
Abstract
The availability of large-scale drug screening data on cell line panels provides a unique opportunity to identify predictive biomarkers for targeted drug efficacy. Analysis of diverse drug data on ~990 cancer cell lines revealed enhanced sensitivity of insulin-like growth factor 1 receptor/ Insulin Receptor (IGF-1R/IR) tyrosine kinase inhibitors (TKIs) in colon cancer cells. Interestingly, β-catenin/TCF(T cell factor)-responsive promoter activity exhibited a significant positive association with IGF-1R/IR TKI response, while the mutational status of direct upstream genes, such as CTNNB1 and APC, was not significantly associated with the response. The β-catenin/TCF activity high cell lines express components of IGF-1R/IR signaling more than the low cell lines explaining their enhanced sensitivity against IGF-1R/IR TKI. Reinforcing β-catenin/TCF responsive promoter activity by introducing CTNNB1 gain-of-function mutations into IGF-1R/IR TKI-resistant cells increased the expression and activity of IGF-1R/IR signaling components and also sensitized the cells to IGF-1R/IR TKIs in vitro and in vivo. Analysis of TCGA data revealed that the stronger β-catenin/TCF responsive promoter activity was associated with higher IGF-1R and IGF2 transcription in human colon cancer specimens as well. Collectively, compared to the mutational status of upstream genes, β-catenin/TCF responsive promoter activity has potential to be a stronger predictive positive biomarker for IGF-1R/IR TKI responses in colon cancer cells. The present study highlights the potential of transcriptional activity as therapeutic biomarkers for targeted therapies, overcoming the limited ability of upstream genetic mutations to predict responses.
Similar content being viewed by others

Introduction
Mutations of cancer genes have been exploited as important drug targets and/or biomarkers in advanced cancer therapies. In particular, gain-of-function mutations in several oncogenes, have been successfully used to improve clinical outcomes by acting as direct drug targets or biomarkers for patient selection [1, 2]. However, mutations in multiple other oncogenes have not been demonstrated to be strong predictive markers for available targeted therapies. Changes in the molecular activity downstream of an oncogenic mutation play key roles in the development and progression of cancer. Mutations that accumulate as a result of genomic instability and dynamic interactions with the microenvironment making the relationship of mutations with downstream signaling networks more complicated [3, 4]. Thus, focusing on the phenotypic events in direct downstream molecules of cancer-related mutations may overcome the challenges in using mutation-oriented approaches to optimize targeted therapies. Quantitative measurement of phenotypic consequences (e.g., transcriptional profiles) of genetic variations could provide improved biomarkers able to select patients most likely to benefit from specific targeted therapies [5].
Upregulation of WNT/β-catenin/TCF signaling through APC or CTNNB1 mutations contributes to tumorigenesis particularly of colon cancers [6]. However, previous efforts have not identified targeted anticancer strategies that would be predicted to be active in tumors with these mutations. Relative WNT/β-catenin/TCF activity measured using a β-catenin/TCF responsive reporter in 85 cell lines showed that upstream mutations (APC/CTNNB1) do not necessarily direct downstream transcriptional activity (WNT/TCF) in many cells [7]. Therefore, we assessed both the molecular activity of the β-catenin/TCF transcription factor and APC/CTNNB1 mutations to identify drugs that could be selective active in colon cancer. Recently, the screening data of 256 anticancer drugs in 990 cancer cells became available from the Genomics of Drug Sensitivity in Cancer (GDSC) data set [8]. We analyzed the association of drug response with the mutation profile of all tested cell lines, using sequence information of cell lines retrieved from the CCLE consortium [4].
Insulin-like growth factor 1 receptor (IGF-1R), a receptor tyrosine kinase, plays an important role in carcinogenesis and metastasis [9, 10]. IGF-1R can form a heterodimer with the insulin receptor (IR) and mediate signaling from insulin, IGF-1 and IGF2 ligands. IGF-1R activity has not been consistently associated with mutations, fusions, or amplification. Several tyrosine kinase inhibitors (TKIs) and monoclonal antibodies directed against IGF-1R have been developed [11, 12]. Multiple IGF-1R inhibitors have been tested in clinical trials but failed, at least partly, as a result of a lack of therapeutic markers to optimize patient selection. In the analysis of GDSC data, we observed the enhanced sensitivity of IGF-1R TKIs against colon cell lines. Interestingly, the strongest association was with TCF transcription activity and not APC/CTNNB1 mutations. This association was confirmed using in vitro and in vivo models, and in human tissues elucidating putative molecular mechanisms. Thus, the present study revealed a novel potential biomarker for IGF-1R TKI therapy in colon cancers. Moreover, the results provide insights into the limitations of genetic alterations as predictors of sensitivity to targeted therapies and highlights the potential of molecular phenotypes, such as transcriptional activity, to predict sensitivity to targeted anticancer drugs.
