
Abstract
Parvalbumin-expressing fast-spiking interneurons (PV-INs) within feedforward microcircuits in the nucleus accumbens (NAc) coordinate goal-directed motivational behavior. Feedforward inhibition of medium spiny projection neurons (MSNs) is initiated by glutamatergic input from corticolimbic brain structures. While corticolimbic synapses onto MSNs are targeted by the psychostimulant, cocaine, it remains unknown whether cocaine also exerts acute neuromodulatory actions at collateralizing synapses onto PV-INs. Using whole-cell patch-clamp electrophysiology, optogenetics, and pharmacological tools in transgenic reporter mice, we found that cocaine decreases thalamocortical glutamatergic drive onto PV-INs by engaging a monoamine-independent mechanism. This mechanism relies on postsynaptic sigma-1 (σ1) activity, leading to the mobilization of intracellular Ca2+ stores that trigger retrograde endocannabinoid signaling at presynaptic type-1 cannabinoid receptors (CB1R). Cocaine-evoked CB1R activity occludes the expression of CB1R-dependent long-term depression (LTD) at this synaptic locus. These findings provide evidence that acute cocaine exposure targets feedforward microcircuits in the NAc and extend existing models of cocaine action on mesolimbic reward circuits.
Similar content being viewed by others


Introduction
Integrated networks of parvalbumin-expressing fast-spiking interneurons (PV-INs) in the nucleus accumbens (NAc) critically contribute to adaptive motivational behavior [1,2,3]. The coordinated activity of PV-INs within feedforward microcircuits gates functional NAc output through D1 and D2 dopamine (DA) receptor-expressing medium spiny projection neurons (MSNs) [4, 5]. PV-IN-directed feedforward inhibition of D1 and D2 MSNs is initiated by concerted glutamatergic input from multiple salience-encoding structures, including the prefrontal cortex (PFC) and mediodorsal thalamus (MDT) [3, 6]. Thalamocortical glutamatergic synapses onto MSNs are an established synaptic substrate for psychostimulant experience, resulting in maladaptive shifts in NAc circuit dynamics [7,8,9,10,11]. While PV-IN output is required for psychostimulant-induced behaviors [12], it remains unknown if psychostimulants, such as cocaine, also exert neuromodulatory actions at collateralizing corticolimbic synapses onto PV-INs within feedforward microcircuits.
Cocaine is a recreationally-abused psychostimulant that enacts cellular and synaptic adaptations throughout the reward network [13,14,15,16]. The canonical molecular mechanism of cocaine action in the NAc is monoamine reuptake inhibition, leading to elevated concentrations of DA, serotonin (5-HT) and norepinephrine (NE) [17,18,19]. Cocaine-induced monoamine signaling contributes to synaptic rearrangements facilitating D1-MSN output to midbrain DA nuclei [7, 8]. However, multiple reports suggest that cocaine engages monoamine-independent signaling through sigma-1 (σ1), an endoplasmic reticulum (ER) chaperone protein regulating intracellular Ca2+ dynamics, membrane excitability, and receptor multimerization [20,21,22,23]. Accordingly, cocaine-induced behavioral responding is blunted following intra-NAc σ1 knockdown and recapitulated by σ1 ligands without altering mesoaccumbens DA signaling [22, 24]. While mechanisms underlying the effects of cocaine have been identified in MSNs, a detailed understanding of how cocaine modulates propagation through NAc feedforward networks is lacking.
In the present study, we examine the acute neuromodulatory actions of cocaine at feedforward glutamatergic synapses onto NAc PV-INs. Using transgenic reporter mice, whole-cell patch-clamp electrophysiology, optogenetics, and ex vivo pharmacology, we found that cocaine decreases thalamocortical glutamatergic drive onto PV-INs through a monoamine- and cholinergic-independent mechanism. Following a detailed pharmacological analysis, our data suggests that cocaine engages postsynaptic σ1 signaling to mobilize intracellular Ca2+ stores. Cocaine-evoked Ca2+ signaling leads to the production of endocannabinoids (eCBs), resulting in retrograde signaling at presynaptic cannabinoid type-1 receptors (CB1R). These findings extend an evolving framework in which cocaine engages complementary effectors in the NAc.
Materials and methods
Animal use
Mice were housed in the Vanderbilt University Medical Center animal care facility in accordance with Institutional Animal Care and Use Committee guidelines. Male mice (6–12 weeks) were bred and housed (3–5/cage, 12-h light-dark cycle) with ad lib access to standard food and water. Cre-inducible STOPfl/fl-tdTomato mice (Ai9, Gt(ROSA)26Sortm9(CAG-tdTomato)Hze) were obtained from The Jackson Laboratory (Stock No.: 007909) and crossed with PV-IRES-Cre mice (PVCre, Pvalbtm1(cre)Arbr/J, Stock No.: 008069), generating PVCre-tdTomatofl-STOP-fl (PVtdT) mice.
Electrophysiology
Whole-cell patch-clamp electrophysiological recordings are described in detail [25, 26]. In brief, recordings were obtained in acute brain slice preparations from PVtdT mice, as described previously [4, 25, 26]. Briefly, mice were euthanized under isoflurane anesthesia and decapitated, after which parasagittal slices (250 μm) containing the NAc core and shell were prepared from whole brain tissue using a Leica Vibratome in oxygenated (95% O2; 5% CO2) ice-cold N-methyl-D-glucamine (NMDG)-based solution (in mM: 2.5 KCl, 20 HEPES, 1.2 NaH2PO4, 25 Glucose, 93 NMDG, 30 NaHCO3, 5.0 sodium ascorbate, 3.0 sodium pyruvate, 10 MgCl2, and 0.5 CaCl2-2H2O). Slices were then recovered in NMDG-based recovery solution for 10–15 min at 31–32 °C before being transferred to a chamber containing artificial cerebral spinal fluid (ACSF, in mM: 119 NaCl, 2.5 KCl, 1.3 MgCl2-6H2O, 2.5 CaCl2-2H2O, 1.0 NaH2PO4-H2O, 26.2 NaHCO3, and 11 glucose; 287–295 mOsm). All electrophysiology experiments were performed using a Scientifica Slicescope Pro System with continuously perfused 28–32 °C ACSF at 2 mL/min. PV-INs in the NAc were visualized using Scientifica PatchVision software and patched with 3–6 MΩ recording pipettes (P1000 Micropipette Puller) filled with K+-based intracellular solution: (in mM: 135 K +-gluconate, 5 NaCl, 2 MgCl2, 10 HEPES, 0.6 EGTA, 3 Na2ATP, 0.4 Na2GTP; 290 mOsm). Experiments examining synaptically-evoked plasticity were performed in a Cs+-based internal solution to minimize space-clamp error (in mM: 120 CsMeSO3, 15 CsCl, 8 NaCl, 10 HEPES, 0.2 EGTA, 10 TEA-Cl, 4.0 Mg-ATP, 0.3 Na-GTP, 0.1 spermine, and 5.0QX 314 bromide). PV-INs were differentiated from MSNs and other neuronal subtypes in the NAc and recorded in voltage-clamp and current-clamp configurations as described previously in [25, 27].
Fast-scan cyclic voltammetry
Mice were sacrificed under isoflurane anesthesia. Using a Leica Vibratome, 250μm-thick sagittal sections containing the NAc core were collected from whole brain tissue in oxygenated (95% O2; 5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mM): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 2.4 CaCl2, 1.2 MgCl2, 25 NaHCO3, 11 glucose, 0.4 L-ascorbic acid, and pH adjusted to 7.4. Slices were transferred to a chamber containing oxygenated ACSF. All experiments were performed using a Scientifica SliceScope Pro System in 32 °C ACSF with a flow rate of 2 ml/min. The carbon fiber microelectrode (100 − 200 μM length, 7 μM radius) and bipolar stimulating electrode were placed in close proximity in the NAc core. A single electrical pulse (750 μA, 4 ms, monophasic) was applied to the tissue every 5 min to evoke dopamine release. Extracellular dopamine was recorded by applying a triangular waveform (−0.4 to +1.2 to −0.4 V vs Ag/AgCl, 400 V/s). Peak evoked dopamine release was collected in ACSF until a stable baseline was established (3 collections <10% variability). We then bath applied 1 μM reserpine and collected peak evoked dopamine release until stable responding was achieved (3 collections <10% variability).
