
Abstract
While blood-based tests are readily available for various conditions, including cardiovascular diseases, type 2 diabetes, and common cancers, Alzheimer’s disease (AD) and other neurodegenerative diseases lack an early blood-based screening test that can be used in primary care. Major efforts have been made towards the investigation of approaches that may lead to minimally invasive, cost-effective, and reliable tests capable of measuring brain pathological status. Here, we review past and current technologies developed to investigate biomarkers of AD, including novel blood-based approaches and the more established cerebrospinal fluid and neuroimaging biomarkers of disease. The utility of blood as a source of AD-related biomarkers in both clinical practice and interventional trials is discussed, supported by a comprehensive list of clinical trials for AD drugs and interventions that list biomarkers as primary or secondary endpoints. We highlight that identifying individuals in early preclinical AD using blood-based biomarkers will improve clinical trials and the optimization of therapeutic treatments as they become available. Lastly, we discuss challenges that remain in the field and address new approaches being developed, such as the examination of cargo packaged within extracellular vesicles of neuronal origin isolated from peripheral blood.
Similar content being viewed by others
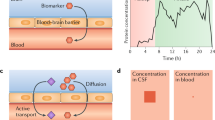


Introduction
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disease that is the leading cause of dementia worldwide [1]. The two neuropathological hallmarks of AD—extracellular amyloid beta (Aβ) deposition and intracellular tau-containing neurofibrillary tangles (NFTs)—begin to change years before symptoms begin, highlighting the need for sensitive and reliable diagnostic tests [2].
The diagnosis of AD has shifted from a syndromal to a biological basis in recent years. As per the clinical guidelines published by the National Institute on Aging and the Alzheimer’s Association (NIA-AA) workgroup in 2011, mild cognitive impairment (MCI) and dementia due to AD may be diagnosed based solely on clinical presentation [3, 4]. Biomarkers of Aβ and neurodegeneration, detected via positron emission tomography (PET), structural magnetic resonance imaging (MRI), or in the cerebrospinal fluid (CSF), were only recommended to diagnose preclinical AD or improve the certainty of the MCI or dementia diagnosis [2, 3]. In 2018, the NIA-AA published guidelines for research that shifted AD diagnosis toward a biological paradigm [5]. This document recognizes three biomarker categories for AD—biomarkers of amyloid deposition (A), pathological tau (T), and neurodegeneration (N) (i.e., AT(N))—that can be detected by neuroimaging or in the CSF, but not in plasma [5]. Furthermore, the severity of the disease may be biologically staged with this ATN profile (i.e., from A-T-(N)- to A+T+(N)+) and clinically staged based on the level of cognitive impairment (i.e., from stage 0 (asymptomatic) to stage 6 (severe dementia)) [5].
In 2024, the AA workgroup released updated research guidelines for the diagnosis and staging of AD [6]. Most notably, the guidelines suggest that AD may be diagnosed if any Core 1 biomarker (i.e., amyloid PET, approved CSF biomarkers, and accurate plasma biomarkers) is abnormal [6]. The inclusion of blood-based biomarkers (BBMs) in disease diagnosis and staging is new to the 2024 guidelines, with previous guidelines based solely on clinical presentation, CSF biomarkers, and neuroimaging. Nonetheless, standardized cut-offs and association with clinical prognosis have not been formally established for BBMs [6].
Here, we review recent developments in neuroimaging and fluid biomarkers and their utility in detecting preclinical and clinical AD and monitoring the effects of drug candidates in interventional trials. We also provide a comprehensive list of AD clinical trials that include neuroimaging or fluid biomarkers as endpoints. Since the detection of AD biomarkers in peripheral blood is a quickly developing field, we provide a list of currently available technologies capable of detecting BBMs. Lastly, we discuss the isolation of neuronal-derived extracellular vesicles (NEVs) in the blood and the analysis of their AD-related biomarker content as an avenue toward detecting brain-specific changes in peripheral samples.