Results and discussions
Positive association of WNT (β-catenin/TCF) activity with IGF-1R/IR drug response in colon cancers
To investigate the association of cancer lineages with cellular responses to targeted drugs, we used high-throughput drug screening data on 990 cancer cell lines derived from the GDSC data set [13]. A cell line enrichment analysis (CLEA) [14] map for 104 targeted drugs that affect 10 major signaling pathways was generated using the categories of 14 major solid tumor types (Fig. 1a). The lineage-based CLEA map separated the drugs into three major groups containing drugs targeting the IGF-1R/IR, ERK-MAPK, and EGFR signaling pathways. Inhibitors targeting IGF-1R/IR and ERK-MAPK signaling showed significant activity in colon and pancreas cancers. IGF-1R/IR inhibitors affecting IGF-1R/IR signaling showed a unique association with colon and pancreatic cancer cell lines. These data suggest that IGF-1R/IR inhibitors may have utility in colorectal cancers. The presence of mutations or amplifications in genes, such as APC, CTNNB1, and EGFR, contributes to the development and progression of colorectal adenocarcinoma (COAD) [15, 16]. To further analyze the efficacy of IGF-1R inhibitors in colon cancer, we classified colon cell lines based on major genotypic aberrations in genes such as APC, CTNNB1, EGFR, BRAF, and KRAS (Fig. 1b and Supplement Fig. 1). The colon cancer cell lines were divided into two groups based on the relative sensitivity to the IGF-1R/IR inhibitors. However, mutations in common colon cancer-associated genes were not sufficient to classify cell sensitivity to IGF-1R/IR inhibitors.

Association of WNT activity with IGF-1R/IR drug response in colon cancers. a Lineage-oriented analysis of 104 targeted drugs on diverse cell lines. The large-scale drug screening data screened on a panel of 990 human cancer cell lines was obtained from the data portal of Genomics of Drug Sensitivity in Cancer 1000 (GDSC1000) (http://www.cancerrxgene.org/gdsc1000/) [13]. We selected 104 targeted drugs, which affect the 10 major signaling pathways—IGF-1R, EGFR, ERK-MAPK, ABL, WNT, p53, RTK, JNK, p38, PI3K, and TOR signaling. The −log(p-value) of the AUC is represented in different colors. Red represents a sensitive response on the lineage, while green represents a resistance (scale bar). The color index on the top represents target pathways of treated drugs. Number in the bracket indicates the total number of cell lines in the lineage. b Analysis of the sensitivity (-logIC50) of four IGF-1R/IR inhibitors in 39 colon cancer cell lines. Median fold-changes in the −logIC50 values were used to generate the heatmap profile. Red represents a sensitive response on each cell line, while green represents a resistance (scale bar). Cellular β-catenin/TCF activity marked as WNT activity is presented by ++; active, +; marginal, −; non-active, and •; unknown. (Supplement Table 1a, b). Cells in a WNT active group were satisfied with >average plus standard deviation of log (β-catenin/TCF4 reporter signal + 10) on 85 cell lines and cells in non-active group were satisfied with <average minus standard deviation of log (β-catenin/TCF4 reporter signal + 10) on 85 cell lines. The status of WNT activity was applicable to a total of 60 and 32 cancer cell lines in GDSC1000 and GSK [53, 54] drug screening data, respectively. Red and blue colored-check marks (√) represent homozygous and heterozygous mutation of indicated genes in each cell line. The cells showed very similar sensitivity to two potent IGF-1R/IR TKIs, BMS754807 and OSI-906 except KM12, which has TPM3-NTRK1 fusion gene and sensitive to TrkA inhibition activity [26] that BMS754807 also has. c The response of IGF-1R inhibitors were compared between 13 WNT active and 21 non-active cell lines. d Comparison of IGF-1R inhibitors with colon cancer drugs (data from GDSC except GSK series and PQIP from GSK data set) for the selectivity to WNT active colon cells. Cell line enrichment analysis (CLEA) [14] was performed using GDSC1000 drug screening data. The prioritization of cell lines included in a given category was analyzed using a receiver operator characteristic (ROC) curve after ranking all of cell lines by the IC50 values. The area under the ROC curve (AUC) is a quantified value to show the association of given cell line lineages or phenotypes with the drug response. The significance (p-value) of the AUC was determined by permutation tests through 1000 repeated randomizations of the ranks. AUC of –log(IC50) for each drug was calculated for WNT active colon, WNT active and non-active cells. *P < 0.05 and **P < 0.01 for the AUC
