cytoskeleton and cytokinesis is involved in polyploid nuclei formation [31, 36]. Considering PLK1, its upregulation in FL-HBx transgenic mice would be responsible for abortive mitosis resulting in the reduction of binucleate hepatocytes and in the formation of polyploid mononuclear hepatocytes (4N). Indeed, PLK1 has been implicated in checkpoint rescue and may participate in DNA damage propagation [37]. Furthermore, in accordance with our data, in HBx immortalized hepatocyte cell line, it was reported that HBx-activation of PLK1, in the G2 phase, reduces DNA damage checkpoint and DNA repair resulting in DNA damage and polyploidy propagation [38]. In the present data, the impact of activation of the p38 MAPK/PLK1 axis was evidenced, using a p38 MAPK inhibitor. We observed a decrease in PLK1 expression and a partial recovery of the Mre11 protein expression and of physiological polyploidy. This partial recovery could be due to an incomplete inhibition of p38 MAPK or to the contribution of other pathways in the abnormal polyploidy identified in FL-HBx transgenic mice.
In FL-HBx transgenic mice, transcription defect of C/EBP-α, AFP, and PepCK proteins, regulated in the course of hepatocyte maturation, indicates the impairment or the delay of hepatocyte maturation. This could be associated with the observed delay, in FL-HBx transgenic mice, in the establishment of liver polyploidy. Indeed, this is in accordance with the suggested role of liver cell polyploidization in terminal differentiation and senescence of hepatocytes [17].
In a previous report, we demonstrated that FL-HBx stimulates hepatocytes to DEN-induced HCC [28]. Therefore we explored the roles of FL-HBx on p38/PLK1 axis during early stages of HCC in association with hepatocyte polyploidy. In FL-HBx transgenic mice, HCC initiation/promotion was clearly associated, as in physiological liver proliferation, with the activation of p38/PLK1 axis. Interestingly, it was reported that increasing phosphorylation of p38 levels in HCC tissues were related with increased tumor size and was proposed as a poor survival predictor for patients with HCC [39, 40]. Moreover, PLK1 upregulation was linked with primary HCC expansion and a poor prognosis [41]. Recently, in hepatocarcinoma and cholangiocarcinoma from Asian patients, PLK1 was highly expressed in a common molecular subtypes [42].
After HCC initiation, we observed that despite the strong induction of DNA damage, evidenced by a larger number of hepatocytes with pH2AX foci, expression of Rad51 and Mre11 and activation of ChK1 were lower. In addition, this was associated with a lower apoptosis induction indicating that FL-HBx promotes the survival of the initiated hepatocytes. In the same line of evidences, the number of 4n polyploid nuclei increased in FL-HBx transgenic treated mice. Thus, DNA damage induced by DEN associated with defects in DNA repair pathways and the disruption of cell cycle checkpoint may participate to chromosomal instability and accelerated progression of HCC in FL-HBx transgenic mice.
Finally, in FL-HBx transgenic mice, overexpression of IL-6 transcripts was evidenced during HCC initiation/promotion. Similarly, in an HBx transgenic mouse model, activation of IL-6/STAT3 and Wnt/β-catenin pathways was associated to activation of hepatic progenitor cells [43]. It was reported that, in chronically HBV-infected patients, IL-6 production was associated with liver dysfunction during liver carcinogenesis [44]. This point out that during HCC initiation/promotion the FL-HBx protein by increasing the number of genetically instable hepatocytes and modulating IL-6 levels, establish a cellular environment favorable for initiated-hepatocytes growth.
Overall, our data provide evidences that during physiological and pathological hepatocytes proliferation, HBx-induced changes in cell cycle regulation lead to abnormal polyploidization in hepatocytes and to propagation of DNA damage (Fig. 7). This was triggered by PLK1 overexpression through p38 and ERK MAPKs pathways. Furthermore, during liver disease, HBx modifies inflammatory surrounding participating to hepatocyte transformation.

Schematic diagram of the proposed mechanism for HBx participates in HCC initiation by fostering DNA damage propagation through activation of PLK1/p38/ERK axis
In conclusion, genetic instability is a characteristic of HBV-associated tumors at which the HBx protein participate and thus increase the risk factor for liver cancer development [1]. However, for a full understanding of HCC development related to HBV- and HBx-induced disease, a broad evaluation of the impact of chromosomal instability, together with the estimation of the precise effect of polyploidy alterations in liver injury are required.