Stereotaxic surgery
6–8-week-old male PVtdT mice were anesthetized using ketamine (75 mg/kg I.P.) and dexdomitor (0.5 mg/kg I.P.). Craniotomies were performed using a drill, AmScope microscope, and World Precision Instruments Aladdin Al-2000 syringe pump hydraulic system. The following coordinates were used based on The Mouse Brain in Stereotaxic Coordinates: PFC (AP 1.4, ML ± 0.5, DV −2.9 mm) and MDT (AP −1.2, ML 0.3, DV −3.00 mm). Injection sites were located using Leica AngleTwo Stereotaxic software. AAV-CaMKII-ChR2-eYFP (Addgene) was injected at 100 nL/min. Mice were revived using antisedan (atipamezole, 0.5 mg/kg I.P.) and treated with ketoprofen (5 mg/kg I.P.) for 3 days post-operatively. ChR2 expression and anatomical specificity were validated empirically according to the expression of eYFP in the medial PFC or paraventricular region of the MDT and the presence of high-fidelity AMPAR-mediated oEPSCs with rapid decay kinetics in NAc PV(+)-INs.
Pharmacology
Cocaine HCl, nifedipine, GDPβS trilithium salt, and citalopram were purchased from Sigma-Aldrich, whereas all other drugs were purchased from Tocris. DO34 and reserpine were generously provided by Sachin Patel (Vanderbilt) and Danny Winder (Vanderbilt), respectively.
Statistics and data analysis
Experiments were analyzed using Clampfit 10.4, Demon voltammetry and analysis software and GraphPad Prism v7.0. Changes in baseline EPSC amplitude, coefficient of variance (CV), and PPR were calculated by comparing mean values during 5 min intervals specified in each time-course to baseline PPR and CV values. All representative traces of EPSCs were obtained by averaging 50–60 sweeps with the designated time-point, thereby ensuring an accurate depiction of the data. Paired or unpaired two-tailed Student’s t tests with a subset of experiments requiring 1-way ANOVA with Sidak’s post-hoc analyses. Power analyses were performed with preliminary data during the acquisition of each new data set. Errors bars depicted in figures represent SEM. For all analyses, α was set as 0.05, with P values < α indicating a statistically significant difference.
Results
Ex vivo cocaine uniformly dampens glutamatergic drive onto NAc PV-INs
To interrogate the acute synaptic effects of cocaine at glutamatergic synapses onto PV-INs, we performed whole-cell patch-clamp electrophysiology in brain slices prepared from male PVCre-tdTomatofl-STOP-fl (PVtdTom) mice (Fig. 1a). Acute brain slices provide a reduced biological system that is amenable to efficient electrophysiological and pharmacological investigation of molecular mechanisms of action of diverse compounds, including cocaine. In line with previous work from our group [25, 27], tdTomato-expressing [PV(+)] cells in the NAc exhibited properties of PV-INs that were distinct from PV(−) MSNs, including a fast-spiking, narrow-complex electrophysiological phenotype (Fig. 1b). Following a stable 10 min electrically-evoked EPSC baseline, cocaine (3 or 10 µM) was bath-applied for 10-min, resulting in a significant decrease in EPSC amplitude at both concentrations that returned to baseline following drug washout (Fig. 1c, d; 65.05 ± 2.89%, n = 7, t(6) = 12.11 (3 µM): 76.32 ± 4.14%, t(4) = 5.72 p < 0.001). Cocaine at 10 µM had no discernible use-dependent effect on AP amplitude when assessed via 800 ms 350 pA somatic current injection (IINJ) (Fig. 1e; ACSF: 74.06 ± 1.30 mV, n = 8, 72.80 ± 1.32 mV, n = 8, t(14) = 0.67, p = 0.508). Additionally, phase-plane spike analysis, whereby the first derivative of membrane potential (dV/dt) is plotted against membrane potential during an AP evoked at 350 pA, revealed no significant difference in AP threshold between cocaine and ACSF conditions (Fig. 1f). Thus, the effects of cocaine at this concentration are not due to an anesthetic effect on voltage-gated Na+ channels (VGNCs).

a Schematic of a sagittal mouse brain section containing the NAc and the electrophysiological recording strategy of PV-INs within feedforward inhibitory microcircuits. b Representative trace of fast-spiking APs elicited via 300 pA somatic current injection with a single AP depicting the narrow-complex AP morphology in PV-INs. c Representative traces and experiment depicting the effects of cocaine on EPSC amplitude in PV-INs. Scale bar: 200 pA/50 ms. d Normalized time-course summary and quantification of EPSC amplitude following cocaine application. e Left: Representative traces of sequential APs elicited via 350 pA somatic current injection in ACSF and cocaine. Scale bar: 50 mV/5 ms. Right: Average AP amplitude in ACSF and cocaine. f Aggregate phase-pane plots of APs elicited at 350 pA in slices perfused with cocaine (bottom, n = 5) or ACSF alone (top, n = 5). Vertical dotted line indicates estimated AP threshold (second derivative, d2V/dt2, of VM at 5% maxima). g Rescaled representative traces of 50 ms PPR obtained in ACSF and cocaine. Scale bars: 50 pA/50 ms. h Average PPR at baseline and in the presence of cocaine superimposed over raw experimental values. i Average CV at baseline and in the presence of cocaine superimposed over raw experimental values. j, k Normalized Ih and Rm averaged across all experiments at t(gray). l Representative traces of sEPSCs in ACSF and cocaine (blue). Scale bar: 20 pA/2 sec. m Average sEPSC frequency in ACSF and cocaine averaged across 5 min epochs. n Average sEPSC amplitude in ACSF and cocaine averaged across 5-min epochs. o Schematic depicting stereotaxic PFC- and MDT-targeting strategy with ChR2 (top) and the synapse being surveyed (bottom). p Representative traces and time-course summary depicting effects of cocaine at PFC-to-PV-IN synapses (red) in the NAc. q Representative traces and time-course summary depicting effects of cocaine at MDT-to-PV-IN synapses (blue) in the NAc. r Quantification of average oEPSC amplitude at t(gray) following cocaine application at PFC and MDT inputs onto NAc PV-INs. Error bars indicate SEM. *p < 0.05.
The cocaine-induced depression was accompanied by an increase in the paired pulse ratio (PPR; baseline: 1.37 ± 0.06, n = 9; PPR cocaine: 1.51 ± 0.05, n = 9, t(8) = 2.601, p = 0.0316) and coefficient of variance (CV; baseline: 0.13 ± 0.01, n = 9; CV cocaine: 0.21 ± 0.02, n = 9, t(8) = 4.407, p = 0.0023), signifying a presynaptic effect on glutamate release probability (Fig. 1g–i). In addition, cocaine decreased spontaneous EPSC (sEPSC) frequency (sEPSC freq. baseline: 13.67 ± 1.40 Hz, n = 15; sEPSC freq. cocaine: 9.02 ± 1.67 Hz, n = 15, t(28) = 2.143, p = 0.0410), without altering sEPSC amplitude (sEPSC amp. baseline: −19.05 ± 1.26 pA, n = 15; sEPSC amp. cocaine: −19.73 ± 0.92 pA, n = 16, t(29) = 0.4375, p = 0.6650), holding current (IHolding; cocaine: 102.20 ± 13.47%, n = 5, t(4) = 0.1634, p = 8781) or membrane resistance (Rm; cocaine: 95.72 ± 12.26%, n = 6, t(5) = 0.3491, p = 0.7472) (Fig. 1j–n). Thus, cocaine decreases glutamatergic synaptic efficacy onto PV-INs through a VGNC-independent presynaptic locus of action. We next asked if thalamo- and corticoaccumbens afferents, shown to support distinct plasticity and motivational outcomes [28,29,30], are differentially sensitive to cocaine. A ChR2-expressing virus was stereotaxically injected into the PFC or MDT of PVtdT mice from which optically-evoked EPSCs (oEPSCs) were recorded from PV-INs, as described previously [3, 28,29,30] (Fig. 1o). Cocaine resulted in decrease in oEPSC amplitude at MDT (53.47 ± 6.81, n = 6, t(5) = 6.829, p < 0.001) and PFC (58.35 ± 6.75%, n = 6, t(5) = 6.168, p < 0.001) synapses. Magnitude of cocaine-induced depression between MDT and PFC was not different (t(10) = 0.5090, p = 0.622), suggesting that expression mechanisms mediating the effects of cocaine shared between inputs can be surveyed using electrical stimulation (Fig. 1p–r).
