
Abstract
Poor oxygenation is a common hallmark of solid cancers that strongly associates with aggressive tumor progression and treatment resistance. While a hypoxia-inducible factor 1α (HIF-1α)-associated transcriptional overexpression of the hepatocyte growth factor (HGF) receptor tyrosine kinase (RTK) MET has been previously documented, any regulation of the HIF-1α system through MET downstream signaling in hypoxic tumors has not been yet described. By using MET-driven in vitro as well as ex vivo tumor organotypic fresh tissue models we report that MET targeting results in depletion of HIF-1α and its various downstream targets. Mechanistically, we provide evidence that MET regulates HIF-1α levels through a protein translation mechanism that relies on phosphorylation modulation of the eukaryotic initiation factor 4G1 (eIF4G1) on serine 1232 (Ser-1232). Targeted phosphoproteomics data demonstrate a significant drop in eIF4G1 Ser-1232 phosphorylation following MET targeting, which is linked to an increased affinity between eIF4G1 and eIF4E. Since phosphorylation of eIF4G1 on Ser-1232 is largely mediated through mitogen-activated protein kinase (MAPK), we show that expression of a constitutively active K-RAS variant is sufficient to abrogate the inhibitory effect of MET targeting on the HIF-1α pathway with subsequent resistance of tumor cells to MET targeting under hypoxic conditions. Analysis of The Cancer Genome Atlas data demonstrates frequent co-expression of MET, HIF-1α and eIF4G1 in various solid tumors and its impact on disease-free survival of non-small cell lung cancer patients. Clinical relevance of the MET-eIF4G1-HIF-1α pathway is further supported by a co-occurrence of their expression in common tumor regions of individual lung cancer patients.
Similar content being viewed by others


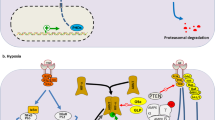
Introduction
Hypoxia is a common biologic hallmark of solid cancers characterized by aggressive manifestations of growth and progression. Uncontrolled tumor growth prevails the capacity of the vasculature to respond to increased oxygen requirements [1] and at the same time, tumor blood vessels are structurally and biologically abnormal, rendering them functionally impaired [2]. As a consequence, the appearance of hypoxic areas in tumors is not necessarily linked to blood vessel proximity [3]. Inadequate oxygenation sustains hypoxia-inducible factor-1 (HIF-1), the master transcriptional activator of the hypoxic response, which induces a series of cellular adaptations favoring tumor progression [4, 5]. In normoxia, the regulatory subunit of HIF-1, HIF-1α, is converted into a hydroxylated form by oxygen-dependent prolyl-hydroxylases (PHDs). This event primes HIF-1α to undergo ubiquitination by the von Hippel-Lindau E3 ubiquitin ligase and subsequent proteasome-dependent degradation [3, 5]. On the other hand, low oxygen conditions inactivate PHDs and subsequently prevent HIF-1α degradation. Stabilized HIF-1α translocates into the nucleus where it forms complex with the constitutively expressed HIF-1β subunit [6]. The formed heterodimer binds to hypoxia-responsive elements on target genes [7, 8], resulting in transactivation of expression of genes involved in key tumorigenic processes such as angiogenesis, cell survival, glucose metabolism and invasion [9]. During particular physiologic conditions (e.g., during embryogenesis), low oxygen tension supports the generation of niches that maintain pluripotent cells [7]. When hypoxia-induced traits such as physiological stemness and invasive growth develop aberrantly, an aggressive malignant phenotype is sustained [7].
Already in 2003, Pennacchietti et al. showed that hypoxia directly induces transcription of the hepatocyte growth factor (HGF) receptor MET [10]. By analysing the human MET promoter, increased transcription of MET was demonstrated to be mediated by two different hypoxia-responsive elements located in the 5′ untranslated region (5′ UTR) [5, 10]. Although MET is implicated in physiological cellular processes such as proliferation and migration and masters the embryonic morphogenetic program referred to as “invasive growth” [7], the MET pathway is at the same time frequently deregulated in human cancer as abnormal MET signaling has been reported in both solid as well as in hematological malignancies [11]. MET was shown to be aberrantly activated via missense mutations, overexpression and amplification, as well as by auto- or paracrine MET/HGF loops [11,12,13]. MET is often overexpressed or amplified in numerous tumor entities such as brain, colorectal, gastric, lung and head and neck tumors [11, 14, 15], frequently associating with unfavorable patient prognosis [11, 14, 15]. Notably, MET transcriptional activation and deregulated signaling are implicated in tumor-related hypoxia [16]. Deregulated MET transduction-associated metastasis may probably stand for a genetic rewiring, which could help tumor cells escape a hypoxic environment [4, 10].
In most of tumor cell lines where MET is aberrantly active due to MET gene amplification, MET inhibition (METi) leads to a cease in proliferation often followed by a massive cell death. So far, this addiction to signaling via the MET receptor has been repeatedly reported in cell cultures derived from gastric and non-small cell lung carcinomas (NSCLC) [17, 18]. It is plausible that this MET-addicted phenotype featuring amplification of the MET gene might serve as a genetic predictor of responses towards antineoplastic therapies [19]. However, the detailed molecular mechanisms underlying MET signaling under clinically-relevant hypoxic conditions remain largely unknown. In order to close this gap, here we aimed at elucidating the molecular effects of the MET receptor tyrosine kinase (RTK) inhibition on MET-overexpressing cancer cells in hypoxic environment. We show that MET receptor targeting in hypoxia leads to a decrease in protein levels of HIF-1α as well as its targets, most probably by interference with HIF-1α translation via modulation of the eukaryotic translation initiation factor eIF4G1 phosphorylation. As the reported association between MET signaling and expression of HIF-1α target genes might be central in cancer progression, METi-based interference with this axis is likely to contribute to the efficacy of MET inhibitors in hypoxic tumors.
Results
MET inhibition decreases expression of HIF-1α and its target genes under hypoxic conditions
Hypoxia is one of the critical determinants of numerous solid tumors, associated both with accelerated progression as well as resistance to anti-cancer treatment modalities including ionizing radiation and molecular targeting [20, 21]. Since various studies reported a crosstalk between the MET RTK and HIF-1α signaling [8, 10, 22], the prime motivation for the current study was to evaluate the impact of inhibition of MET on hypoxia-associated pathways. As HIF-1α is the master transcriptional activator of the hypoxic response, we examined its expression following MET targeting in a set of four MET-addicted human cancer cell lines. The NSCLC line EBC-1 and three gastric cancer cell lines GTL-16, SNU-638 and KATOII were exposed to hypoxic conditions (1.5% O2) and 24 h later treated with 100 nM of the specific anti-MET small molecule inhibitor tepotinib (EMD1214063; Merck, Darmstadt, Germany) for 24 h. Interestingly, the protein levels of both HIF-1α and its transcriptional target carbonic anhydrase IX (CAIX) substantially decreased following METi in all tested cell lines (Fig. 1a). Furthermore, this observed decrease in HIF-1α protein levels could be detected as early as 5 min after exposing the cells to tepotinib (Supplementary Figure 1A) and also by employing 300 nM of PHA665752, another specific MET small molecule inhibitor (Supplementary Figure 1B).