Materials and methods
Animals
As previously described, in FL-HBx transgenic mice the full-length HBx was under the control of viral regulatory elements [6] (see supplementary material). Experiments were executed on male heterozygous transgenic mice and littermate controls at the indicated age. Then, animals were randomly allocated in each groups. The characteristics of Alb-uPA+/+/SCID mice were previously described [45] (see supplementary material). The animals were kept in pathogen-free conditions, housed on a 12-h light–dark cycle and fed a normal diet with free access to water in the animal facility (Pitié-Salpetrière faculty). Animals were well-maintained in agreement with European Union procedures on animal care (Directive 86/609/EEC) and the sample size was estimated in accordance with the statistical test.
Animal experiments
For liver development (days 13–90 postnatal) analysis, on days 13–25, mice received an intraperitoneal injection of BrdU (100 mg/Kg) and were killed two hours after. For BrdU accumulation assays, animals were injected twice (days 17–20). The p38 inhibitor SB203580 (5 mg/kg, InvivoGen) was injected intraperitoneal at the indicated schedule. uPA+/+/Scid/RAG/GammaC mice were injected intrasplenic with human hepatocytes of the same batch (BD Bioscience, repopulation-efficacy around 20–30%) and infected with HBV as previously described [45]. For the studies of carcinogenesis initiation, 15-day-old mice were intraperitoneally injected with diethyl nitrosamine (DEN) (25 mg/kg). For DMSO/SB203580 and PBS/DEN treatments, the genotypes of animals were determined after administration of drugs. The mice were euthanized at the indicated schedule and the liver was perfused with FCS (2% in PBS). Liver samples were embedded in paraffin after overnight fixation in paraformaldehyde (4%), frozen in OCT after overnight fixation in sucrose (30%) (Sakura Finetek) or frozen in liquid nitrogen. After staining liver sections (3–5 µm) were scanned (Hamamatsu NanoZoomer scanner). All animal procedures were approved by the local animal care and use committee (Agreement A75-14-08; Nos.02890 and 02891), in agreement with the French ethic procedures (Article L-1245-2 of the Huriet law).
Statistical analysis
The number of samples to be analyzed have been predetermined based on the request of the statistical test used. For each analysis, when required, the Gaussian distribution of data was achieved with Shapiro–Wilk normality test. Exclusion criteria have been pre-established and applied only when the distribution is not normal because of one outlier. Statistical significance was evaluated in two-tailed unpaired Student’s t-tests (used to analysed means in normally distributed populations), Mann–Whitney U-tests (used when the assumption of the t-test is not met), or two-way ANOVA (used to analysis three or more variables), in Prism 6 (GraphPad software). When required, the variance was compared based on the statistical test used (Anova).
References
Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64:S84–101.
Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol. 2011;26:144–52.
Bandopadhyay M, Sarkar N, Datta S, Das D, Pal A, Panigrahi R, et al. Hepatitis B virus X protein mediated suppression of miRNA-122 expression enhances hepatoblastoma cell proliferation through cyclin G1-p53 axis. Infect Agent Cancer. 2016;11:40.
Qiao L, Leach K, McKinstry R, Gilfor D, Yacoub A, Park JS, et al. Hepatitis B virus X protein increases expression ofp21(Cip-1/WAF1/MDA6) and p27(Kip-1) in primary mouse hepatocytes, leading to reduced cell cycle progression. Hepatology. 2001;34:906–17.
Wu BK, Li CC, Chen HJ, Chang JL, Jeng KS, Chou CK, et al. Blocking of G1/S transition and cell death in the regenerating liver of Hepatitis B virus X protein transgenic mice. Biochem Biophys Res Commun. 2006;340:916–28.
Quetier I, Brezillon N, Duriez M, Massinet H, Giang E, Ahodantin J, et al. Hepatitis B virus HBx protein impairs liver regeneration through enhanced expression of IL-6 in transgenic mice. J Hepatol. 2013;59:285–91.
Na TY, Ka NL, Rhee H, Kyeong D, Kim MH, Seong JK, et al. Interaction of hepatitis B virus X protein with PARP1 results in inhibition of DNA repair in hepatocellular carcinoma. Oncogene. 2016;35:5435–45.
Bontron S, Lin-Marq N, Strubin M. Hepatitis B virus X protein associated with UV-DDB1 induces cell death in the nucleus and is functionally antagonized by UV-DDB2. J Biol Chem. 2002;277:38847–54.
Qadri I, Fatima K, Abde LHH. Hepatitis B virus X protein impedes the DNA repair via its association with transcription factor, TFIIH. BMC Microbiol. 2011;11:48.
Yue D, Zhang Y, Cheng L, Ma J, Xi Y, Yang L, et al. Hepatitis B virus X protein (HBx)-induced abnormalities of nucleic acid metabolism revealed by (1)H-NMR-based metabonomics. Sci Rep. 2016;6:24430.
Kim S, Lee HS, Ji JH, Cho MY, Yoo YS, Park YY, et al. Hepatitis B virus X protein activates the ATM-Chk2 pathway and delays cell cycle progression. J Gen Virol. 2015;96:2242–51.