The activation of canonical WNT signaling is important in colon cancer progression and metastasis [17], which is mediated through the transcriptional activity of β-catenin and facilitates the activation of many target genes through TCF. However, WNT activation is not uniformly associated with the mutational status of APC and CTNNB1 in colon cancer suggesting additional levels of regulation [18]. To investigate the association of β-catenin/TCF activity with the sensitivity to IGF-1R inhibitors, we classified the colon cancer cell lines based on published data obtained for TOP flash promoter signal from 85 cancer cell lines [7] (Supplement Table 1a, b). Interestingly, the colon cancer cell line classified according to β-catenin/TCF activity did not show a strong association with mutations in APC and CTNNB1 but was strongly associated with the sensitivity to IGF-1R inhibitors (Fig. 1b). Thus, we compared the efficacy of IGF-1R/IR inhibitors in β-catenin/TCF active and β-catenin/TCF-non-active cell lines (Fig. 1c). Sensitivity to IGF-1R inhibitors was significantly higher in β-catenin/TCF active cells compared to that in non-active cells, except for GSK529, which had limited activity in all of the cell lines (data not shown). Moreover, we performed a CLEA analysis to compare the sensitivity and selectivity of 7 IGF-1R inhibitors with 2 clinically used therapeutic agents (5-fluorouracil and cetuximab, the chemotherapy drugs and EGFR targeted drug respectively) for colon cell lines (Fig. 1d). The response to most IGF-1R inhibitors was significantly associated with WNT activity, particularly for colon cancer. Furthermore, the efficacy of 5-fluorouracil nor cetuximab was not associated with β-catenin/TCF activity, showing that β-catenin/TCF active cells were not more vulnerable to treatment in general. Therefore, systematic analyses using large-scale cell line-based drug screening data collectively suggested that the inhibition of IGF-1R in β-catenin/TCF active cell lines represents a promising therapeutic approach that could be effective in colon cancer patients with a positive biomarker.
WNT active colon cancer cells are more sensitive to IGF-1R/IR TKIs than the WNT inactive colon cancer cells
Since the association between IGF-1R/IR TKI sensitivity and β-catenin/TCF signaling activity was derived from publicly available data from the GDSC and previously published studies, we examined whether these data were reproducible in the present study. To this end, we selected 6 cell lines: three β-catenin/TCF-active cell lines (SW480, Colo205, and HT-55) and three-inactive/moderate cell lines (SNU-C2A, KM12, and RKO) (Supplement Table 1a, b).
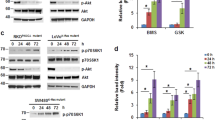
β-catenin/TCF transcriptional activity is stronger in those active cell lines compared to in those sensitive cells, as expected [17] (Fig. 2a). The IC50 values for the IGF-1R/IR TKI, GSK183705A, in the inactive cells were much higher than that of active cells, showing that the association between IGF-1R/IR TKI sensitivity and β-catenin/TCF activity assessed from the bioinformatics analyses was reproducible (Fig. 2b). mRNA expression of the IGF-1R and/or IR receptors were high in Colo205 cells, and SW480 cells (Fig. 2c). Furthermore mRNA for IGF-1 and/or IGF-2 ligands were more in the active cells (Fig. 2c). Increased IR-A mediates proliferation/survival signaling rather than metabolic/differentiation signaling [19, 20]. Endogenously activated IGF-1R was detected in the active cells and significantly inhibited by IGF-1R/IR TKI (Fig. 2b, d). Collectively, β-catenin/TCF active cells demonstrated relatively higher expression of IGF-1R/IR signaling components, suggesting that these cells could be dependent on this signaling pathway for their proliferation/survival compared to inactive cells. IGF-1R/IR may activate β-catenin through AKT [21]. However, IGF-1R/IR TKI failed to suppress the β-catenin activity in the resistant and sensitive cells demonstrating that the differential IGF-1R/IR TKI sensitivity is not mediated through β-catenin (Supplement Fig. 2).