Cocaine engages a monoamine- and cholinergic-independent synaptic mechanism
Cocaine exerts heterosynaptic actions on glutamatergic and GABAergic synapses by inhibiting the reuptake of monoamines [18]. We recently found that NE transporter (NET)-regulated NE release decreases PV-IN-directed feedforward inhibition via α2-adrenoreceptors (ARs) [27]. Thus, we suspected that cocaine facilitates NE signaling by examining the pharmacological interaction between NET and cocaine (Fig. 2a). Consistent with our previous findings, superfusion of selective NET inhibitor, tomoxetine (TMx, 10 µM), markedly depressed EPSC amplitude (Fig. 2b–d). Once the TMx effect stabilized for ≥10-min, cocaine was incorporated into the superfusate, resulting in a decrease in EPSC amplitude surprisingly similar to control conditions (TMx: 72.89 ± 5.93%, n = 6, ACSF vs. cocaine-TMx, p = 0.4842) (Fig. 2b–e, h). In addition, prior application of α-AR antagonist, phentolamine (1 µM), or β-AR antagonist, propranolol (1 µM), had no effect on the magnitude of the cocaine-induced depression (cocaine in phentol: 73.57 ± 6.26%, n = 5, ACSF vs. phentol, p = 0.4584; cocaine in prop: 77.01 ± 1.74%, n = 6, ACSF vs. prop, p = 0.1608) (Fig. 2f–h). Therefore, despite α-AR-dependent NE signaling within this microcircuit, cocaine engages a rapid, NE- and NET-independent mechanism (Fig. 2h; 1-way ANOVA, Sidak’s post-hoc analysis, F(3,20) = 1.49, p = 0.2474) to reduce glutamatergic synaptic efficacy onto PV-INs.

a Schematic of candidate transporter-regulated monoaminergic systems mediating the effects of cocaine at PV-IN synapses in the NAc. b, c Representative experiment and traces of EPSCs obtained during a TMx- cocaine occlusion experiment. Scale bar: 200 pA/20 ms. d Normalized time-course summary depicting the effects of cocaine following TMx. e Time-course summary renormalized following the stabilization of the TMx-induced synaptic depression. f Representative traces of EPSCs depicting the effects of cocaine in phentolamine (green circles) or propranolol (black circles). Scale bar: 200 pA/20 ms. g Normalized time-course summary of EPSCs following cocaine application in phentolamine or propranolol. h Quantification of average EPSC amplitude at t(gray) following each NE-related pharmacological manipulation. i Representative traces of EPSCs depicting the effects of cocaine in GBR12783. Scale bar: 200 pA/20 ms. j Normalized time-course summary of EPSCs following cocaine application in GBR12783. k Representative traces of EPSCs depicting the effects of cocaine in sulpiride (green circles) or SCH23390 (black circles). Scale bar: 50 pA/20 ms. l Normalized time-course summary of EPSCs following cocaine application in sulpiride or SCH23390. m Representative traces of EPSCs depicting the effects of cocaine in citalopram. Scale bar: 100 pA/20 ms. n Normalized time-course summary of EPSCs following cocaine application in citalopram. o Quantification of average EPSC amplitude at t(gray) following each DA- and 5-HT-related pharmacological manipulation. p Schematic depicting reserpine inhibition of vesicular monoamine transporter, VMAT. q Representative traces of EPSCs depicting the effects of cocaine in slices incubated in VMAT inhibitor, reserpine. Scale bars: 50 pA/20 ms. Note: A prolonged (20 min) EPSC baseline was obtained to ensure monoamine removal from presynaptic terminals. r Normalized time-course summary of EPSCs following cocaine application in reserpine. s Quantification of average EPSC amplitude at t(gray) following cocaine in reserpine. Error bars indicate SEM. *p < 0.05.
To determine if cocaine instead recruits DA signaling, we pursued a similar experimental strategy in which cocaine was superfused in the continuous presence of selective DA transporter (DAT) inhibitor, GBR12783 (1 µM). Similar to NET, DAT blockade failed to occlude the cocaine-induced depression in EPSC amplitude (cocaine in GBR: 66.35 ± 4.34%, n = 6, ACSF vs. GBR, p = 0.9986) (Fig. 2i, j, o). The cocaine-induced depression was also unaffected by D1-like receptor antagonist, SCH 23390 (1 µM; cocaine in sulp: 66.26 ± 5.42%, n = 5, ACSF vs. sulp, p = 0.9677), or D2-like receptor antagonist, sulpiride (4 µM; cocaine in SCH: 75.24 ± 4.89%, n = 5, ACSF vs. SCH, p = 0.3122), suggesting that cocaine also engages a DA and DAT-independent synaptic mechanism (Fig. 2k, l, o). Finally, we assessed the contribution of 5-HT transporter (SERT)-regulated 5-HT signaling by examining the effects of cocaine in ACSF containing selective 5-HT reuptake inhibitor (SSRI), citalopram (4 µM). Citalopram also had no effect on the cocaine-induced decrease in EPSC amplitude (cocaine in citalopram: 62.89 ± 6.35%, n = 5, ACSF vs. citalopram, p = 0.9940). Along with NE findings, lack of effect on average EPSC amplitude following each DA- and 5-HT-related pharmacological manipulation (1-way ANOVA, Sidak’s post-hoc analysis, F(4,23) = 1.18, p = 0.3465) suggests that the neuromodulatory actions of cocaine do not rely on the blockade of monoamine reuptake transporters for NE, DA, and 5-HT in the NAc (Fig. 2m–o).
These data align with recent work from our group showing that pharmacological blockade of SERT or DAT alone does not acutely modulate glutamatergic synaptic strength onto PV-INs [27]. To rule out indiscriminate actions of cocaine on all monoamine systems in the NAc, we bath-applied cocaine in slices incubated for 2 h in a monoamine-depleting vesicular monoamine transporter (VMAT) inhibitor, reserpine (10 µM) (Fig. 2p). A 20-min EPSC baseline was obtained prior to cocaine application to ensure complete evacuation of terminal monoamine stores [31]. The cocaine-induced depression in reserpinized slices was indistinguishable from control condition (cocaine in reserpine: 68.44 ± 5.44%, n = 7, t(6) = 5.801, p = 0.0012), strongly supporting a monoamine-independent mechanism of action at this synapse (Fig. 2q–s). To validate that reserpine-induced VMAT blockade diminishes evoked DA release in the NAc, we superfused reserpine while measuring electrically-evoked peak DA currents (eDA) via fast-scan cyclic voltammetry (FSCV). Reserpine application abolished eDA, confirming that the synaptic effects of cocaine in reserpinized slices are monoamine-independent (Supplementary Fig. S1a–c). We next asked if cocaine engages a cholinergic interneuron microcircuit mechanism, as shifts in acetylcholine (ACh) release could elicit circuit-specific adaptations in glutamatergic transmission in the NAc and striatum [32,33,34,35,36]. However, prior application of mAChR antagonist, atropine (10 µM), or pan-nicotinic ACh receptor (nAChR) blocker, mecamylamine (MA, 1 µM), had no effect on the cocaine-induced decrease in EPSC amplitude (cocaine in MA: 65.41 ± 4.16%, n = 4, ACSF vs. MA, p = 0.9958; cocaine in atropine: 65.88 ± 4.81%, n = 5, ACSF vs. atropine, p = 0.9794, 1-way ANOVA, Sidak’s post-hoc analysis, F(2,13) = 0.1517, p = 0.9850) (Supplementary Fig. S2a–c). Taken together, these data suggest that cocaine engages a monoamine-and ACh- independent mechanism at glutamatergic synapses onto PV-INs.