Cheng B, Zheng Y, Guo X, Wang Y, Liu C. Hepatitis B viral X protein alters the biological features and expressions of DNA repair enzymes in LO2 cells. Liver Int. 2010;30:319–26.
Forgues M, Difilippantonio MJ, Linke SP, Ried T, Nagashima K, Feden J, et al. Involvement of Crm1 in hepatitis B virus X protein-induced aberrant centriole replication and abnormal mitotic spindles. Mol Cell Biol. 2003;23:5282–92.
Yun C, Cho H, Kim SJ, Lee JH, Park SY, Chan GK. Mitotic aberration coupled with centrosome amplification is induced by hepatitis B virus X oncoprotein via the Ras-mitogen-activated protein/extracellular signal-regulated kinase-mitogen-activated protein pathway. Mol Cancer Res. 2004;2:159–69.
Rakotomalala L, Studach L, Wang WH, Gregori G, Hullinger RL, Andrisani O. Hepatitis B virus X protein increases the Cdt1-to-geminin ratio inducing DNA re-replication and polyploidy. J Biol Chem. 2008;283:28729–40.
Studach LL, Rakotomalala L, Wang WH, Hullinger RL, Cairo S, Buendia MA, et al. Polo-like kinase 1 inhibition suppresses hepatitis B virus X protein-induced transformation in an in vitro model of liver cancer progression. Hepatology. 2009;50:414–23.
Gentric G, Desdouets C. Polyploidization in liver tissue. Am J Pathol. 2014;184:322–31.
Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–10.
Toyoda H, Bregerie O, Vallet A, Nalpas B, Pivert G, Brechot C, et al. Changes to hepatocyte ploidy and binuclearity profiles during human chronic viral hepatitis. Gut. 2005;54:297–302.
Guidotti JE, Bregerie O, Robert A, Debey P, Brechot C, Desdouets C. Liver cell polyploidization: a pivotal role for binuclear hepatocytes. J Biol Chem. 2003;278:19095–101.
Kudryavtsev BN, Kudryavtseva MV, Sakuta GA, Stein GI. Human hepatocyte polyploidization kinetics in the course of life cycle. Virchows Arch B Cell Pathol Incl Mol Pathol. 1993;64:387–93.
Margall-Ducos G, Celton-Morizur S, Couton D, Bregerie O, Desdouets C. Liver tetraploidization is controlled by a new process of incomplete cytokinesis. J Cell Sci. 2007;120:3633–9.
Celton-Morizur S, Merlen G, Couton D, Margall-Ducos G, Desdouets C. The insulin/Akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. J Clin Invest. 2009;119:1880–7.
Duncan AW, Hanlon Newell AE, Smith L, Wilson EM, Olson SB, Thayer MJ, et al. Frequent aneuploidy among normal human hepatocytes. Gastroenterology. 2012;142:25–8.
Pandit SK, Westendorp B, de Bruin A. Physiological significance of polyploidization in mammalian cells. Trends Cell Biol. 2013;23:556–66.
Gentric G, Maillet V, Paradis V, Couton D, L’Hermitte A, Panasyuk G, et al. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J Clin Invest. 2015;125:981–92.
Kawai H, Suda T, Aoyagi Y, Isokawa O, Mita Y, Waguri N, et al. Quantitative evaluation of genomic instability as a possible predictor for development of hepatocellular carcinoma: comparison of loss of heterozygosity and replication error. Hepatology. 2000;31:1246–50.
Quetier I, Brezillon N, Revaud J, Ahodantin J, DaSilva L, Soussan P, et al. C-terminal-truncated hepatitis B virus X protein enhances the development of diethylnitrosamine-induced hepatocellular carcinogenesis. J Gen Virol. 2015;96:614–25.
Schmit TL, Ahmad N. Regulation of mitosis via mitotic kinases: new opportunities for cancer management. Mol Cancer Ther. 2007;6:1920–31.
Awad MM, Enslen H, Boylan JM, Davis RJ, Gruppuso PA. Growth regulation via p38 mitogen-activated protein kinase in developing liver. J Biol Chem. 2000;275:38716–21.
Kurata S. Selective activation of p38 MAPK cascade and mitotic arrest caused by low level oxidative stress. J Biol Chem. 2000;275:38716–21.
Tarn C, Zou L, Hullinger RL, Andrisani OM. Hepatitis B virus X protein activates the p38 mitogen-activated protein kinase pathway in dedifferentiated hepatocytes. J Virol. 2002;76:9763–72.
He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 2013;155:384–96.
Pandey V, Kumar V. HBx protein of hepatitis B virus promotes reinitiation of DNA replication by regulating expression and intracellular stability of replication licensing factor CDC6. J Biol Chem. 2012;287:20545–54.