WNT active colon cancer cells are more sensitive to IGF-1R/IR TKIs than the WNT inactive colon cancer cells. a The comparison of β-catenin/TCF promoter activity and target gene expression in the six representative colon cancer cells selected from Supplement Table 1. The relative β-catenin/TCF-responsive TOP Flash activity was shown. Three cell lines, HT-55, SW480, and COLO205, showed higher TOP Flash promoter activity than other three cell lines, RKO, KM12, and SNU-2CA. One of the main β-catenin/TCF target Axin2 mRNA expression is also shown. The quantitative RT-PCR was done using the primers listed in supplement Table 2. b The relative sensitivity against GSK1838705A of the three strong and three weak β-catenin/TCF activity cell lines demonstrated that the TCF active cells show higher sensitivity than the TCF inactive cells. c The increased expression of IGF-1R signaling components in the β-catenin/TCF active and inactive cells. The β-catenin/TCF activity is associated to the higher expression of IGF-1R, IRS, IGF-1, IGF2, or IR mRNA. The electrophoresis of IR RT-PCR products showed that the Colo205 and SW480 mainly expressed the IR A. d The IGF axis modulation by IGF-1R/IR TKI GSK1838705A in the two resistant and two sensitive cell lines. The mean value for triplicated is shown with SEM. *p < 0.01 to two resistant cell lines. #p < 0.05 to two resistant cell lines. Students’ t-test, two sides
Activation of WNT/β-catenin/TCF signaling sensitized KM12 cell lines to IGF-1R/IR TKIs through increased expression of IGF-1R signaling components
We next examined whether enforced β-catenin/TCF activation could lead to IGF-1R/IR TKI sensitivity in a β-catenin/TCF-inactive, IGF-1R/IR TKI-insensitive, cell line. KM12 showed the lowest TCF promoter activity (Supplement Table 1a, b). Furthermore, KM12 were highly resistant to IGF-1R/IR TKI (Fig. 2b). We overexpressed an oncogenic CTNNB1 mutant [22] in KM12 cells to generate KM12-MT cells, which increased β-catenin in total cell lysates and the nucleus and activated the β-catenin/TCF-responsive transcription (Fig. 3a, b) Strikingly, mutant β-catenin transfection increased sensitivity of KM12 cells to IGF-1R/IR TKIs (Fig. 3c) while the vector transfection did not change it (data not shown). The downregulation of the IGF-1R using siRNA suppressed the survival of KM12-MT cells (Fig. 3d). Consistently, mutant β-catenin increased expression of the IGF-1R, the mediator protein IRS1 and the ligands IGF-1 and IGF-2 (Fig. 3e). In addition, mutant β-catenin also increased the protein levels of IR, IRS, and p-AKT (Fig. 3f). Collectively, ectopically expressed oncogenic β-catenin activated the β-catenin/TCF signaling pathway in KM12 cells and induced IGF-1R dependency likely through the expression of IGF-1R signaling components. Currently, the detailed mechanism is not clear. However, Wnt activation of IRS1 [23] and epigenetics activation of IGF2 [24] expression may be related.

Activation of β-catenin/TCF signaling sensitized KM12 cell lines to IGF-1R/IR TKIs through increasing the expression of IGF-1R signaling components. a Introduction of a mutant β-catenin, E[beta]P, into KM12 cell line increased nuclear β-catenin and enhanced TOP Flash activity. The nuclear fraction, Nu; total cell lysate, TCL. b Increased transcription of β-catenin/TCF target genes by E[beta]P introduction. c Mutant β-catenin induced sensitization of KM12 cells to IGF-1R/IR TKIs, GSK1838705A, and OSI-906. d IGF-1R is required for the survival of KM12-MT cells. SiRNA against IGF-1R suppressed the survival of KM12-MT but did not that of KM12. e Increased expression of the IGF-1R signaling components by the mutant β catenin. f The increased expression of IGF-1R signaling components, receptor (IGF-1R and IR), the ligands (IGF1 and IGF2) and the key mediator (p-AKT). *P < 0.01 compared to KM12. Students’ t-test, two sides. g Activation of IGF-1R/IR signaling axis in KM12 cells by the CTNNB1 mutant. In the serum-starved condition, basal level of p-AKT, and the downstream mediators. The activation of the IGF-1R signaling by Insulin, IGF1, and IGF2 are prominent in KM12-MT. h The enhanced molecular responses to IGF-1R/IR TKI in KM12-MT. The molecules in IGF axis were tested after adding the IGF-1R/IR TKI. The downstream of p-IGF-1R, p-AKTs were well decreased by the IGF-1R/IR TKI in the KM12-MT while rather increased in KM12. The pIGF-1R without induction was too weak to detect by western blotting. The Src was activated (p-Y416) by IGF-1R/IR TKI in KM12 but was deactivated (p-Y527) in KM12-MT. The mean value for triplicated is shown with SEM *P < 0.01 Students’ t-test, two sides