Cocaine mobilizes intracellular Ca2+ stores
Having ruled out neurotransmitter systems mediating the canonical effects of cocaine, we next addressed the requirement for Ca2+ by superfusing cocaine in ACSF containing cell-permeant Ca2+ chelator, BAPTA-AM (20 µM). Prior application of BAPTA-AM completely blocked the cocaine-induced depression in EPSC amplitude (cocaine in BAPTA-AM: 103.70 ± 5.82%, n = 5, p = 0.5559) (Fig. 3a, b, i). Additionally, intracellular loading of cell-impermeant BAPTA (20 mM) in the patch pipette blocked the cocaine-induced decrease in EPSC amplitude (cocaine in BAPTA: 99.60 ± 10.62%, n = 6, ACSF vs. BAPTA p = 0.0012), indicating that cocaine modulates Ca2+ signaling specifically in PV-INs to elicit a change in glutamatergic synaptic strength (Fig. 3a, b, i). A source of Ca2+ mediating synaptic plasticity throughout the striatal network is L-type voltage-gated Ca2+ channels (VGCCs, CaV1.x) [32, 36,37,38]. To determine if L-type VGCCs contribute to the effects of cocaine, we bath-applied cocaine while voltage-clamping PV-INs at −90 mV, a subthreshold membrane potential for L-type VGCC activation [36, 38]. Hyperpolarizing PV-INs to −90 mV had no effect on the cocaine-induced decrease in EPSC amplitude (Fig. 3c, d, i; cocaine at −90 mV: 61.77 ± 8.56%, n = 3, ACSF vs. −90 mV, p = 0.9991). Similarly, prior application of selective L-type VGCC blocker, nifedipine (30 µM), failed to prevent the effects of cocaine (cocaine in nifed: 65.59 ± 2.99%, n = 7, ACSF vs. nifed, p > 0.9999), indicating that cocaine does not require Ca2+ influx via L-type VGCCs to elicit a change in glutamatergic transmission onto PV-INs (Fig. 3c, d, i). In contrast, diminishing intracellular Ca2+ stores by inhibiting endoplasmic reticulum (ER)-embedded Ca2+ -ATPase (SERCA) with thapsigargin (1 µM) abolished the cocaine-induced depression (Fig. 3e, f, i; cocaine in thap: 89.37 ± 5.50%, n = 5, ACSF vs. thap, p = 0.0422). Thapsigargin and intracellular BAPTA also prevented the cocaine-induced increase in PPR and CV (Fig. 3i). Thus, Ca2+ liberated from intracellular stores is a proximal effector mediating the acute synaptic effects of cocaine at glutamatergic synapses onto PV-INs.

a Left: Representative traces of EPSCs depicting the effects of cocaine in PV-INs dialyzed with BAPTA. Scale bars: 50 pA/20 ms. Right: Representative traces of EPSCs depicting the effects of cocaine in the presence of BAPTA-AM. Scale bar: 20 pA/20 ms. b Normalized time-course summary of EPSCs following cocaine application in PV-INs dialyzed with BAPTA (green circles) or in slices incubated in BAPTA-AM (black circles). c Left: Representative traces of EPSCs obtained at −90 mV (black circles) depicting the effects of cocaine at a hyperpolarized command Vm. Right: Representative traces of EPSCs depicting the effects of cocaine in the presence of nifedipine (green circles). Scale bars: 50 pA/20 ms. d Normalized time-course summary of EPSCs following cocaine application at −90 mV and in nifedipine. e Representative traces of EPSCs depicting the effects of cocaine in slices incubated and continuously perfused with thapsigargin. Scale bars: 100 pA/20 ms. f Normalized time-course summary of EPSCs following cocaine application in thapsigargin. g Representative traces of EPSCs depicting the effects of cocaine in PV-INs loaded with GDPβS. Scale bars: 100 pA/20 ms. h Normalized time-course summary of EPSCs following cocaine application in PV-INs loaded with. i Quantification of average EPSC amplitude at t(gray) following each pharmacological manipulation. j Normalized average PPR (left) and CV (right) superimposed over experimental values in cocaine (pooled: thapsigargin and BAPTA conditions). k Representative traces of EPSCs depicting the effects of cocaine in BD1063 or BD1047. Scale bars: 50 pA/20 ms. l Normalized time-course summary of EPSCs following cocaine application in BD1063 or BD1047. m Quantification of average EPSC amplitude at t(gray) following cocaine in BD1063 or BD1047. n Quantification of average PPR (left) and CV (right) superimposed over experimental values in cocaine with BD1063 or BD1047. o Representative traces of EPSCs depicting the effects of PRE-084. Scale bars: 100 pA/20 ms. p Normalized time-course summary of EPSCs during PRE-084 wash-on. q Quantification of average EPSC amplitude at t(gray) following PRE-084 (white bar) and PRE-084 + BD1063 (dark bar). r Quantification of average PPR (left) and CV (right) superimposed over experimental values at baseline and in PRE-084. s Representative experiment and traces of EPSCs depicting cocaine occlusion following bath-application of PRE-084. t Renormalized time-course summary of EPSCs during cocaine application in the presence of PRE-084. u Quantification of average EPSC amplitude at t(gray) following cocaine in PRE-084. Error bars indicate SEM. *p < 0.05.
Cocaine-induced effector function requires σ1 and eCB signaling
Downstream activation of G-protein-coupled receptors (GPCRs) often mediates cocaine-induced changes in synaptic transmission [39,40,41]. Thus, we assessed whether cocaine broadly recruits postsynaptic G-protein signaling by including non-hydrolyzable GDP analog, GDPβS (1 mM), in the intracellular solution of the patch pipette. Interestingly, the cocaine-induced decrease in EPSC amplitude persisted in the presence of GDPβS (Fig. 3g–i; cocaine in GDP: 70.39 ± 3.41%, n = 5, p = 0.0010; 1-way ANOVA, F(5,27) = 8.868, Sidak’s post-hoc analysis, p < 0.0001), pointing to an intracellular effector that does not require G-protein signaling. These data are reminiscent of recent work showing that cocaine engages non-GPCR ER chaperone protein, σ1, to reduce the intrinsic excitability of D1-MSNs in the NAc [20]. Similar to other psychostimulants and clinically-used psychotropic medicines, cocaine is a σ1 agonist with micromolar (≤2 µM) affinity [42, 43]. σ1 also chaperones IP3 receptors (IP3Rs) within the ER reticular network to regulate cellular Ca2+ homeostasis [21, 44,45,46]. Considering data that cocaine triggers intracellular Ca2+ independently of G-protein or monoamine signaling, we hypothesized that cocaine instead engages postsynaptic σ1 signaling. To begin to test this hypothesis, we perfused slices with selective σ1 antagonist, BD1063 (2 µM). BD1063 had no effect on basal EPSC amplitude (data not shown) but completely blocked the cocaine-induced decrease in EPSC amplitude and associated rise in PPR and CV (Fig. 3k–n). Experiments were repeated and pooled with σ1 antagonist, BD1047 (1 µM), to rule out anomalous properties of BD1063, providing initial pharmacological evidence that σ1 is required for the effects of cocaine at synapses onto PV-INs (cocaine in σ1 antag: 98.97 ± 1.88%, n = 7, t(6) = 0.5511, p = 0.6015; PPR baseline: 1.33 ± 0.08, PPR cocaine in σ1 antag: 1.32 ± 0.09, n = 7, t(6) = 0.06683, p = 0.9489; CV baseline: 0.21 ± 0.01; CV cocaine in σ1antag.: 0.19 ± 0.012, n = 7, t(6) = 1.422, p = 0.2049). To determine if σ1 activation alone recapitulates the effects of cocaine, we superfused σ1-selective agonist, PRE-084 (5 µM). PRE-084 resulted in a slow-onset decrease in EPSC amplitude (PRE-084: 56.34 ± 5.30%, n = 7, t(6) = 2.283, p = 0.0012) that was accompanied by an increase in PPR and CV (PPR baseline: 1.20 ± 0.07, PPR PRE: 1.48 ± 0.14, n = 7, t(6) = 3.134, p = 0.0451; CV baseline: 0.23 ± 0.02; CV PRE: 0.39 ± 0.04, n = 7, t(6) = 3.263, p = 0.0172), mirroring the effects of cocaine (Fig. 3o–r). The PRE-084-induced depression was concentration-sensitive and σ1-dependent, as a high concentration of PRE-084 (20 µM) potentiated EPSC amplitude (data not shown). In addition, the synaptic depression triggered by PRE-084 was σ1-mediated, as the effects were abolished by prior application of BD1063, consistent with the high specificity of PRE-084 for σ1 (Fig. 3q) [47]. Prior application of PRE-084 also occluded any subsequent depression evoked by cocaine (Fig. 3s–u; cocaine in PRE: 95.59 ± 6.51%, n = 5, t(4) = 0.6777, p = 0.535), encouraging a model in which σ1 mediates the cocaine-induced decrease in glutamatergic synaptic strength onto PV-INs in the NAc.