Gentric G, Desdouets C. Liver polyploidy: Dr Jekyll or Mr Hide? Oncotarget. 2015;6:8430–1.
Tormos AM, Rius-Perez S, Jorques M, Rada P, Ramirez L, Valverde AM, et al. p38alpha regulates actin cytoskeleton and cytokinesis in hepatocytes during development and aging. PLoS ONE. 2017;12:e0171738.
Parrilla A, Cirillo L, Thomas Y, Gotta M, Pintard L, Santamaria A. Mitotic entry: The interplay between Cdk1, Plk1 and Bora. Cell Cycle. 2016;15:3177–82.
Studach L, Wang WH, Weber G, Tang J, Hullinger RL, Malbrue R, et al. Polo-like kinase 1 activated by the hepatitis B virus X protein attenuates both the DNA damage checkpoint and DNA repair resulting in partial polyploidy. J Biol Chem. 2010;285:30282–93.
Wang SN, Lee KT, Tsai CJ, Chen YJ, Yeh YT. Phosphorylated p38 and JNK MAPK proteins in hepatocellular carcinoma. Eur J Clin Invest. 2012;42:1295–301.
Wang Y, Cui R, Zhang X, Qiao Y, Liu X, Chang Y, et al. SIRT1 increases YAP- and MKK3-dependent p38 phosphorylation in mouse liver and human hepatocellular carcinoma. Oncotarget. 2016;7:11284–98.
He ZL, Zheng H, Lin H, Miao XY, Zhong DW. Overexpression of polo-like kinase1 predicts a poor prognosis in hepatocellular carcinoma patients. World J Gastroenterol. 2009;15:4177–82.
Chaisaingmongkol J, Budhu A, Dang H, Rabibhadana S, Pupacdi B, Kwon SM, et al. Common molecular subtypes among Asian hepatocellular carcinoma and cholangiocarcinoma. Cancer Cell. 2017;32:57–70 e53.
Wang C, Yang W, Yan HX, Luo T, Zhang J, Tang L, et al. Hepatitis B virus X (HBx) induces tumorigenicity of hepatic progenitor cells in 3,5-diethoxycarbonyl-1,4-dihydrocollidine-treated HBx transgenic mice. Hepatology. 2012;55:108–20.
Wong VW, Yu J, Cheng AS, Wong GL, Chan HY, Chu ES, et al. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int J Cancer. 2009;124:2766–70.
Brezillon N, Brunelle MN, Massinet H, Giang E, Lamant C, DaSilva L, et al. Antiviral activity of Bay 41-4109 on hepatitis B virus in humanized Alb-uPA/SCID mice. PLoS ONE. 2011;6:e25096.
Acknowledgements
We thank S. Berissi and N. Gadessaud (Inserm US24/CNRS UMS3633) for tissue processing and slide staining, S. Morosan and O. Bregerie (UMS28, phénotyage du petit animal, UPMC-Paris VI) for animal care.
Funding
This work was supported by grants from the Agence Nationale de Recherches sur le Sida et les Hépatites Virales (ANRS) (AO 2014-1 16032), Ligue Contre le Cancer (2016, RS16/75-7 and 2017 RS17/75-23), Institut National de la Santé et de la Recherche Médicale (Inserm) and Université Pierre et Marie Curie (UPMC). J.A. was supported by an ANRS doctoral grant.
Author contributions
DK, JA, and CD contributed to conception of the study and design. JA, MB, CC, and JM performed experiments. All authors contributed to the interpretation of results. DK, JA, and CD drafted the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ahodantin, J., Bou-Nader, M., Cordier, C. et al. Hepatitis B virus X protein promotes DNA damage propagation through disruption of liver polyploidization and enhances hepatocellular carcinoma initiation. Oncogene 38, 2645–2657 (2019). https://doi.org/10.1038/s41388-018-0607-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0607-3
- Springer Nature Limited
This article is cited by
-
Morphological and Transcriptomic Analysis of the Supplemental Boron in the Liver of Ostrich Chicks
Biological Trace Element Research (2023)
-
Long noncoding RNA RP11-241J12.3 targeting pyruvate carboxylase promotes hepatocellular carcinoma aggressiveness by disrupting pyruvate metabolism and the DNA mismatch repair system
Molecular Biomedicine (2022)
-
Effects of Aurora kinase A on mouse decidualization via Stat3-plk1-cdk1 pathway
Reproductive Biology and Endocrinology (2021)
-
SLC38A4 functions as a tumour suppressor in hepatocellular carcinoma through modulating Wnt/β-catenin/MYC/HMGCS2 axis
British Journal of Cancer (2021)
-
Polyploidy in liver development, homeostasis and disease
Nature Reviews Gastroenterology & Hepatology (2020)