We further examined whether the induced expression of IGF-1R components indeed activated IGF-1R signaling (Fig. 3g). The IGF signaling axis (p-IGF-1R/IR and the surrogate marker of it, p-AKT) of KM12-MT cells was more sensitive to IGF-1R ligands, insulin, IGF-1, and IGF-2 compared with KM12 cells. Increased basal levels of p-AKT (T308) in KM12-MT cells suggested that IGF-1R signaling in these cells was activated under serum-starved conditions, likely through autocrine and paracrine pathways (Fig. 3g). AKT phosphorylation was decreased in KM12-MT cells by GSK1838705A in a dose-dependent manner but increased in KM12 cells. Mutual phosphorylation between SRC and IGF-1R was reported to control IGF-1R TKI resistance [25]. Src was constitutively activated (p-SRC Y416) in KM12-MT. Inhibition of IGF-1R did not alter p-SRC(Y416) levels in KM12-MT but did slightly increase p-SRC(Y416) in the parental line. However, the induction of p-SRC(Y527; inhibitory form) by GSK1838705A suggested that SRC was inactivated in response to IGF-1R/IR inhibition in KM12-MT cells. The combined treatment with IGF-1R/IR TKI and a Src inhibitor on KM12 did not lead synergistic activity (data not shown). Collectively, these data showed that treatment with IGF-1R/IR TKI only effective in KM12-MT cells (Fig. 3h). We also find that the blocking of APC gene in KM12 also sensitized the cells to IGF-1R TKI and it is probably through of IGF-1R components expression (Supplement Fig. 3)
Elevated β-catenin/TCF activity driven sensitivity to IGF-1R TKI is preserved in vivo
We subsequently examined whether the increased sensitivity resulting from the ectopic activation of β-catenin/TCF signaling in tumor cells was reproducible in vivo (Fig. 4a). KM12 and KM12-MT xenografts were generated in SCID mice followed by treatment with IGF-1R/IR TKI. Treatment with GSK1838705A did not delay the growth of KM12 tumors but did significantly suppress the growth of KM12-MT tumors, demonstrating enhanced sensitivity to IGF-1R/IR TKI. Blood glucose levels were not significant altered, suggesting the effects of the TKI were on IGF-1R rather than insulin signaling (data not shown). The regrowth of KM12-MT treated with GSK705A may be due to the transition to original TrkA dependency [26] of parental cells.

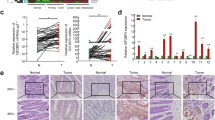
Elevated β-catenin/TCF activity is associated to IGF-1R/IR signaling in mouse and in human. a Activated β-catenin/TCF sensitized resistant colon cancer cell line KM12 to IGF-1R/IR TKIs in vivo. 5 × 105 cells of KM12 or KM12-MT were mixed with an equal volume of Matrigel (BD Biosciences, Oxford, UK) and subcutaneously injected into the flanks of NOD/SCID mice. The GSK1838705A (GSK705) or vehicle were treated to the tumor baring mice, with 10 mg per kg daily oral injection for 14 days [55]. The KM12 tumors (Top) did not show responses to the drug while the KM12-MT tumor (Bottom) growth were significantly suppressed by the treatment of GSK705 suggesting the in vivo sensitization was led by β-catenin. The dissected tumor mass also showed no difference in the KM12 cells but significant decrease in the KM12-MT cells by the treatment of GSK705 (right). *P < 0.01 compared to control. Students’ t-test, two sides. The mice were randomized for treatment, with investigator blinded for tumor measurements. b Positive association of β-catenin/TCF transcriptional activity with IGF-1R signaling in colon cancers. Differential expression of IGF-1R and IGF2 along expression level of Wnt-regulated genes in 328 TCGA COAD tissue samples. The tumor tissue samples were classified into “high” and “low” groups (25th and 75th percentiles) along expression level of LEF1, CCND1, LGR5, AXIN2, TCF7, and MYC genes, known targets of β-catenin/TCF signaling, respectively. In a box plot, each horizontal line of the rectangle indicates the first quartile, median, and third quartile from the bottom to top. A vertical line extended from the rectangle means the minimum and maximum values, except outliers (the supplement methods). The colon tumors highly expressing β-catenin/TCF target genes express more the IGF-1R and IGF2 significantly except MYC expressing tumors. Students’ t-test, two sides
β-catenin/TCF activity is associated with IGF-1R and IGF2 expression in human colon cancer tissues
To confirm the relevance of association between the IGF-1R TKI sensitivity and β-catenin/TCF activity, we extended our analysis to clinical tumor samples. The expression of β-catenin/TCF targets and IGF-1R/IR signaling components, acquired from TCGA RNA sequencing data in COAD tissue samples were analyzed (Fig. 4b). IGF-1R and IGF2 genes were significantly overexpressed in tissues showing higher expression of β-catenin/TCF target genes than the lower expressing tissues. Interestingly, higher expression of Myc was associated with IGF-1R overexpression but not with IGF2 suggesting the various human tissues may use slightly different ways to activate IGF-1R/IR signaling.