While our data implicate a cocaine effector pathway involving postsynaptic σ1 and intracellular Ca2+ signaling in PV-INs, PPR, CV and sEPSC analyses converge on a VGNC-independent effect on glutamate release probability. A bridge between σ1-induced Ca2+ signaling and a presynaptic locus of action is the eCB system. Phasic shifts in intracellular Ca2+ levels promote the production of arachidonic acid-derived eCBs, such as 2-arachidonylglycerol (2-AG) and anandamide (AEA), that act on presynaptic type-1 cannabinoid receptors (CB1R) [48, 49]. We recently found that glutamatergic synapses onto PV-INs are heavily regulated by Ca2+ -sensitive eCB signaling at presynaptic CB1Rs [25]. Furthermore, a recent report in the VTA suggests that cocaine reduces synaptic transmission by promoting 2-AG release through σ1 [23]. Therefore, we postulated that cocaine -induced σ1 signaling leads to the Ca2+ -dependent production of eCBs, resulting in presynaptic CB1R activity.
CB1R activity decreases glutamatergic drive at both PFC [35, 50] and MDT inputs to the NAc (Supplementary Fig. S3). To address the involvement of eCBs in mediating the synaptic effects of cocaine, we bath-applied cocaine in the presence of CB1R inverse agonist, AM251 (2 µM) (Fig. 4a, b). The cocaine-induced depression in EPSC amplitude was completely blocked by AM251 (cocaine in AM251: 96.94 ± 4.29%, n = 6, ACSF vs. AM251, p < 0.001) and occluded by a suprathreshold concentration of CB1/2R agonist, WIN 55-212 (5 µM; cocaine in WIN: 92.63 ± 5.36%, n = 4, ACSF vs. WIN, p = 0.003), supporting the involvement of the eCB system (Fig. 4c–e; 1-way ANOVA, Sidak’s post-hoc analysis, F(2,13) = 29.44, p < 0.0001). If cocaine engages presynaptic CB1R signaling, then glutamatergic synapses onto PV-INs should exhibit increased sensitivity to CB1R blockade in the presence of cocaine. In ACSF alone, AM251 evoked a significant increase in EPSC amplitude (ACSF: 116.80 ± 4.59%, n = 6); consistent with prior work showing that tonic CB1R signaling negatively regulates glutamatergic transmission onto PV-INs [25]. However, in the presence of cocaine, AM251 evoked a significantly greater increase in EPSC amplitude relative to ACSF (cocaine: 149.91 ± 9.42%, n = 5, ACSF vs. cocaine, p = 0.0062), indicating that cocaine engages presynaptic CB1R activity. To discern further whether cocaine-induced CB1R activity is mediated by σ1, we bath-applied AM251 in slices perfused with both cocaine and BD1063. Consistent with a σ1-dependent mechanism, BD1063 blocked the cocaine-facilitated increase in EPSC amplitude unmasked with AM251 without altering basal CB1R tone (cocaine + BD: 117.70 ± 3.35%, n = 5, cocaine vs. cocaine + BD, p = 0.0101), suggesting that cocaine augments CB1R activity in a σ1-dependent manner (Fig. 4f–h; 1-way ANOVA, Sidak’s post-hoc analysis, F(2,13) = 9.059, p = 0.0034).

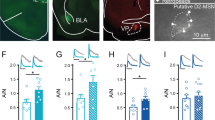
a Representative traces of EPSCs depicting the effects of cocaine in AM251. Scale bars: 100 pA/50 ms. b Normalized time-course summary of EPSCs following cocaine application in AM251. c Representative experiment and traces (top) of EPSCs depicting the effects of cocaine following superfusion WIN 55-212. Scale bars: 100 pA/20 ms. d Time-course summary renormalized following the stabilization of the WIN 55-212-induced synaptic depression. e Quantification of average EPSC amplitude at t(gray) following cocaine in AM251 or WIN 55-212 (occlusion). f Representative traces of EPSCs depicting the effects of AM251 in ACSF (black circles), cocaine (green circles), or cocaine + BD1063 (open circles). Scale bars: 50 pA/20 ms. g Normalized time-course summary of EPSCs following AM251 in ACSF, cocaine or cocaine + BD1063. h Quantification of average EPSC amplitude at t(gray) following AM251 in each pharmacological manipulation. i Representative experiments of EPSCs during a 10-sec ISI baseline followed by a transition to a 3-sec ISI time-locked with the application of ACSF or PRE-084. j Normalized time-course summary of EPSCs during the 10-sec ISI baseline and 3-sec ISI transition in ACSF (open circles) or PRE-084 (green circles). k Quantification of average EPSC amplitude at t(gray) following 3-sec ISI transition. l Schematic depicting pharmacological manipulation of 2-AG synthesis and AEA degradation via DAGL and FAAH, respectively. m Representative traces of EPSCs depicting the effects of cocaine in slices incubated in URB597 (open circles) or DO34 (filled circles). Scale bars: 100 pA/50 ms (URB)/20 ms (DO34). n Normalized time-course summary of EPSCs following cocaine application in URB597 or DO34. o Quantification of average EPSC amplitude at t(gray) following cocaine in each eCB manipulation. Error bars indicate SEM. *p < 0.05.
Since CB1R-induced plasticity is often activity-dependent and the synaptic effects of isolated σ1 activation evolved gradually over time, we speculated that cocaine-associated σ1-induced plasticity at this synapse proceeds through an activity-dependent eCB release step. To test this hypothesis, we accompanied the application of PRE-084 with a shortened interstimulus interval (ISI) from 10-sec to 3-sec, a protocol adapted from prior analyses of activity-dependent eCB production in the striatum [51, 52]. In slices perfused with ACSF alone, EPSC amplitude, on average, remained stable during the transition to a 3-sec ISI (ACSF3-sec ISI: 98.17 ± 1.78%, n = 4, p = 0.981), indicating that a 3-sec ISI results in minimal readily releasable pool (RRP)-associated synaptic rundown at this synapse. However, time-locked application of PRE-084 during the 3-sec ISI epoch resulted in a significant decrease in EPSC amplitude (Fig. 4i–k; PRE3-sec ISI: 76.84 ± 6.82%, n = 7, p < 0.001; 1-way ANOVA, ACSF vs. PRE at 3-sec ISI, F(3, 18) = 49.99, p < 0.0001). Taken together, these findings suggest that cocaine engages intracellular σ1 in PV-INs, resulting in a decrease in synaptic strength that is expedited by activity within the feedforward network.