Impact of MET inhibition on HIF-1α expression, its targets transcription and glucose metabolism under hypoxic conditions. a GTL-16, EBC-1, SNU-638 and KATOII cells were exposed for 48 h to hypoxia (1.5% O2). They underwent a treatment with 100 nM tepotinib after the first 24 h of exposure. 24 h later, the cells were lysed and total cell proteins were subjected to Western blotting with a specific antibody against phospho-MET, HIF-1α or CAIX. b GTL-16 tumor xenografts developed in RAG2−/−; γc−/− mice were extracted, exposed as organotypic tissue slices to the same conditions as in (a) and processed for IHC. Representative images (100× magnification) of phospho-MET, HIF-1α and GLUT-1 staining in organotypic tissue slices. c Quantification of CAIX and GLUT-1 mRNAs by Real-Time PCR analysis under normoxia or hypoxia (1.5% O2) in control GTL-16 cells and cells treated by 100 nM tepotinib for 16 or 24 h. P values are calculated by student’s t test. **P = 0.0079; ***P < 0.0001. d Levels of TCA cycle and glycolysis-related metabolites in GTL-16 cells altered by hypoxia with or without concomitant METi, as described in (a), were determined by non-targeted metabolomics measurements (Hx—hypoxia, Nx—normoxia, EMD—METi (tepotinib)). e Quantitative Real-Time PCR analysis of ALDOC (left panel) and PFKFB-4 (right panel) gene expression in GTL-16 cells exposed to the same conditions as in (c). P values are calculated by student’s t test, ***P < 0.0001
Next, we wished to extend this observation to a 3D ex vivo model. To that end, EBC-1 and GTL-16 cells were subcutaneously injected into the flank of RAG2−/−; γc−/− mice to form tumors that were extracted and further cultured as organotypic tissue culture slices. One day after establishing the ex vivo cultures, tissue slices were exposed to hypoxia (1.5% O2), the next day they were treated with 100 nM of tepotinib for 24 or 72 h and finally, they were processed for immunohistochemical staining to assess the activation of the MET and HIF-1α signaling pathways. We could show that MET targeting decreases the protein level of HIF-1α and its target glucose transporter 1 (GLUT-1) in the 3D ex vivo model analogously to the in vitro findings (Fig. 1b).
In order to evaluate if the observed decreased protein levels of HIF-1α target genes upon METi are consequence of a decrease in HIF-1α-mediated transactivation of genes, GTL-16 cells exposed for 24 h to hypoxia (1.5% O2) were treated with 50 nM tepotinib, harvested 16 and 24 h later and isolated RNA was analyzed by Real-Time PCR. Consistent with the previous observations, transcription of the two HIF-1α target genes, CAIX and GLUT-1, was decreased following METi (Fig. 1c), thus underlining the functional consequences of the decreased HIF-1α total protein levels.
Impact of METi on GLUT-1 is further associated with changes in glycolysis-related metabolome and decreased levels of aldolase C (ALDOC) and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 (PFKFB-4)
As it was previously shown that a HIF-1α-mediated increase in the expression level of GLUT-1 serves cancer cells as an important mechanism that mediates glucose uptake under hypoxic conditions [23], we set to examine a possible functional change in glucose metabolism upon METi and performed non-targeted mass spectrometry measurements of the cellular metabolome in GTL-16 in hypoxic conditions (1.5% O2) with and without 100 nM tepotinib for 24 h. Interestingly, the expected increase in metabolites involved in tricarboxylic acid cycle and glycolysis under hypoxia as compared to normoxic conditions seems to be largely reversed by METi (Fig. 1d). In this respect, we could detect a significant decrease in levels of 338.9889 hexose bisphosphate (fold change of −1.659, p-value 2.99 × 10−10 in METi versus control under hypoxia) corresponding to fructose-1,6-bisphosphate (F1, 6BP) and/or fructose-2,6-bisphosphate (F2, 6BP). These two metabolites are products of ALDOC and PFKFB-4, respectively. Intriguingly, the expression of PFKFB-3 and PFKFB-4, enzymes implicated in the regulation of glycolysis in cancer cells as well as in their proliferation and survival, is strongly induced by HIF-1α through active HIF-1α-binding sites in their promoter regions and their expression under hypoxic conditions has been correlated with enhanced expression of VEGF and GLUT-1 mRNA as well as with increased levels of HIF-1α [24,25,26]. Thus, to determine if the observed decrease in hexose bisphosphate levels upon METi under hypoxic conditions is possibly a result of reduced transcription of HIF-1α targets, we investigated mRNA expression of both ALDOC and PFKFB-4 in GTL-16 cells treated with 50 nM tepotinib for 16 or 24 h in normoxic (21% O2) and hypoxic conditions (1.5% O2). Indeed, although ALDOC and PFKFB-4 mRNA levels are both elevated following hypoxia, prolonged METi (16 or 24 h) significantly decreases their expression (Fig. 1e), most probably following the decrease of the HIF-1α protein.
Targeting MET does not interfere with HIF-1α gene transcription and protein stability but affects its translation
At this point, we wished to investigate whether the decreased HIF-1α protein levels upon METi might possibly reflect MET-mediated decrease in HIF-1α gene transcription. Real-time PCR analysis was performed on RNA isolated from EBC-1 and GTL-16 cells exposed to hypoxia (1.5% O2) 24 h prior to treatment with 50 nM tepotinib for additional 24 h. As shown in Fig. 2a, METi leads rather to a slight increase of HIF-1α mRNA in both tested cell lines, indicating that HIF-1α transcription is not negatively affected by tepotinib treatment.

Targeting MET inhibits translation of the transcript encoding HIF-1α protein without affecting HIF-1α mRNA expression and protein stability. a GTL-16 and EBC-1 cells were exposed for 48 h to hypoxia (1.5% O2). After the first 24 h of exposure, the cells were treated with 50 nM tepotinib and 24 h later harvested. Quantification of HIF-1α mRNA was performed by Real-Time PCR analysis. P values are calculated by student’s t test. ***P1 = 0.0002. ***P2 < 0.0001; **P = 0.0069. b, c GTL-16 and EBC-1 cells were incubated in hypoxia (1.5% O2). 24 h after the start of the incubation, the cells underwent a treatment with 100 nM tepotinib and (b) 100 μM DMOG (PHDi—prolyl-hydroxylase inhibition) or (c) 10 μM MG-132 (Pi—proteasome inhibition) for additional 24 h. Cell lysates were assayed by Western blot showing a HIF-1α decrease following METi combined with (b) PHDi or (c) Pi. d GTL-16 and EBC-1 cells were exposed to hypoxia (1.5% O2). After 24 h, they were treated with 100 nM tepotinib and/or 50 μM CHX (TRi—translation inhibition) during the indicated time points. Whole-cell lysates were subjected to Western blotting using specific antibodies against phospho-MET and HIF-1α following METi and TRi treatment during the indicated time points. e GTL-16 and EBC-1 cells were pretreated with 100 nM tepotinib for 24 h and 100 μM DMOG was added for the indicated times. Western blot analysis showes HIF-1α protein accumulation following inhibition of PHD and MET
As HIF-1α protein expression and transcription of its target genes both decrease following inhibition of the MET receptor in MET-addicted cancer cell lines and at the same time, METi does not affect transcription of the HIF-1α gene itself as inferred from stable or slightly elevated HIF-1α mRNA levels following tepotinib treatment, we hypothesized that the observed METi-mediated decrease in HIF-1α protein levels and function may be caused by decreased stability or reduced translation of HIF-1α. To test whether METi affects the stability of HIF-1α protein, we first inhibited the activity of PHD, a key enzyme responsible for HIF-1α degradation, by employing 100 µM of the PHD inhibitor DMOG in combination with 100 nM of tepotinib during 24 h. As expected, DMOG alone stabilizes HIF-1α protein levels under normoxic conditions (21% O2) (Fig. 2b). However, when PHD is inhibited in the presence of the MET inhibitor tepotinib, HIF-1α protein amount nevertheless decreases (Fig. 2b), indicating that METi most probably does not impact HIF-1α stability.