Neuroimaging biomarkers – MRI
In AD, the earliest site of atrophy is seen in the medial temporal lobe, moving to the parietal, frontal, and cingulate cortices with advancing disease [7, 8]. In vivo measurements of brain mass and ventricular volume began in the 1970s with computed tomography, which was used to support a syndromal diagnosis of AD [9]. MRI was subsequently established as a measure of neurodegeneration in regions known to be vulnerable to AD neuropathology [10]. Brain atrophy detected by MRI is related to symptom severity in a specific topographic manner [11]. MRI measures of the entorhinal cortex, superior temporal sulcus, and anterior cingulate can, with moderate accuracy, distinguish between patients with AD, MCI to AD converters, MCI non-converters, and healthy controls [12]. Hippocampal texture and morphology on MRI brain scans have also shown some success in predicting the conversion of MCI to AD [13, 14]. However, AD progresses non-linearly with atrophy rates. The rate of hippocampal atrophy accelerates at a greater rate than cortical atrophy and a deceleration of cortical atrophy has been observed in later AD stages [15, 16]. These variations must be considered when assessing the efficacy of disease-modifying interventions or when providing prognostic information. Further, MRI neuroimaging is mainly based on atrophy and other non-specific features. Non-AD neurodegenerative diseases such as Lewy body dementia and vascular dementia also show increased atrophy rates compared to healthy controls [17]. For these reasons, MRI is useful in a patient’s clinical workup but not sufficient to diagnose MCI or dementia due to AD. Instead, MRI may be more useful as a measure of neurodegeneration for biomarker profiling or monitoring treatment effects in clinical trials. Indeed, in a survey of interventional trials of AD drugs (with US Food and Drug Administration (FDA) phase, 3, phase 4, or approved status) listing biomarkers as primary or secondary endpoints, we found that structural or functional MRI was used in 61.8% (Tables 1 and 2; Supplementary Table 1). The use of MRI as endpoint in clinical trials has increased in recent years due to the need to monitor amyloid-related imaging abnormalities (ARIA), a common adverse event associated with amyloid-targeting immunotherapies (Fig. 1) [18].

Data obtained from information compiled in Supplementary Table 1. Figure created in GraphPad Prism version 10.2.3.
Neuroimaging biomarkers – PET
The development of amyloid PET tracers provided the AD field with a specific biomarker to detect and quantify brain beta-amyloidosis. One of the first amyloid-imaging PET tracers developed was 11C-labeled Pittsburgh Compound-B (PIB) [19]. Post-mortem studies demonstrated that, compared to controls, AD patients show elevated retention of PIB in cortical areas consistent with Aβ deposition patterns [19, 20]. Subsequent longitudinal studies have shown conflicting results, with some reporting a positive association between PIB retention and progression to AD in MCI patients, and others finding no differences in PIB retention between controls, MCI, and AD patients [21,22,23]. Three second-generation 18F-labeled amyloid tracers with longer half-lives have been FDA- and European Medicines Agency (EMA)-approved for clinical use: 18F-florbetapir, 18F-florbetaben, and 18F-flutemetamol [24]. These tracers correlate with brain amyloid burden and successfully predict AD progression in patients with MCI [25,26,27,28,29].
Much progress has been made in the development and application of tau PET tracers. Tau imaging may be a stronger predictor of cognitive dysfunction than Aβ imaging, especially in normal cognition and amyloid-positive MCI [30,31,32]. In 2020, the FDA approved the first PET tracer, 18F-Flortaucipir, for imaging tau in cognitively impaired adults [33]. 18F-Flortaucipir-PET may predict longitudinal changes in cognitive impairment more strongly than MRI or amyloid PET [32]. While this tracer overcomes some limitations of other first-generation tau tracers, such as nonspecific white matter binding and high bone uptake, it still faces problems with off-target binding and detecting preclinical AD [33]. Second-generation tau PET tracers (e.g., PI-2620, MK-6240, and RO-948) are superior in terms of off-target binding but are still in the process of achieving clinical validity and utility [34, 35].