Having established that cocaine engages eCB signaling through CB1Rs, we next asked if cocaine promotes 2-AG or AEA production by pharmacologically manipulating their synthesis and degradation, respectively (Fig. 4l). Pre-incubating slices for 2 h in the selective DAG lipase (DAGL) inhibitor, DO34 (1 µM) previously shown to inhibit 2-AG signaling at CB1Rs [53], had no significant effect on the cocaine-induced decrease in EPSC amplitude (cocaine in DO34: 67.84 ± 3.41%, n = 4). However, pre-incubation in URB597 (1 µM), an inhibitor of the AEA degradative enzyme fatty acid amide hydrolase (FAAH), significantly attenuated the magnitude of the cocaine depression (cocaine in URB: 88.12 ± 3.36%, n = 7), mirroring tonic AEA-dependent occlusion of CB1R described previously (see Discussion, [25]) (Fig. 4m–o; t(9) = 3.916, p = 0.0035). Although these findings implicate AEA signaling, taken together, our data suggest that cocaine triggers 2-AG-independent eCB signaling at CB1Rs to reduce glutamatergic transmission onto PV-INs.
Cocaine occludes CB1R-dependent long-term plasticity
Cocaine exposure modulates the expression of CB1R-dependent long-term depression (LTD) at glutamatergic synapses in the NAc [30, 54, 55]. We recently found that low-frequency stimulation (LFS: 5 min, 10 Hz) triggers Ca2+ entry via Ca2+ -permeable AMPARs (CP-AMPARs) at PV-IN synapses, leading to the induction of CB1R-dependent LTD [25, 27]. Given data here endorsing an eCB-dependent mechanism mediating the actions of cocaine, we speculated that ex vivo cocaine exposure also alters the expression of LFS-induced LTD at PV-IN synapses (Fig. 5a). Indeed, pre-incubating slices in cocaine for 1-hr abolished the expression of LFS-induced LTD at PV-IN synapses relative to ACSF controls (Fig. 5b–d; Post-LFS cocaine: 98.39 ± 5.92%, n = 10; Post-LFS ACSF: 48.02 ± 4.10%, n = 7, t(15) = 6.386, p = <0.001). To examine whether cocaine occludes LFS-induced LTD by recruiting σ1, we repeated these experiments in slices incubated in both cocaine and BD1063. The integrity of LFS-induced LTD was unaffected by BD1063 alone, indicating that basal σ1 activity does not contribute to the expression of this LTD (Fig. 5e–g). However, BD1063 prevents loss of LFS-induced LTD in cocaine-treated slices, supporting a convergent mechanism whereby cocaine occludes CB1R-dependent plasticity at PV-INs by triggering σ1-dependent eCB signaling (Fig. 5e–g; (Post-LFS BD vs baseline: 69.30 ± 5.75%, n = 4, p = 0.0276; Post-LFS cocaine + BD vs. baseline: 72.40 ± 8.77%, n = 7, p = 0.0119; 1-way ANOVA, Post-LFS in BD vs. cocaine + BD, F(3, 18) = 7.851, p = 0.0051)).Taken together, these data offer insight into ways in which acute cocaine experience co-opts endogenous mechanisms of plasticity at feedforward synapses in the NAc.

a Schematic and timeline of ex vivo electrophysiological recordings in slices incubated in cocaine or ACSF ± BD1063. b Representative traces of EPSCs pre- and post-LFS in cocaine (green circles) vs. ACSF-treated slices (black circles). c, d Normalized time-course summary and quantification of EPSCs pre- and post-LFS in cocaine vs. ACSF-treated slices. e Representative traces of EPSCs pre- and post-LFS in cocaine + BD1063 (green circles) vs. BD1063 alone (open circles). f, g Normalized time-course summary and quantification of EPSCs pre- and post-LFS in cocaine + BD1063 vs. BD1063-treated slices. All scale bars: 50 pA/20 s.
Discussion
Here, we interrogate synaptic mechanisms by which cocaine, a recreationally-used psychostimulant with abuse liability, modulates feedforward glutamatergic drive onto PV-INs in the NAc. We report that cocaine recruits a non-canonical monoamine-independent mechanism to reduce thalamocortical glutamatergic transmission onto PV-INs. Our data suggests that cocaine engages presynaptic CB1R signaling by targeting intracellular σ1, leading to the Ca2+ -sensitive, activity-dependent production of eCBs. Furthermore, acute ex vivo cocaine exposure occludes synaptically-evoked CB1R-dependent LTD at glutamatergic synapses onto PV-INs (Supplementary Fig. S4). These data highlight a synaptic substrate for cocaine within PV-IN microcircuits and extend existing heuristics of cocaine action with the mesolimbic reward network.
Monoamine-independent actions of cocaine at PV-IN synapses
We found that a subanesthetic concentration of cocaine (3–10 µM) reduces feedforward glutamatergic drive onto PV-INs that is comparable between thalamo- and cortico-accumbens inputs. A similar phenomenon described at synapses onto MSNs relies on extracellular elevations in DA and D1-like/adenosine A1 receptor signaling [41, 56]. However, at synapses onto PV-INs, the effects of cocaine were impervious to DAT, D1-, D2-like receptor blockade, and monoamine depletion, indicating a DA-independent mechanism. Moreover, despite our recent report of a α2-AR-mediated effect following NET blockade at this synapse, the effects of cocaine at PV-IN synapses remained intact following multiple pharmacological manipulations of NE signaling [27]. While the cocaine-induced depression occurred rapidly, plasticity elicited by NET blockade evolved gradually over time, pointing to a separate mechanism of modulation whereby NE accumulates at the synapse to elicit heterosynaptic plasticity [27]. Acute involvement of the 5-HT system also fails to explain the actions of cocaine, as prior SSRI application, which elicits heterosynaptic effects on synaptic transmission at striatal and pallidal synapses, had no effect on the cocaine-induced decrease in synaptic efficacy onto PV-INs [57,58,59]. These data shed light on a complex neuromodulatory environment in which synapse-specific monoamine microdomains may cooperatively regulate NAc circuit function. In addition, these data encourage future studies that disentangle acute synaptic effects of cocaine from the sustained actions mediated by adjacent cocaine-avid monoamine transporters, such as NET.
Cocaine-induced behavioral and cellular adaptations via σ1
The monoamine-independent actions of cocaine at glutamatergic synapses onto PV-INs prompted us to consider less “traditional” pharmacological targets of cocaine, including ER chaperone protein, σ1. σ1 is increasingly implicated in the reinforcing properties of cocaine [45]. For example, self-administration of cocaine transfers to selective σ1 ligands, including the σ1 agonist used in our studies (PRE-084) [60, 61]. Similar to a NAc-specific σ1 knockdown strategy, systemic administration of D1- and D2-like DA receptor antagonists reduces but does not block the induction of locomotor sensitization to cocaine, supporting the involvement of complementary effector systems mediating the behavioral response to cocaine [22, 24]. Additionally, cocaine-induced MSN hypoactivity is mediated by monoamine-independent intracellular σ1 signaling [20]. It remains unclear how cocaine interacts temporally with established DA receptor-dependent effects on MSN output, or whether both mechanisms are occurring in parallel [62, 63]. Nevertheless, it is becoming increasingly evident that σ1 activity contributes to cocaine-induced behavioral and cellular adaptations.
Our findings extend this model by showing that the cocaine-induced decrease in synaptic efficacy onto PV-INs is blocked, reproduced, and occluded by σ1 antagonists and agonists, respectively. In the VTA, σ1 modulates extracellular vesicle release through regulation of the GTPase ADP-ribosylation factor 6 (ARF6), a process that further requires GTPase-activating proteins. In contrast to VTA dopamine neurons, the effects of cocaine on NAc PV-INs were resistant to G-protein-disabling agent [23]. Thus, the present study indicates cocaine, via σ1 mobilized eCB release through a different mechanism – unlikely to involve GDP to GTP conversion. Our data discourages but does not rule out alternative possibilities of a PV-IN-independent circuit interaction or parallel presynaptic mechanism operating in the presence of intracellularly-loaded GDPβSs. While we do not directly identify molecular interactions by which σ1 signaling decreases synaptic efficacy, postsynaptic Ca2+ chelation and ER Ca2+ -ATPase inhibition, but not L-type VGCC antagonists, blocked the cocaine-induced decrease in synaptic strength, indicating that store-associated intracellular Ca2+ release is a probable proximal step in the cocaine effector pathway. One possibility is that cocaine-induced σ1 signaling results in the mobilization of intracellular Ca2+ stores from the ER through molecular chaperoning of ER-resident IP3Rs, as cocaine elicits IP3R-induced Ca2+ transients in dissociated NAc MSNs via σ1 signaling [44]. Alternatively, cocaine may engage σ1-associated client proteins at the ER interface to redistribute intracellular Ca2+ stores [21]. An intriguing additional finding from our study is that σ1-induced changes in synaptic strength were expedited by a shortened ISI, suggesting that increased synaptic activity contributes to synaptic alterations elicited by σ1. While our data identifies nodes within an effector arm underlying the cocaine-induced reduction in glutamatergic synaptic strength, these possibilities encourage future analyses of the intracellular transduction events underlying cocaine and σ1 signaling in PV-INs in the NAc.