It is somewhat surprising that TCF activity, but not APC/CTNNB1 mutations, was significantly associated with IGF-1R/IR TKI efficacy. APC/ CTNNB1 mutations or epigenetic suppression of APC [27] is observed in more than 80% of colon cancer patients [28] and believed to lead upregulation of β-catenin/TCF activity via the canonical pathway. However, β-catenin/TCF target genes are expressed in diverse level in colon cancer tissues, consistent with nuclear β-catenin accumulation [29]. Further, non-canonical regulation of WNT signaling through calcium may suppress TCF activity, even in the presence of those mutations [30]. β-catenin activation may also be mediated by non-canonical pathways [31]. Consistently, β-catenin/TCF signaling activity showed extreme variation among cell lines with mutations in APC/CTNNB1 [7]. In an alternative mechanism, APC mutants regulate Hippo-YAP signaling independent of β-catenin stabilization [32]. Thus, APC/CTNNB1 mutations may not fully capture β-catenin/TCF activity.
Discrepancies between the predictive value of genotype and phenotype may exist in other well-established oncogenic signaling networks. Although many driver mutations are critical for early stages of carcinogenesis, they may not be essential in later stages when the cancer is established [33]. The development of resistance mutations in cancers treated with targeted therapeutic agents (e.g., EGFR TKI) in downstream signaling components [34, 35], other growth signaling pathways [36] or independent signaling pathways [37, 38] contributes to cancer evolution and crosstalk among oncogenic signaling cascades. The existence of cancer MR (master regulator) gene would explain the mechanism that the different repertoire of mutations leads similar transcriptional consequences [39] as well.
Multiple studies have shown that IGF-1R signaling is associated with the risk and prognosis of malignancy in a variety of tissue types [40,41,42,43,44], including colon cancers [45, 46]. The early success of clinical trials targeting this signaling pathway using monoclonal antibodies [47, 48] and small molecules [49, 50] were exciting, although failures were reported in subsequent clinical trials [47, 51]. Indeed, stage II/III clinical trials for IGF-1R targeting of unselected populations of refractory colorectal cancers did not demonstrate marked activity [52]. Therefore, the positive association between β-catenin/TCF activity and IGF-1R/IR TKI sensitivities may provide an approach to enrich patients likely to benefit from IGF-1R/IR TKI in clinical trials. Currently, the use of transcriptional activity for biomarker may be not easy. However, the fast development of genomics technology may help us to use transcriptomic phenotype of tumor cells as a biomarker in the near future.
We demonstrated that, rather than any mutation profiles, β-catenin/TCF activity is a better positive biomarker for the efficacy of IGF-1R/IR TKIs on colon cancer cells based on recently reported public data [8] and the in vitro and in vivo experiments with human tumor tissue analysis conducted in the present study. Mechanistically, we demonstrated that β-catenin/TCF transcriptional activation led to the transcriptional induction of IGF-1R pathway members, resulting in the increased dependency of these cells on IGF-1R signaling. This novel biomarker may be useful for the selection of patients for future clinical trials for IGF-1R/IR TKI-based therapies. In addition, it also highlights the potential of transcriptional activity as therapeutic biomarkers for targeted therapies, overcoming the limited ability of upstream genetic mutations to predict responses.
References
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500.
Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22.
Michor F, Iwasa Y, Nowak MA. Dynamics of cancer progression. Nat Rev Cancer. 2004;4:197–205.
Saunders NA, Simpson F, Thompson EW, Hill MM, Endo-Munoz L, Leggatt G, et al. Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives. EMBO Mol Med. 2012;4:675–84.