In order to strengthen this assumption, we next interfered with proteosomal degradation of HIF-1α using 10 µM of the proteasome inhibitor MG-132. Analogously to the interference with PHD activity, proteasome inhibition prevented degradation of HIF-1α and led to its stabilization while METi in the presence of MG-132 was still capable of reducing protein levels of HIF-1α (Fig. 2c). Thus, as despite blocking either of two distinct components involved in HIF-1α degradation the HIF-1α protein levels remain reduced upon METi, the disruption of MET signaling seems to affect HIF-1α protein levels independently of its stability.
To test whether METi interferes with the translation of HIF-1α, general de novo protein synthesis was inhibited using 50 µM of cycloheximide (CHX). GTL-16 and EBC-1 cells were first exposed to hypoxia (1.5% O2) and 24 h later, the cells were treated with 50 µM CHX alone or in combination with 100 nM tepotinib for various time points. As expected, treatment with CHX alone reduced HIF-1α protein levels in time, indicating the desired block in HIF-1α protein synthesis (Fig. 2d, Supplementary Figure 1). Interestingly, when the cells were treated with both CHX and tepotinib, the pattern of HIF-1α decrease remained the same (Fig. 2d, Supplementary Figure 2). This equal half-life of HIF-1α protein observed when the cells are treated with CHX alone or in combination with METi suggests that METi may possibly have an inhibitory effect on translation of the transcript encoding HIF-1α protein.
As an additional way to examine the effect of METi on HIF-1α protein synthesis, we determined the accumulation rate of HIF-1α protein by using DMOG. GTL-16 and EBC-1 cells were pretreated with 100 nM of tepotinib for 24 h and 100 µM DMOG was added at the indicated times. When PHD is inhibited by DMOG, HIF-1α is not anymore degraded but constantly translated and therefore rapidly accumulates (Fig. 2e). However, the addition of tepotinib together with DMOG leads to a significant reduction in the HIF-1α accumulation, thus strengthening the notion that blocking MET signaling inhibits HIF-1α translation (Fig. 2e).
MET signaling influences HIF-1α translation through MAPK-dependent phosphorylation of eIF4G1 on Ser-1232
The observations that targeting MET does not affect HIF-1α protein half-life following inhibition of de novo protein synthesis and considerably reduces the accumulation rate of HIF-1α protein in time strongly imply that MET targeting has an inhibitory effect on HIF-1α translation. In order to get deeper insights into this effect, we aimed at identifying particular players that are implicated in the synthesis of HIF-1α protein and are affected by METi. Badura et al reported in 2012 that HIF-1α translation displays a strong dependence on eIF4G1 [27]. Interestingly, although eIF4G1 protein levels are not affected by tepotinib treatment, HIF-1α reduction observed following METi correlates with a decrease in phosphorylation of eIF4G1 on Ser-1232 (Fig. 3a). Interestingly, METi-mediated decrease in eIF4G1 Ser-1232 phosphorylation could be detected also in normoxic conditions (21% O2) (Supplementary Figure 3A) and, in addition to Western blot analysis, we could confirm this observation in EBC-1 and GTL-16 cells as well as in 3 additional MET-addicted cancer cell lines (SNU-638, H1993 and Hs746T) also by the targeted mass spectrometry-based quantitative phosphoproteomic method selected reaction monitoring (SRM) (Fig. 3b, Supplementary Figure 3B).

MET signaling influences HIF-1α translation through phosphorylation of eIF4G1 on Ser-1232. a GTL-16 and EBC-1 cells underwent a treatment with 100 nM tepotinib during exposure to hypoxia (1.5 O2). Whole cell lysates were subjected to Western blotting using specific antibodies against eIF4G1/phospho-eIF4G1, phospho-MET and HIF-1α. b SRM measurements of eIF4G1 phosphorylation on Ser-1232 following 50 nM tepotinib treatment during 24 h under normoxic conditions (21% O2). c EBC-1 organotypic tissue slices were exposed to the same conditions as in (a) and processed for IHC. Phospho-eIF4G1 (peIF4G1), HIF-1α, phospho-MET (pMET), CAIX and GLUT-1 staining in representative images (100× magnification). d Pulldown of total eIF4G1 protein on lysates of GTL-16 cells incubated in hypoxia (1.5 O2) and treated for 24 h with 100 nM tepotinib was followed by Western blotting using a specific antibody against eIF4E (WCL - whole cell lysates; IP - immunoprecipitation)
Furthermore, we could validate that the reduced HIF-1α protein abundance and the reduced mRNA expression of its transcriptional targets CAIX and GLUT-1 correlate with decreased phosphorylation of eIF4G1 on Ser-1232 following METi also in the 3D ex vivo model (Fig. 3c, Supplementary Figure 4).
Interestingly, the link between MET, HIF-1α, and eIF4G1 seems to be relevant also in cells that express non-phosphorylated MET protein as activation of MET by 50 ng/ml HGF leads to increase in both eIF4G1 Ser-1232 phosphorylation and HIF-1α expression after only 7 min (Supplementary Figure 5).
eIF4G1 is a part of the eukaryotic initiation factor complex F4 (eIF4F), together with eIF4E and eIF4A [28]. While eIF4E binds to the mRNA cap, eIF4A acts as an ATP-dependent RNA helicase needed to unwind mRNA secondary structure, and eIF4G1 is a scaffold protein upon which ribosomes and the other eukaryotic initiation factors assemble [27]. By binding to eIF4A, eIF4G1 inhibits its helicase activity. Following phosphorylation on Ser-1232, the HEAT-2 domain of eIF4G1 dissociates from eIF4A, reconstituting its helicase activity [29]. In a study with Saccaromyces cerevisiae, Tarun et al reported that binding of eIF4E to eIF4G represses eIF4G from mediating uncapped-mRNA translation [30]. To further investigate over the interaction between eIF4G1 and eIF4E in eukaryotic cells applied to our system, we assessed by immunoprecipitation the interaction status between these two eukaryotic initiation factors in response to tepotinib treatment. We could show that METi (24 h, 100 nM tepotinib) under hypoxic conditions coincides with increased interaction between eIF4G1 and eIF4E (Fig. 3d), thus providing a novel possible consequence of MET signaling-mediated eIF4G1 Ser-1232 phosphorylation.
In 2013 Dobrikov et al reported that activation of MAPK signaling results in phosphorylation of eIF4G1 at Ser-1232 by the ERK1/2 kinase [29]. In order to investigate the plausible role of the MAPK (RAS-RAF-MEK-ERK1/2) pathway located downstream of the MET receptor in eIF4G1 phosphorylation and subsequent HIF-1α translation, we made use of GTL-16 cells that ectopically express the G12D K-RAS mutation (GTL-16 G12D K-RAS cells). GTL-16 G12D K-RAS cells were incubated for 24 h in hypoxia prior to treatment with 100 nM tepotinib for another 24 h. Our data indicate that constitutive K-RAS activation caused by the K-RAS G12D activating mutation enables ERK1/2 to keep phosphorylating eIF4G1 despite METi (Fig. 4a). As a consequence, the effect of tepotinib in these cells is bypassed and HIF-1α translation is reconstituted, thus proving the role of RAS-MAPK signaling in METi-related HIF-1α decrease. Importantly, stable HIF-1α levels could be observed upon METi also in GTL-16 cells expressing H-RAS G12V mutation that analogously constitutively activates the MAPK pathway (Supplementary Figure 6).