The eCB system is recruited by cocaine via σ1
Interestingly, cocaine consistently and selectively altered metrics of presynaptic release probability, including PPR, CV and sEPSC frequency. Having recently established that presynaptic CB1Rs gate glutamatergic synaptic strength onto PV-INs in the NAc [25, 27], we postulated that cocaine interacts functionally with the eCB system, as described previously [14, 16]. Indeed, convergent pharmacological data support a model in which cocaine triggers 2-AG-independent eCB signaling at presynaptic CB1Rs, as the cocaine-induced decrease in synaptic efficacy were abolished by blocking and saturating presynaptic CB1R activity with AM251 and WIN 55-212, respectively. Although AEA is a probable eCB candidate, experiments performed in the presence of FAAH inhibitor URB597 are difficult to interpret in a synaptic milieu constrained by tonic AEA signaling. A more definitive result from our study is the failure of DAGL inhibition to block the cocaine-induced depression, aligning with previous work showing that AEA, but not 2-AG, participates in tonic and phasic eCB signaling at glutamatergic synapses onto PV-INs [25]. A similar but distinct mechanism in the VTA suggests that cocaine decreases GABAergic transmission onto DA neurons via σ1-dependent eCB signaling, supporting a putative role of the eCB system and σ1 in mediating cocaine-induced synaptic transmission in the mesolimbic network [23, 64].
PV-IN function in the NAc
A key question prompted by our results and others is the behavioral importance of glutamatergic drive onto PV-INs in motivational behavior. We recently found that pharmacologically reducing glutamatergic drive onto PV-INs in the NAc enhances basal locomotor output [25]. Additionally, strengthening amygdalo-accumbens transmission onto PV-INs gates the acquisition of cocaine self-administration [3]. Thus, one possibility is that cocaine-induced CB1R activity at PV-IN synapses reduces feedforward drive onto MSNs, initiating the recruitment of distinct MSN ensembles. A persistent reduction in glutamatergic drive onto PV-INs could theoretically mount a homeostatic increase in membrane responsiveness, resulting in the increased PV-IN excitability observed following chronic cocaine exposure [65]. Clearly, future studies are needed to define circuit and molecular interactions through which PV-INs gate cocaine-induced behavioral outcomes.
References
Pisansky MT, Lefevre EM, Retzlaff CL, Trieu BH, Leipold DW, Rothwell PE. Nucleus accumbens fast-spiking interneurons constrain impulsive action. Biol Psychiatry. 2019;86:836–47.
Tepper JM, Koos T, Ibanez-Sandoval O, Tecuapetla F, Faust TW, Assous M. Heterogeneity and diversity of striatal GABAergic interneurons: update 2018. Front Neuroanat. 2018;12:91.
Yu J, Yan Y, Li KL, Wang Y, Huang YH, Urban NN, et al. Nucleus accumbens feedforward inhibition circuit promotes cocaine self-administration. Proc Natl Acad Sci USA. 2017;114:E8750–9.
Manz KM, Baxley AG, Zurawski Z, Hamm HE, Grueter BA. Heterosynaptic GABAB receptor function within feedforward microcircuits gates glutamatergic transmission in the nucleus accumbens core. J Neurosci. 2019;39:9277–93.
Scudder SL, Baimel C, Macdonald EE, Carter AG. Hippocampal-evoked feedforward inhibition in the nucleus accumbens. J Neurosci. 2018;38:9091–104.
Wright WJ, Schluter OM, Dong Y. A feedforward inhibitory circuit mediated by CB1-expressing fast-spiking interneurons in the nucleus accumbens. Neuropsychopharmacology. 2017;42:1146–56.
Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron. 2012;76:790–803.
Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, Luscher C. Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature. 2014;509:459–64.
Pascoli V, Turiault M, Luscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2011;481:71–5.
Stuber GD, Sparta DR, Stamatakis AM, van Leeuwen WA, Hardjoprajitno JE, Cho S, et al. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature. 2011;475:377–80.
Terrier J, Luscher C, Pascoli V. Cell-type specific insertion of GluA2-lacking AMPARs with cocaine exposure leading to sensitization, cue-induced seeking, and incubation of craving. Neuropsychopharmacology. 2016;41:1779–89.
Wang X, Gallegos DA, Pogorelov VM, O’Hare JK, Calakos N, Wetsel WC, et al. Parvalbumin interneurons of the mouse nucleus accumbens are required for amphetamine-induced locomotor sensitization and conditioned place preference. Neuropsychopharmacology. 2018;43:953–63.
Hearing M, Graziane N, Dong Y, Thomas MJ. Opioid and psychostimulant plasticity: targeting overlap in nucleus accumbens glutamate signaling. Trends Pharm Sci. 2018;39:276–94.
Jedynak J, Hearing M, Ingebretson A, Ebner SR, Kelly M, Fischer RA, et al. Cocaine and amphetamine induce overlapping but distinct patterns of AMPAR plasticity in nucleus accumbens medium spiny neurons. Neuropsychopharmacology. 2016;41:464–76.
Luscher C. The emergence of a circuit model for addiction. Annu Rev Neurosci. 2016;39:257–76.
Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–23.
Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S, et al. Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci USA. 2006;103:9333–8.
Cunningham KA, Callahan PM. Monoamine reuptake inhibitors enhance the discriminative state induced by cocaine in the rat. Psychopharmacology. 1991;104:177–80.
Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–23.
Delint-Ramirez I, Garcia-Oscos F, Segev A, Kourrich S. Cocaine engages a non-canonical, dopamine-independent, mechanism that controls neuronal excitability in the nucleus accumbens. Mol Psychiatry. 2020;25:680–91.
Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610.
Kourrich S, Hayashi T, Chuang JY, Tsai SY, Su TP, Bonci A. Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell. 2013;152:236–47.
Nakamura Y, Dryanovski DI, Kimura Y, Jackson SN, Woods AS, Yasui Y, et al. Cocaine-induced endocannabinoid signaling mediated by sigma-1 receptors and extracellular vesicle secretion. Elife. 2019;8.
White FJ, Joshi A, Koeltzow TE, Hu XT. Dopamine receptor antagonists fail to prevent induction of cocaine sensitization. Neuropsychopharmacology. 1998;18:26–40.
Manz KM, Ghose D, Turner BD, Taylor A, Becker J, Grueter CA, et al. Calcium-permeable AMPA receptors promote endocannabinoid signaling at parvalbumin interneuron synapses in the nucleus accumbens core. Cell Rep. 2020;32:107971.
Manz KM, Siemann JK, McMahon DG, Grueter BA. Patch-clamp and multi-electrode array electrophysiological analysis in acute mouse brain slices. STAR Protoc. 2021;2:100442.
Manz KM, Coleman BC, Grueter CA, Shields BC, Tadross MR, Grueter BA. Noradrenergic signaling disengages feedforward transmission in the nucleus accumbens shell. J Neurosci. 2021;41:3752–63.
Joffe ME, Grueter BA. Cocaine experience enhances thalamo-accumbens N-Methyl-D-aspartate receptor function. Biol Psychiatry. 2016;80:671–81.