Zenonos K, Kyprianou K. RAS signaling pathways, mutations and their role in colorectal cancer. World J Gastrointest Oncol. 2013;5:97–101.
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2016;36:1461–1473.
Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151:1457–73.
Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–61.
Pollak M. Insulin, insulin-like growth factors and neoplasia. Best Pract Res Clin Endocrinol Metab. 2008;22:625–38.
Pollack MN. Insulin, insulin-like growth factors, insulin resistance, and neoplasia. Am J Clin Nutr. 2007;86:s820–22.
Qu X, Wu Z, Dong W, Zhang T, Wang L, Pang Z, et al. Update of IGF-1 receptor inhibitor (ganitumab, dalotuzumab, cixutumumab, teprotumumab and figitumumab) effects on cancer therapy. Oncotarget. 2017;8:29501–18.
Tognon CE, Sorensen PH. Targeting the insulin-like growth factor 1 receptor (IGF1R) signaling pathway for cancer therapy. Expert Opin Ther Targets. 2012;16:33–48.
Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166:740–54.
Kim N, He N, Kim C, Zhang F, Lu Y, Yu Q, et al. Systematic analysis of genotype-specific drug responses in cancer. Int J Cancer. 2012;131:2456–64.
Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–4.
Spano JP, Fagard R, Soria JC, Rixe O, Khayat D, Milano G. Epidermal growth factor receptor signaling in colorectal cancer: preclinical data and therapeutic perspectives. Ann Oncol. 2005;16:189–94.
Ormanns S, Neumann J, Horst D, Kirchner T, Jung A. WNT signaling and distant metastasis in colon cancer through transcriptional activity of nuclear beta-Catenin depend on active PI3K signaling. Oncotarget. 2014;5:2999–3011.
Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–8.
Beneit N, Fernandez-Garcia CE, Martin-Ventura JL, Perdomo L, Escribano O, Michel JB, et al. Expression of insulin receptor (IR) A and B isoforms, IGF-IR, and IR/IGF-IR hybrid receptors in vascular smooth muscle cells and their role in cell migration in atherosclerosis. Cardiovasc Diabetol. 2016;15:161.
Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999;19:3278–88.
Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, et al. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–9.
Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci USA. 1997;94:10330–4.
Bommer GT, Feng Y, Iura A, Giordano TJ, Kuick R, Kadikoy H, et al. IRS1 regulation by Wnt/beta-catenin signaling and varied contribution of IRS1 to the neoplastic phenotype. J Biol Chem. 2010;285:1928–38.
Zhong H, Fazenbaker C, Chen C, Breen S, Huang J, Yao X, et al. Overproduction of IGF-2 drives a subset of colorectal cancer cells, which specifically respond to an anti-IGF therapeutic antibody and combination therapies. Oncogene. 2017;36:797–806.
Min HY, Yun HJ, Lee JS, Lee HJ, Cho J, Jang HJ, et al. Targeting the insulin-like growth factor receptor and Src signaling network for the treatment of non-small cell lung cancer. Mol Cancer. 2015;14:113.
Tatematsu T, Sasaki H, Shimizu S, Okuda K, Shitara M, Hikosaka Y, et al. Investigation of neurotrophic tyrosine kinase receptor 1 fusions and neurotrophic tyrosine kinase receptor family expression in non-small-cell lung cancer and sensitivity to AZD7451 in vitro. Mol Clin Oncol. 2014;2:725–30.
Jass JR, Whitehall VL, Young J, Leggett BA. Emerging concepts in colorectal neoplasia. Gastroenterology. 2002;123:862–76.
Takayama T, Ohi M, Hayashi T, Miyanishi K, Nobuoka A, Nakajima T, et al. Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology. 2001;121:599–611.
Hlubek F, Brabletz T, Budczies J, Pfeiffer S, Jung A, Kirchner T. Heterogeneous expression of Wnt/beta-catenin target genes within colorectal cancer. Int J Cancer. 2007;121:1941–8.
Najdi R, Syed A, Arce L, Theisen H, Ting JH, Atcha F, et al. A Wnt kinase network alters nuclear localization of TCF-1 in colon cancer. Oncogene. 2009;28:4133–46.
Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell. 2008;133:340–53.
Cai J, Maitra A, Anders RA, Taketo MM, Pan D. β-catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015;29:1493–506.
Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–24.
Agaram NP, Wong GC, Guo T, Maki RG, Singer S, Dematteo RP, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008;47:853–9.
Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci USA. 2012;109:E2127–2133.
Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88.