Constitutive activation of MAPK pathway bypasses the effect of MET inhibition. a GTL-16 cells stably expressing the G12D K-RAS activating mutation were treated for 24 h with 100 nM tepotinib under hypoxic conditions (1.5% O2). Cell lysates were subjected to Western blot analysis showing stable phosphorylation of eIF4G1 (peIF4G1) and ERK (pERK) as well as expression of HIF-1α despite METi. b Organotypic tissue slices generated from GTL-16 pBabe and GTL-16 G12D K-RAS xenografts, respectively, were exposed to the same conditions as in (a). Representative images (100× magnification) of phospho-MET (pMET), HIF-1α, phospho-eIF4G1 (peIF4G1), GLUT-1 and phospho-ERK (pERK) staining. (c) Representative images (10× magnification; left panel) and quantification (right panel; (**P = 0.0034) of viable (calcium AM-positive (green)) and dead (ethidium homodimer-positive (red)) GTL-16 parental and K-RAS-mutated cells after having been treated for 72 h with 100 nM tepotinib under hypoxic conditions (1.5% O2)
To investigate if the constitutively active RAS-MAPK signaling has the capacity to revert METi-induced HIF-1α decrease in tissues, the 3D ex vivo model employing GTL-16 cells ectopically expressing the empty vector (GTL-16 pBabe cells) or the construct encoding the G12D K-RAS mutation was used. Although expression levels of HIF-1α and GLUT-1 as well as phosphorylation of eIF4G1 expectedly decreased following tepotinib treatment (final concentration of 100 nM) of organotypic tissue slices resulting from GTL-16 pBabe tumors, HIF-1α, GLUT-1 and phosphorylation of eIF4G1 remained stable after tepotinib treatment of GTL-16 K-RAS G12D or H-RAS G12V tissue slices (Fig. 4b, Supplementary Figure 7). Importantly, inhibition of the MAPK pathway by AZD6244 in K-RAS G12D or H-RAS G12V tumors resulted in decrease in eIF4G1 Ser-1232 phosphorylation as well as in HIF-1α and CAIX protein levels (Supplementary Figure 7).
To study the impact of decreased phosphorylation of eIF4G1 and subsequent HIF-1α protein decrease on cell fitness under hypoxia (1.5% O2), we assessed cell viability 72 h after treatment with 100 nM tepotinib in GTL-16 parental as well as in GTL-16 G12D K-RAS cells. Consistently with the reported mechanistic considerations, tepotinib treatment in GTL-16 cells significantly decreased cell survival under hypoxia down to 25,3% as compared to the untreated counterparts (Fig. 4c). At the same time, survival rate of GTL-16 cells overexpressing the G12D mutant K-RAS variant following tepotinib administration was significantly higher (70,2% of the untreated cells) as compared to the parental cells (Fig. 4c). Similarly, the impact of METi on the decrease in cell migration in GTL-16 and EBC-1 cells seems to be pronounced under hypoxic conditions as compared to normoxia (Supplementary Figure 8), another observation possibly connected to MET signaling-related regulation of HIF-1α function.
Co-expression of MET, HIF-1α and eIF4G1 in patients’ samples
Based on our in vitro and ex vivo data, we sought to assess the potential translational relevance of co-alterations in expression levels of MET, HIF-1α, and eIF4G1 in patients with four representative types of solid tumors (breast, esophageal, head and neck and lung) known to potentially display deregulated MET signaling [11, 31]. By extracting mutation, copy number variation and mRNA overexpression data (summarized as ‘overexpression’) from the TCGA database, we found that overexpression of HIF-1α tended to significantly co-occur with overexpression of MET and/or eIF4G1 especially in invasive breast cancer and lung adenocarcinoma specimens (Table 1). In addition, co-overexpression of all the three genes (MET, HIF-1α, and eIF4G1) in patients with lung adenocarcinoma was significantly associated with lower disease-free survival (Fig. 5a).

The MET-HIF-1α-eIF4G1 axis in clinical samples. a Correlation between survival and MET, HIF-1α and eIF4G1 mutation, copy number variation and mRNA overexpression in a TCGA cohort of 230 lung adenocarcinoma patients. P-value was calculated by log-rank test, *P = 0.048. b Co-occurrence between expression/phosphorylation of MET and eIF4G1 as well as expression of HIF-1α in a NSCLC cohort of 566 patients. c Schematic representation (upper panel) and representative images (100× magnification) (lower panel) of MET/phospho-MET, eIF4G1/phospho-eIF4G1, HIF-1α, and Ki-67 co-occurrence in four different tumor regions of three representative patients selected from the NSCLC cohort
In order to gain further translational insights into the potential relevance of the MET—HIF-1α proposed network in patients with lung cancer, we assessed MET/phospho-MET, eIF4G1/phospho-eIF4G1 and HIF-1α protein levels in a TMA containing a cohort of NSCLC tissues from 566 patients treated at the University Hospital Basel and University Hospital Inselspital Bern (Bern/Basel NSCLC cohort). Interestingly, in 55% of the cases when MET is phosphorylated (n = 9 (5 adenocarcinomas and 4 squamous cell carcinomas (SCCs))), also phosphorylation of eIF4G1 occurs (75% of adenocarcinoma and 50% of SCC cases) and in 20% of these cases HIF-1α is expressed at the same time (Fig. 5b). In addition, we could observe that MET-positive tumors (n = 38 (27 adenocarcinomas, six SCCs, three large-cell carcinomas (LCCs), one pleomorphic carcinoma and one undefined)) tend to express eIF4G1 — in 60% of MET-expressing tissues, co-expression of MET with either phosphorylated or total eIF4G1 protein occurs (70% adenocarcinomas, 13% SCCs, 13% LCCs and 4% undefined) and in 10% of these cases, HIF-1α can be detected as well (Fig. 5b).
As solid tumors often present with both hypoxic and non-hypoxic regions [1], we deepened our analysis by assessing MET/phospho-MET, eIF4G1/phospho-eIF4G1, HIF-1α and Ki-67 protein levels in a TMA containing biopsies from four different tumor regions of 75 patients of the Inselspital Bern selected from the Bern/Basel NSCLC cohort. In Fig. 5c three different types of tumors included in this cohort are shown: patient 1 tumor is an example of a relatively homogenous expression of MET/phospho-MET, eIF4G1/phospho-eIF4G1 and HIF-1α, while the other two (patient 2 and mostly patient 3) are in this respect examples of differently heterogenic tumors. Interestingly, MET/phospho-MET, eIF4G1/phospho-eIF4G1 and HIF-1α tend to be co-expressed in the same tumor regions (Fig. 5c). In patient 3 tumor samples, Ki-67 expression very well correlates with the presence of the MET protein, underlining the importance of MET expression in a proliferative aggressive tumor phenotype. Taken together, our results suggest that hypoxic tumors or tumors with hypoxic areas that co-express MET and eIF4G1 and are found in patients have the potential to respond to prospective MET receptor-targeted therapies utilizing small molecule inhibitors or other anti-MET approaches.