Manz KM, Becker JC, Grueter CA, Grueter BA. Histamine H3 receptor function biases excitatory gain in the nucleus accumbens. Biol Psychiatry. 2021;89:588–99.
Turner BD, Rook JM, Lindsley CW, Conn PJ, Grueter BA. mGlu1 and mGlu5 modulate distinct excitatory inputs to the nucleus accumbens shell. Neuropsychopharmacology. 2018;43:2075–82.
Yorgason JT, Zeppenfeld DM, Williams JT. Cholinergic interneurons underlie spontaneous dopamine release in nucleus accumbens. J Neurosci. 2017;37:2086–96.
Augustin SM, Chancey JH, Lovinger DM. Dual dopaminergic regulation of corticostriatal plasticity by cholinergic interneurons and indirect pathway medium spiny neurons. Cell Rep. 2018;24:2883–93.
Gipson CD, Reissner KJ, Kupchik YM, Smith AC, Stankeviciute N, Hensley-Simon ME, et al. Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc Natl Acad Sci USA. 2013;110:9124–9.
Leyrer-Jackson JM, Holter M, Overby PF, Newbern JM, Scofield MD, Olive MF et al. Accumbens cholinergic interneurons mediate cue-induced nicotine seeking and associated glutamatergic plasticity. eNeuro. 2021;8.
Mateo Y, Johnson KA, Covey DP, Atwood BK, Wang HL, Zhang S, et al. Endocannabinoid actions on cortical terminals orchestrate local modulation of dopamine release in the nucleus accumbens. Neuron. 2017;96:1112–26.e1115.
Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, et al. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–52.
Mathur BN, Tanahira C, Tamamaki N, Lovinger DM. Voltage drives diverse endocannabinoid signals to mediate striatal microcircuit-specific plasticity. Nat Neurosci. 2013;16:1275–83.
Patton MH, Padgett KE, McKeon PN, Qadir H, Patton MS, Mu C, et al. TrkB-dependent disinhibition of the nucleus accumbens is enhanced by ethanol. Neuropsychopharmacology. 2019;44:1114–22.
Dobbs LK, Kaplan AR, Lemos JC, Matsui A, Rubinstein M, Alvarez VA. Dopamine regulation of lateral inhibition between striatal neurons gates the stimulant actions of cocaine. Neuron. 2016;90:1100–13.
Matsui A, Alvarez VA. Cocaine inhibition of synaptic transmission in the ventral pallidum is pathway-specific and mediated by serotonin. Cell Rep. 2018;23:3852–63.
Nicola SM, Kombian SB, Malenka RC. Psychostimulants depress excitatory synaptic transmission in the nucleus accumbens via presynaptic D1-like dopamine receptors. J Neurosci. 1996;16:1591–604.
Chen Y, Hajipour AR, Sievert MK, Arbabian M, Ruoho AE. Characterization of the cocaine binding site on the sigma-1 receptor. Biochemistry. 2007;46:3532–42.
Sharkey J, Glen KA, Wolfe S, Kuhar MJ. Cocaine binding at sigma receptors. Eur J Pharm. 1988;149:171–4.
Barr JL, Deliu E, Brailoiu GC, Zhao P, Yan G, Abood ME, et al. Mechanisms of activation of nucleus accumbens neurons by cocaine via sigma-1 receptor-inositol 1,4,5-trisphosphate-transient receptor potential canonical channel pathways. Cell Calcium. 2015;58:196–207.
Katz JL, Hong WC, Hiranita T, Su TP. A role for sigma receptors in stimulant self-administration and addiction. Behav Pharm. 2016;27:100–15.
Su TP, Hayashi T. Cocaine affects the dynamics of cytoskeletal proteins via sigma(1) receptors. Trends Pharm Sci. 2001;22:456–8.
Motawe ZY, Abdelmaboud SS, Cuevas J, Breslin JW. PRE-084 as a tool to uncover potential therapeutic applications for selective sigma-1 receptor activation. Int J Biochem Cell Biol. 2020;126:105803.
Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–86.
Brown SP, Brenowitz SD, Regehr WG. Brief presynaptic bursts evoke synapse-specific retrograde inhibition mediated by endogenous cannabinoids. Nat Neurosci. 2003;6:1048–57.
Deroche MA, Lassalle O, Castell L, Valjent E, Manzoni OJ. Cell-type- and endocannabinoid-specific synapse connectivity in the adult nucleus accumbens core. J Neurosci. 2020;40:1028–41.
Adermark L, Lovinger DM. Retrograde endocannabinoid signaling at striatal synapses requires a regulated postsynaptic release step. Proc Natl Acad Sci USA. 2007;104:20564–9.
Chen M, Wan Y, Ade K, Ting J, Feng G, Calakos N. Sapap3 deletion anomalously activates short-term endocannabinoid-mediated synaptic plasticity. J Neurosci. 2011;31:9563–73.
Marcus DJ, Bedse G, Gaulden AD, Ryan JD, Kondev V, Winters ND, et al. Endocannabinoid signaling collapse mediates stress-induced amygdalo-cortical strengthening. Neuron. 2020;105:1062–76.e1066.
Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–25.
Huang CC, Liang YC, Lee CC, Hsu KS. Cocaine withdrawal impairs mGluR5-dependent long-term depression in nucleus accumbens shell neurons of both direct and indirect pathways. Mol Neurobiol. 2015;52:1223–33.
Corkrum M, Covelo A, Lines J, Bellocchio L, Pisansky M, Loke K, et al. Dopamine-evoked synaptic regulation in the nucleus accumbens requires astrocyte activity. Neuron. 2020;105:1036–47.e1035.
Dolen G, Darvishzadeh A, Huang KW, Malenka RC. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature. 2013;501:179–84.
Heifets BD, Salgado JS, Taylor MD, Hoerbelt P, Cardozo Pinto DF, Steinberg EE, et al. Distinct neural mechanisms for the prosocial and rewarding properties of MDMA. Sci Transl Med. 2019;11.
Mathur BN, Capik NA, Alvarez VA, Lovinger DM. Serotonin induces long-term depression at corticostriatal synapses. J Neurosci. 2011;31:7402–11.
Garces-Ramirez L, Green JL, Hiranita T, Kopajtic TA, Mereu M, Thomas AM, et al. Sigma receptor agonists: receptor binding and effects on mesolimbic dopamine neurotransmission assessed by microdialysis. Biol Psychiatry. 2011;69:208–17.
Hiranita T, Mereu M, Soto PL, Tanda G, Katz JL. Self-administration of cocaine induces dopamine-independent self-administration of sigma agonists. Neuropsychopharmacology. 2013;38:605–15.
Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–66.
Lahiri AK, Bevan MD. Dopaminergic transmission rapidly and persistently enhances excitability of D1 receptor-expressing striatal projection neurons. Neuron. 2020;106:277–90.e276.
Wang H, Treadway T, Covey DP, Cheer JF, Lupica CR. Cocaine-induced endocannabinoid mobilization in the ventral tegmental area. Cell Rep. 2015;12:1997–2008.
Winters BD, Kruger JM, Huang X, Gallaher ZR, Ishikawa M, Czaja K, et al. Cannabinoid receptor 1-expressing neurons in the nucleus accumbens. Proc Natl Acad Sci USA. 2012;109:E2717–25.
Funding
This study was supported by National Institute on Drug Abuse (NIDA) grant R01DA040630 (to B.A.G). KMM is supported by National Institute of General Studies T32 GM108554.
Author information
Authors and Affiliations
Contributions
Conceptualization, KMM and BAG; Methodology, KMM, BCC, ANJ, DG, and BAG; Investigation, KMM, BCC, ANJ and DG; Resources, BAG, SP; Writing – original draft, KMM; Writing – Reviewing & Editing, KMM and BAG; Supervision, BAG; Funding Acquisition, BAG.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Manz, K.M., Coleman, B.C., Jameson, A.N. et al. Cocaine restricts nucleus accumbens feedforward drive through a monoamine-independent mechanism. Neuropsychopharmacol. 47, 652–663 (2022). https://doi.org/10.1038/s41386-021-01167-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01167-3
- Springer Nature Switzerland AG