Kubo T, Yamamoto H, Lockwood WW, Valencia I, Soh J, Peyton M, et al. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer. 2009;124:1778–84.
Wang L, Hu H, Pan Y, Wang R, Li Y, Shen L, et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS ONE. 2014;9:e88291.
Califano A, Alvarez MJ. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat Rev Cancer. 2017;17:116–30.
Jones RA, Campbell CI, Wood GA, Petrik JJ, Moorehead RA. Reversibility and recurrence of IGF-IR-induced mammary tumors. Oncogene. 2009;28:2152–62.
Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351:1393–6.
Ma J, Giovannucci E, Pollak M, Stampfer M. RESPONSE: Re: Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J Natl Cancer Inst. 1999;91:2052.
Ma J, Pollak M, Giovannucci E, Chan JM, Tao Y, Hennekens C, et al. A prospective study of plasma levels of insulin-like growth factor I (IGF-I) and IGF-binding protein-3, and colorectal cancer risk among men. Growth Horm IGF Res. 2000;10(Suppl A):S28–29.
Yu H, Spitz MR, Mistry J, Gu J, Hong WK, Wu X. Plasma levels of insulin-like growth factor-I and lung cancer risk: a case-control analysis. J Natl Cancer Inst. 1999;91:151–6.
Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenburg S, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299:1753–5.
Wu Y, Yakar S, Zhao L, Hennighausen L, LeRoith D. Circulating insulin-like growth factor-I levels regulate colon cancer growth and metastasis. Cancer Res. 2002;62:1030–5.
Di Cosimo S, Sathyanarayanan S, Bendell JC, Cervantes A, Stein MN, Brana I, et al. Combination of the mTOR inhibitor ridaforolimus and the anti-IGF1R monoclonal antibody dalotuzumab: preclinical characterization and phase I clinical trial. Clin Cancer Res. 2015;21:49–59.
Tap WD, Demetri G, Barnette P, Desai J, Kavan P, Tozer R, et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol. 2012;30:1849–56.
Jones RL, Kim ES, Nava-Parada P, Alam S, Johnson FM, Stephens AW, et al. Phase I study of intermittent oral dosing of the insulin-like growth factor-1 and insulin receptors inhibitor OSI-906 in patients with advanced solid tumors. Clin Cancer Res. 2015;21:693–700.
Puzanov I, Lindsay CR, Goff L, Sosman J, Gilbert J, Berlin J, et al. A phase I study of continuous oral dosing of OSI-906, a dual inhibitor of insulin-like growth factor-1 and insulin receptors, in patients with advanced solid tumors. Clin Cancer Res. 2015;21:701–11.
Fassnacht M, Berruti A, Baudin E, Demeure MJ, Gilbert J, Haak H, et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. Lancet Oncol. 2015;16:426–35.
Sclafani F, Kim TY, Cunningham D, Kim TW, Tabernero J, Schmoll HJ et al. A randomized phase ii/iii study of dalotuzumab in combination with cetuximab and irinotecan in chemorefractory, KRAS wild-type, metastatic colorectal cancer. J Natl Cancer Inst. 2015; 107:djv258.
McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci USA. 2007;104:19936–41.
Greshock J, Bachman KE, Degenhardt YY, Jing J, Wen YH, Eastman S, et al. Molecular target class is predictive of in vitro response profile. Cancer Res. 2010;70:3677–86.
Sabbatini P, Korenchuk S, Rowand JL, Groy A, Liu Q, Leperi D, et al. GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol Cancer Ther. 2009;8:2811–20.
Acknowledgements
This work was supported by the National Research Foundation of Korea, [NRF-2018R1A2B6009313 to EJ], [NRF-2017R1A2B2007745, and NRF-2016R1A5A1011974 to SY] and [NRF-2012M3A9B6055466, NRF-2015R1D1A1A01056594, and NRF-2011-0030074 to W-YK].
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
These authors contributed equally: Hani Lee and Nayoung Kim
Rights and permissions
About this article

Cite this article
Lee, H., Kim, N., Yoo, Y.J. et al. β-catenin/TCF activity regulates IGF-1R tyrosine kinase inhibitor sensitivity in colon cancer. Oncogene 37, 5466–5475 (2018). https://doi.org/10.1038/s41388-018-0362-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0362-5
- Springer Nature Limited
This article is cited by
-
Canonical WNT/β-catenin signaling upregulates aerobic glycolysis in diverse cancer types
Molecular Biology Reports (2024)
-
Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: a challenge for cancer therapy
Journal of Hematology & Oncology (2020)