Discussion
Tumor-associated hypoxia is of high clinical relevance as it is an important determinant of local tumor progression and systemic dissemination and is therefore considered as an adverse prognostic factor associated with decreased disease-free survival for various cancers [32, 33]. Solid tumors develop hypoxic regions as a consequence of poor blood supply due to aberrant neovascularization. Lesions with deprived oxygenation undergo a series of cellular adaptations, of which the major one is activation of the HIF system, which leads to upregulation of several genes encoding for proteins supporting cellular survival in low-oxygen conditions. Under low-oxygen conditions, a considerable amplification of signaling by various RTKs has been observed as well [33]. This phenomenon is mediated through several distinct mechanisms, such as transcriptional activation [10, 34], enhanced mRNA stability [35] or delayed RTK endocytosis [36]. In 2003, Pennacchietti et al. found that HIF-1α activates transcription of the MET gene whose promoter contains two HIF-1-responsive elements [10]. Concomitant to the increased expression, an upregulation of signaling activity of MET has been linked to a deregulated MET-dependent invasive program, a finding that could correlate with an increased metastatic potential of hypoxic tumors [10]. Inversely, the HIF system was also found to be activated by various RTKs signaling through induction of its transcription [37] or translation [38, 39], increase of the protein stability [40, 41] or enhancement of the DNA-binding and transactivation ability [42, 43]. In the case of the MET receptor, Neill et al. could show that abrogation of the HGF/MET signaling axis using decorin, a small leucine-rich proteoglycan, leads to reduced HIF-1α and VEGF-A expression in breast carcinoma cells [22]. Despite these findings, however, the exact mechanisms underlying the specificities of MET signaling in hypoxia are still far from being clear.
In this study, we elucidated the effect of METi on MET-overexpressing tumor cell lines under hypoxic conditions. METi was accomplished by the highly specific ATP-competitive tyrosine kinase inhibitor tepotinib. Following tepotinib treatment, a decrease in protein levels of HIF-1α and its transcriptional target CAIX was observed in a panel of MET-addicted tumor cell lines. Analogously to the in vitro observations, tepotinib treatment decreased the protein levels of HIF-1α and GLUT-1, another classic HIF-1α target gene, in a 3D ex vivo model. As following METi, the HIF-1α mRNA levels seem to be stable or rather increasing, we hypothesized that an alternative molecular mechanism to transcription (e.g., protein stability or translation) is causing the decrease in HIF-1α protein levels. In this respect we could show that HIF-1α protein stability is not affected by tepotinib as documented by a clear decrease of HIF-1α protein levels upon inhibition of either PHD or proteasome, two essential components involved in degradation of HIF-1α. Interestingly, the time-course decrease of HIF-1α protein following treatment with CHX alone and along with tepotinib was the same, indicating that inhibition of translation and MET targeting both lead to the same endpoint. To further strengthen this assumption, the accumulation rate of HIF-1α protein was determined by inhibiting PHD to block HIF-1α protein degradation and thereby to cause HIF-1α protein accumulation. As expected, this treatment led to a rapid accumulation of HIF-1α protein in time. However, the addition of tepotinib led to a considerable reduction in the HIF-1α protein accumulation, adding another proof to the concept that METi has an inhibitory effect on HIF-1α protein synthesis.
We next aimed at dissecting the molecular mechanisms underlying the METi-related HIF-1α impaired translation. Badura et al reported in 2012 that translation of HIF-1α and a subset of other mRNAs involved in cell survival is selectively increased by eIF4G1 activity [27]. Here, we could show by Western blot, mass spectrometry via SRM and in a 3D ex vivo model that METi considerably decreases phosphorylation of eIF4G1 on Ser-1232.
Moreover, we could show that decreased eIF4G1 phosphorylation caused by METi correlates with increased interaction between eIF4G1 and eIF4E. The formation of the translation preinitiation complex requires interaction of eIF4G with eIF4E, eIF4A, and eIF3. eIF4G comprises two binding sites for eIF4A and one single binding site for eIF4E, eIF3, Poly(A)-binding protein, MAPK-signal integrating kinase (MNK1) and RNA, which interact independently of each other [44]. How interaction of eIF4G1 with each of these components is modulated has not yet been described so far. Here we propose for the first time Ser-1232 phosphorylation to play a role in the regulation of eIF4G1-eIF4E interaction. In yeast, increased interaction between eIF4G and eIF4E has been shown to repress eIF4G from mediating uncapped-mRNA translation [30]. Interestingly, HIF-1α mRNA is known to have strong internal ribosome entry site (IRES) activity which corresponds to a cap-independent translation [27]. In response to cellular stress, cap-dependent protein synthesis is down-regulated. But a subset of mRNAs, involved in survival, growth arrest and DNA damage response, maintains translation during cell stress responses. Such mRNAs contain both a cap and an IRES in their 5′ UTR. In IRES-mediated translation, eIF4G binds to secondary structures in the IRES, independently of eIF4E. This is a potential explanation of how the METi-mediated increased interaction between eIF4G1 and eIF4E impairs the translation of HIF-1α. In view of these facts, our current data reveal the mechanistic similarity in uncapped-mRNA translation between yeast and eukaryotic cells as well as underline the critical role that MET signaling possibly plays in hypoxic tumor cells response.
As phosphorylation of eIF4G1 on Ser-1232 was reported to be executed by ERK [29], we assumed that the MAPK pathway activated via MET plays a central role in translation progression of HIF-1α in MET-expressing hypoxic tumor cells. Indeed, using a gastric cancer cell line model in which the MAPK pathway is constitutively active due to the overexpression of mutated K-RAS, we were able to bypass the effect of tepotinib and to reconstitute HIF-1α translation. Taken together, our current data suggest that MET blockade by tepotinib, which inhibits the MAPK pathway activation [45, 46], prevents ERK from phosphorylating eIF4G1 on Ser-1232 whose interaction with other members of the preinitiation complex is modulated. As a consequence, HIF-1α translation is hindered, HIF-1α protein levels decrease and the transcription of all pro-survival HIF-1α target genes is impaired. Consistently with this proposed mechanism (Fig. 6), the survival and proliferation of cells with mutant K-RAS overexpression following tepotinib treatment is higher as compared to the survival and proliferation of parental cells. These observations are of high importance as they are, although rather indirectly, reflecting the relevance of MET signaling in HIF-1α translation for one of the biological features of tumor cells. At the same time it is important to note that as MET itself has been previously reported to be a target gene of HIF-1α [10] and we show that MET in turn increases the abundance of HIF-1α protein, a potential positive feedback loop that amplifies the effects of both HIF and MET signaling might exist in tumor cells.

Schematic representation of the MET-eIF4G1-HIF-1α regulation axis. MET autophosphorylation leads to activation of the Ras-Raf-MEK1/2-ERK1/2 axis and to a subsequent ERK1/2-mediated phosphorylation of eIF4G1 on Ser-1232. This phosphorylation disrupts the association of eIF4G1 with eIF4E and thus possibly stimulates translation of the HIF-1α protein. As a result, transcription of HIF-1α targets occurs under hypoxic conditions
From clinical perspective, TCGA data indicate that alterations in expression levels of MET, HIF-1α and/or eIF4G1 in patients with distinct types of solid tumors (e.g., breast, esophageal, head and neck and lung cancer) are common and tend to co-occur. In the lung adenocarcinoma dataset, patients with tumors with co-overexpression of all of the three components of the newly described molecular axis (e.g., MET, HIF-1α, and eIF4G1) were found to have a significantly lower disease-free survival as compared to patients with tumors with normal expression of any of these three proteins, warranting further exploration of the role of MET targeting in hypoxic tumors.
Analysis of a separate NSCLC patient cohort revealed that tumors with activated MET signaling display at the same time phosphorylation of eIF4G1 and often also overexpression of HIF-1α. This observation underlines the compatibility of the experimental results with clinical cases. When analyzing four different regions of individual tumors belonging to the same cohort, we could observe differential expression of MET, eIF4G1 and HIF-1α within the individual tumors. Importantly, however, in three selected representative homogenous or heterogenous tumors, MET/phospho-MET, eIF4G1/phospho-eIF4G1 and HIF-1α tend to be co-expressed in the same tumor regions and their expression seems to correlate with proliferating tumor areas as inferred from Ki-67 staining positivity.
Taken together, in this study we revealed the existence of a new branch of the intricate crosstalk between hypoxia-related signaling and MET receptor activity. This emerging molecular interplay between MET activity and hypoxic tumor state, each correlated with a bad prognosis and increased patient mortality [9, 11], supports the rationale of employing MET targeting for the treatment of hypoxic solid tumors with MET expression. The finding that targeting MET diminishes HIF-1α translation through impairment of eIF4G1 activity necessarily implies further investigation of MET signaling and translation of particular mRNAs during stress responses in cancer cells. In addition, our study indicates that phosphorylation of eIF4G1 at Ser-1232 modulates the interaction between eIF4G1 and eIF4E. The potential role of this phosphorylation in modulating the interaction with other components of the eIF4F complex encourages for further investigation.
Material and methods
Cell lines and culture conditions
The human gastric carcinoma cell lines GTL-16, SNU-638, KATOII and Hs746T were obtained from Dr. Paolo Comoglio (Medical School University of Torino, Italy), Korean Cell Line Bank, Dr. Morag Park (Cancer Research Centre, McGill University, Montreal, Canada) and Dr. Silvia Giordano (University of Torino, Torino, Italy), respectively. The human lung carcinoma cell lines EBC-1 and H1993 were kindly provided by Dr. Silvia Giordano and Dr. Sunny Zachariah (UT Southwestern Medical Center, Dallas, TX), respectively. All the cells were cultured in RPMI (GIBCO, Invitrogen) with 5–10% fetal calf serum (Sigma) and antibiotic-antimycotic (GIBCO). GTL-16 cell lines that ectopically express the G12D and G12V mutations of K-RAS were maintained as described in [47]. Normal oxygen culturing conditions were 21% O2, 5% CO2 at 37 °C in a humidified incubator, hypoxia 1.5% O2 and 5% CO2 humidified environment (microaerophilic system (Ruskinn, Biotrace International, UK)). Generally, the cells were maintained in culture and used for the experiments for up to 1 month.
Inhibitors
Tepotinib (EMD1214063) (Merck KGaA, Darmstadt, Germany) and MG-132 (Sigma) were dissolved in DMSO. Dimethyloxaloylglycine (DMOG) (Sigma) and cycloheximide (CHX) (Sigma) were dissolved in sterile water or in PBS, respectively. Working concentrations were prepared in the corresponding medium.
RNA isolation and real-time PCR
Total RNA extraction was performed using Trizol reagent (Roche) and mRNA reverse transcription using the Omniscript RT Kit (Qiagen). Quantitative PCR of HIF-1α, CAIX and GLUT-1 (a 7900HT fast Real-time PCR system (Applied Biosystems)) was assessed by a TaqMan assay (Applied Biosystems). The mean CT resulted from triplicates, mRNA levels were normalized to hydroxymethylbilane synthase (Microsynth, Switzerland), expression changes were determined by ΔΔCT calculations.
Protein extraction and western blot analysis
Cell pellets were lysed in urea buffer (HEPES, urea, sodium orthovanadate, sodium pyrophosphate, β-glycerol-phosphate) followed by sonication. Tissues were homogenized prior to the urea lysis. In all experiments, 50 µg of total proteins (BioRad protein quantification reagent (Bio-Rad Laboratories, Inc.)) were resolved by SDS-PAGE and transferred to PVDF membranes. Primary antibodies were directed against phospho-MET (Tyr-1234/1235) (polyclonal, cell signaling technologies (CST)), HIF-1α (clone 54, BD Technologies), CAIX (polyclonal, Novus Biologicals), eIF4G1 (polyclonal, CST), phospho-eIF4G1 (Ser-1232) (polyclonal, Novus Biologicals), phospho-ERK (polyclonal, Millipore) and β Actin (clone C4, Millipore). Detection of horseradish peroxidase-conjugated secondary antibodies was accomplished using an ECL kit (AmershamPharmacia Biotech, Little Chalfont, UK).
Co-immunoprecipitation
To detect eIF4G1-eIF4E interaction, an antibody against eIF4G1 (polyclonal, CST) was incubated with Dynabeads Protein G (ThermoFisher Scientific). The antibody-coated beads were incubated with total protein lysates. Bound material was collected using a DynaMag magnet. Immunoprecipitated complexes were analyzed by SDS-PAGE and probed by the anti-eIF4E specific antibody (polyclonal, Bethyl).
Cell viability assay
Living and dead cells were discriminated using the Live/Dead assay kit (molecular probes) as previously described [46].
Metabolites extraction
Cells were seeded in 6-well plates in quadruplicates. Upon treatments, they were washed (75 mM ammonium carbonate (Sigma), pH 7.4), quenched by liquid nitrogen and stored at −80 °C. Extraction of metabolites was performed twice on ice by 400 μL extraction solution (2:2:1 acetonitrile:methanol:water; −20 °C) for 10 min. Extracts were collected and centrifuged (4 °C, 13,000 rpm, 2 min). Supernatant was stored at −20 °C until measurements.
Metabolomics measurements
Samples for metabolomics were analyzed by flow injection—time-of-flight mass spectrometry analysis on an Agilent 6550 Q-TOF instrument in negative ionization mode as previously described [48]. Ions were putatively annotated based on accurate masses using a tolerance of 0.001 Da and the KEGG hsa database. Matches of the ion annotation are showed in Supplementary Table 1. Significantly altered metabolites were found by an unpaired, heteroscedastic t-test. P-values were corrected for multiple hypothesis testing using the Benjamini-Hochberg procedure [49].
3D ex vivo studies
EBC-1, GTL-16 parental cells and GTL-16 cells ectopically expressing the G12D mutation of K-RAS as well as the empty vector, respectively, were injected subcutaneously into the flank of 5 weeks and 21 weeks old RAG2-/-; γc-/- mice (1,5 × 106 cells/animal). One month after injection, developed tumors were harvested and cut into 350 µm thin slices. The resulting organotypic tissue slices were further cultured in normal cell culture medium under hypoxic or normoxic conditions as indicated. Animal experimentations were approved by the veterinary committee of Canton of Bern (Switzerland).
Tumor tissue microarrays (TMAs)
The cores of formalin-fixed paraffin-embedded organotypic tissue slices were punched using biopsy punch needles (EZ-TMA Tissue Microarray Kits, IHC world). A 1mm-diameter cylinder of tissue was removed and re-embedded into a recipient paraffin block. Two separate cores were selected per sample. Sections cut from the TMA block were mounted on microscope slides. A TMA block encompassing lung cancer samples of 566 patients diagnosed in the Basel University Hospital/Inselspital Bern (TMA1) was obtained from the Basel University Hospital, Basel, Switzerland. A heterogeneity TMA (TMA2) block constructed using 450 lung cancer and normal tissue samples originating from 75 selected Inselspital Bern patients included in TMA1 (2 normal and 4 tumor tissue samples (retrieved from distinct tumor regions) per patient included) was obtained from the Institute of Pathology, University of Bern, Bern, Switzerland.
Immunohistochemistry
Organotypic tissue slices were fixed in 10% formalin overnight at 4 °C. Phospho-MET, HIF-1α, CAIX, GLUT-1, and phospho-ERK staining was performed by the Translational Research Unit (TRU) platform of the University of Bern (Bern, Switzerland). To stain for the other epitopes, sections were deparaffinized and rehydrated and antigen retrieval was performed in Tris-EDTA buffer (pH 9). Following blocking of endogenous peroxidase and incubating the sections in goat serum, primary antibodies for MET (clone D1C2, CST), phospho-eIF4G1 (polyclonal, Novus Biologicals) and total eIF4G1 (polyclonal, CST) were applied and detected with the Vectastain ABC Kit (Vector Laboratories) and 3,3′-Diaminobenzidine (DAB) (Sigma-Aldrich). Slides were counterstained with haematoxylin. Images were obtained with a Pannoramic 250 Flash III Scanner (3D Histech) at 100× magnification. The staining results originating from the lung tumor cohort (TMA1) were blindly scored by three independent investigators, without any prior knowledge of relevant clinical data. The tumors were categorized as either positive or negative in expression of the indicated proteins (cytoplasmic and membrane localization of MET/phospho-MET, cytoplasmic localization of eIF4G1/phospho-eIF4G1 and nuclear localization of HIF-1α were considered (HIF-1α scores of these TMAs were reported previously [50])).
Selected reaction monitoring (SRM)
The cells were washed, scraped and spun down for 5 min at 1000 rpm, and resulting pellets stored at −80 °C. Prior measurements, the cells were resuspended in 8 M urea solution containing 0.1 M ammonium bicarbonate and pellets were disrupted by sonicating for 10 min. Supernatants were centrifuged (12,000 rpm for 10 min) and protein concentration was determined (BCA Protein Assay (Pierce)). Disulfide bonds were reduced with 5 mM tris(2-carboxyethyl)phosphine at 37 °C for 30 min and free thiols were alkylated with 10 mM iodoacetamide at room temperature for 30 min in the dark. The solution was subsequently diluted with 0.1 M ammonium bicarbonate to a final concentration of 1.5 M urea and digested overnight at 37 °C with sequencing-grade modified trypsin (Promega) at a protein-to-enzyme ratio of 50:1. The digestion was stopped by acidification with formic acid to a final pH < 3. Peptides were desalted on a C18 Sep-Pak cartridge (Waters) and dried under vacuum. Phosphopeptides were isolated from 1 mg of total peptide mass with TiO2. A mix containing heavy-labeled synthetic peptides was used to resuspend samples prior to MS analysis. Heavy and light transitions were targeted by scheduled SRM to monitor the co-elution of endogenous (light) peptides and the spiked-in (heavy) surrogates. Development and validation of SRM assays was performed using heavy-labeled, unpurified synthetic versions of the target peptides. To extract their SRM coordinates, we first performed shotgun analysis on mixtures of the synthetic peptides. For each target peptide, the ten most intense SRM transitions were selected using Skyline. For assay refinement, the five most intense transitions for each peptide were selected and the peptide retention time for scheduled SRM acquisition was annotated. Samples were measured on a triple quadrupole/ion trap mass spectrometer (5500 QTrap, ABSciex). Chromatographic separation of peptides was achieved with an Eksigent Nano LC system (Eksigent Technologies) connected to a 15-cm fused-silica emitter with 75-µm inner diameter (BGB Analytik) packed in-house with a Magic C18 AQ 3-µm resin (Michrom BioResources). The peptide mixtures were analyzed by LC-MS/MS and separated with a linear gradient of acetonitrile/water from 2 to 40% acetonitrile in 55 min at a flow rate of 300 nl/min. SRM peaks were manually inspected with Skyline. Relative quantification of the target peptides was obtained by calculating the ratio between the SRM peaks of the endogenous and reference peptides.
The Cancer Genome Atlas (TCGA) data analysis
cBioPortal (http://www.cbioportal.org/public-portal/cross_cancer.do) was used to obtain data on expression levels of MET, HIF-1α, and eIF4G1, and the eventual correlation between co-alterations in expression (overexpression) and clinical outcome. Overexpression for a given sample was defined as having a z-score superior to +1.5.
Statistics
Data for each treatment group were represented as means ± SD as indicated and compared to evaluate significance using the Student t-test (GraphPad Prism (version 5.03)). P-values < 0.05 were considered statistically significant. (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). Survival data from TCGA was compared with the log-rank test (significance set at P < 0.05).
References
Brahimi-Horn MC, Chiche J, Pouyssegur J. Hypoxia and cancer. J Mol Med. 2007;85:1301–7.
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science . 2005;307:58–62.
Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47.
Michieli P. Hypoxia, angiogenesis and cancer therapy: to breathe or not to breathe? Cell Cycle. 2009;8:3291–6.
Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell . 2001;107:1–3.
Clara CA, Marie SK, de Almeida JR, Wakamatsu A, Oba-Shinjo SM, Uno M, et al. Angiogenesis and expression of PDGF-C, VEGF, CD105 and HIF-1alpha in human glioblastoma. Neuropathology. 2014;34:343–52.
Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6:637–45.
Ide T, Kitajima Y, Miyoshi A, Ohtsuka T, Mitsuno M, Ohtaka K, et al. Tumor-stromal cell interaction under hypoxia increases the invasiveness of pancreatic cancer cells through the hepatocyte growth factor/c-Met pathway. Int J Cancer J Int du Cancer. 2006;119:2750–9.
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32.
Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–61.
Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005;225:1–26.
Liu X, Yao W, Newton RC, Scherle PA. Targeting the c-MET signaling pathway for cancer therapy. Expert Opin Investig Drugs. 2008;17:997–1011.
Zeng ZS, Weiser MR, Kuntz E, Chen CT, Khan SA, Forslund A, et al. c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett. 2008;265:258–69.
Okuda K, Sasaki H, Yukiue H, Yano M, Fujii Y. Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008;99:2280–5.
Seiwert TY, Jagadeeswaran R, Faoro L, Janamanchi V, Nallasura V, El Dinali M, et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009;69:3021–31.
Cosse JP, Michiels C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anti-Cancer Agents Med Chem. 2008;8:790–7.
Smolen GA, Sordella R, Muir B, Mohapatra G, Barmettler A, Archibald H, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci Usa. 2006;103:2316–21.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science . 2007;316:1039–43.
Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–16.
Vordermark D, Horsman MR. Hypoxia as a biomarker and for personalized radiation oncology: recent results. Cancer Res. 2016;198:123–42.
Boeckx C, Van den Bossche J, De Pauw I, Peeters M, Lardon F, Baay M, et al. The hypoxic tumor microenvironment and drug resistance against EGFR inhibitors: preclinical study in cetuximab-sensitive head and neck squamous cell carcinoma cell lines. BMC Res Notes. 2015;8:203.
Neill T, Painter H, Buraschi S, Owens RT, Lisanti MP, Schaefer L, et al. Decorin antagonizes the angiogenic network: concurrent inhibition of Met, hypoxia inducible factor 1alpha, vascular endothelial growth factor A, and induction of thrombospondin-1 and TIMP3. J Biol Chem. 2012;287:5492–506.
Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001;276:9519–25.
Minchenko OH, Tsuchihara K, Minchenko DO, Bikfalvi A, Esumi H. Mechanisms of regulation of PFKFB expression in pancreatic and gastric cancer cells. World J Gastroenterol. 2014;20:13705–17.
Minchenko O, Opentanova I, Minchenko D, Ogura T, Esumi H. Hypoxia induces transcription of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase-4 gene via hypoxia-inducible factor-1alpha activation. FEBS Lett. 2004;576:14–20.
Obach M, Navarro-Sabate A, Caro J, Kong X, Duran J, Gomez M, et al. 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J Biol Chem. 2004;279:53562–70.
Badura M, Braunstein S, Zavadil J, Schneider RJ. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc Natl Acad Sci Usa. 2012;109:18767–72.
Pelletier J, Graff J, Ruggero D, Sonenberg N. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 2015;75:250–63.
Dobrikov MI, Dobrikova EY, Gromeier M. Dynamic regulation of the translation initiation helicase complex by mitogenic signal transduction to eukaryotic translation initiation factor 4G. Mol Cell Biol. 2013;33:937–46.
Tarun SZ Jr., Sachs AB. Binding of eukaryotic translation initiation factor 4E (eIF4E) to eIF4G represses translation of uncapped mRNA. Mol Cell Biol. 1997;17:6876–86.
Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GFMet. metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25.
Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–13.
Gluck AA, Aebersold DM, Zimmer Y, Medova M. Interplay between receptor tyrosine kinases and hypoxia signaling in cancer. Int J Biochem Cell Biol. 2015;62:101–14.
Thangasamy A, Rogge J, Ammanamanchi S. Recepteur d’origine nantais tyrosine kinase is a direct target of hypoxia-inducible factor-1alpha-mediated invasion of breast carcinoma cells. J Biol Chem. 2009;284:14001–10.
Ikeda E, Achen MG, Breier G, Risau W. Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J Biol Chem. 1995;270:19761–6.
Paatero I, Seagroves TN, Vaparanta K, Han W, Jones FE, Johnson RS, et al. Hypoxia-inducible factor-1alpha induces ErbB4 signaling in the differentiating mammary gland. J Biol Chem. 2014;289:22459–69.
Cheng JC, Klausen C, Leung PC. Hypoxia-inducible factor 1 alpha mediates epidermal growth factor-induced down-regulation of E-cadherin expression and cell invasion in human ovarian cancer cells. Cancer Lett. 2013;329:197–206.
Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004.
Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A. EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res. 2006;66:3197–204.
Paatero I, Jokilammi A, Heikkinen PT, Iljin K, Kallioniemi OP, Jones FE, et al. Interaction with ErbB4 promotes hypoxia-inducible factor-1alpha signaling. J Biol Chem. 2012;287:9659–71.
Skuli N, Monferran S, Delmas C, Lajoie-Mazenc I, Favre G, Toulas C, et al. Activation of RhoB by hypoxia controls hypoxia-inducible factor-1alpha stabilization through glycogen synthase kinase-3 in U87 glioblastoma cells. Cancer Res. 2006;66:482–9.
Takacova M, Bullova P, Simko V, Skvarkova L, Poturnajova M, Feketeova L, et al. Expression pattern of carbonic anhydrase IX in Medullary thyroid carcinoma supports a role for RET-mediated activation of the HIF pathway. Am J Pathol. 2014;184:953–65.
Gariboldi MB, Ravizza R, Monti E. The IGFR1 inhibitor NVP-AEW541 disrupts a pro-survival and pro-angiogenic IGF-STAT3-HIF1 pathway in human glioblastoma cells. Biochem Pharmacol. 2010;80:455–62.
Villa N, Do A, Hershey JW, Fraser CS. Human eukaryotic initiation factor 4G (eIF4G) protein binds to eIF3c, -d, and -e to promote mRNA recruitment to the ribosome. J Biol Chem. 2013;288:32932–40.
Medova M, Pochon B, Streit B, Blank-Liss W, Francica P, Stroka D, et al. The novel ATP-competitive inhibitor of the MET hepatocyte growth factor receptor EMD1214063 displays inhibitory activity against selected MET-mutated variants. Mol Cancer Ther. 2013;12:2415–24.
Francica P, Nisa L, Aebersold DM, Langer R, Bladt F, Blaukat A, et al. Depletion of FOXM1 via MET Targeting Underlies Establishment of a DNA Damage-Induced Senescence Program in Gastric Cancer. Clin Cancer Res. 2016;22:5322–36.
Leiser D, Medova M, Mikami K, Nisa L, Stroka D, Blaukat A, et al. KRAS and HRAS mutations confer resistance to MET targeting in preclinical models of MET-expressing tumor cells. Mol Oncol. 2015;9:1434–46.
Fuhrer T, Heer D, Begemann B, Zamboni N. High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Anal Chem. 2011;83:7074–80.
Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B. 1995;57:289–300.
Berezowska S, Galvan JA, Langer R, Bubendorf L, Savic S, Gugger M, et al. Glycine decarboxylase and HIF-1alpha expression are negative prognostic factors in primary resected early-stage non-small cell lung cancer. Virchows Arch. 2017;470:323–30.
Acknowledgements
We cordially thank Dr. Nicola Zamboni (ETH Zürich, Switzerland) for his help with metabolomics measurements and data analysis.
Funding
This work has been supported by Stiftung für klinisch-experimentelle Tumorforschung (grant to YZ) and by Stiftung zur Krebsbekämpfung (grant Nr.374/2016 to MM). YZ received a drug donation from Merck / EMD Serono. Merck / EMD Serono has reviewed the publication, and the views and opinions described in the publication do not necessarily reflect those of Merck.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
AB is listed as a co-inventor on all patents related to Merck’s c-Met inhibitor referred to in this manuscript. The remaining authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Glück, A.A., Orlando, E., Leiser, D. et al. Identification of a MET-eIF4G1 translational regulation axis that controls HIF-1α levels under hypoxia. Oncogene 37, 4181–4196 (2018). https://doi.org/10.1038/s41388-018-0256-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0256-6
- Springer Nature Limited
This article is cited by
-
An oncogene addiction phosphorylation signature and its derived scores inform tumor responsiveness to targeted therapies
Cellular and Molecular Life Sciences (2023)
-
MicroRNA-495: a therapeutic and diagnostic tumor marker
Journal of Molecular Histology (2023)
-
Correction: An oncogene addiction phosphorylation signature and its derived scores inform tumor responsiveness to targeted therapies
Cellular and Molecular Life Sciences (2023)
-
CircTMTC1 contributes to nasopharyngeal carcinoma progression through targeting miR-495-MET-eIF4G1 translational regulation axis
Cell Death & Disease (2022)
-
Processing body (P-body) and its mediators in cancer
Molecular and Cellular Biochemistry (2022)