Despite advances in PET tracers, PET remains very expensive and is not part of the routine clinical assessment of AD in most of the world. Additionally, amyloid PET cannot differentiate AD from other amyloid-positive diseases such as Lewy body dementia [36]. The half-lives of PET tracers are relatively short (e.g., ~20 min for 11C and 110 min for 18F) so they must be produced and used in the same facility [24]. Nonetheless, PET is commonly used as an endpoint in 36.8% of AD interventional trials that list fluid or neuroimaging biomarkers as primary or secondary outcomes (Tables 1 and 2; Supplementary Table 1). PET is also the most frequently used endpoint in trials of amyloid immunotherapies (Fig. 2).

Data obtained from information compiled in Supplementary Table 1. Figure created in GraphPad Prism version 10.2.3.
Fluid-based biomarkers
The core AD biomarkers that can be detected in biofluids include those in the Aβ (e.g., Aβ42/Aβ40) and tau (e.g., phosphorylated tau (p-tau)) categories. Non-core biomarkers include those belonging to the categories of neurodegeneration (e.g., neurofilament light chain (NfL)) and inflammation (e.g., glial fibrillary acidic protein (GFAP)). The core fluid biomarkers can be used to diagnose, stage, and monitor AD, while non-core biomarkers can complement disease staging and monitoring or identify co-pathologies [6]. Given the unspecific nature and broader applicability of biomarkers such as NfL and GFAP [37, 38], our focus here will be on fluid Aβ- and tau-related biomarkers.
Of the two main fluid sources of biomarkers for AD, CSF and blood, CSF offers the advantage of having direct contact with the brain and thus being enriched in central nervous system (CNS)-specific proteins. Standard immunochemical assays are sensitive enough and commonly used to quantify Aβ42, total tau (t-tau), and p-tau in the CSF. To date, the Elecsys® (Roche) CSF tests for Aβ42, p-tau181, and t-tau and the Lumipulse® (Fujirebio) CSF test for Aβ42/Aβ40 have been approved by the FDA for diagnostic use. Biomarker ratios measured by both tests show similarly high concordance with amyloid PET status and clinical diagnosis [39, 40]. Moreover, CSF biomarkers are used in 32.4% of AD interventional trials that list fluid or neuroimaging biomarkers as endpoints (Tables 1 and 2; Supplementary Table 1).
Detecting CNS-derived biomarkers in the blood is far more challenging. Due to the blood-brain barrier (BBB) and the high blood to CSF volume ratio, their concentration in the periphery is usually a small fraction of what can be measured in the CSF. Advancements in ultrasensitive immunoassay technologies have only recently allowed for the reliable detection of CNS-derived proteins in the blood. One such technology is the Single Molecule Array (SiMoA®), an automated bead-based enzyme-linked immunosorbent assay (ELISA) with an innovative detection step. Using femtoliter wells to confine individual beads, it can detect signals produced by a single captured molecule of the analyte, increasing sensitivity dramatically in well-optimized assays. SiMoA can detect Aβ42, Aβ40, p-tau, and t-tau in the blood at sub-femtomolar concentrations [41, 42]. Numerous assays developed on the SiMoA platform, such as the ALZpath, Eli Lilly, and Janssen SiMoA kits for plasma p-tau217 and the ADx Neuroscience SiMoA kit for plasma p-tau181 have shown excellent performance detecting amyloid positivity [43, 44]. Several other types of immunoassays currently in use also rely on ultrasensitive detection steps, including those based on chemiluminescence (Fujirebio Lumipulse®; Siemens ADVIA®) or electrochemiluminescence (ECLIA; Roche Elecsys®; Meso Scale Discovery). In parallel, mass spectrometry (MS)-based techniques have also achieved detection of BBMs with high diagnostic accuracy. MS typically requires much more complex sample processing, but it does not rely on an antibody or enzymatic reaction for its detection step. For certain analytes, particularly plasma Aβ42/Aβ40, immunoprecipitation coupled to MS (IP-MS) has shown better performance than immunoassays for detecting brain Aβ burden [45]. To facilitate inter-assay and inter-laboratory consistency, the Standardization of Alzheimer’s Blood Biomarkers group has developed a standard operating procedure for pre-analytical sample handling for some of the most common BBMs across several platforms [46].
A selection of assay kits currently capable of detecting AD-related biomarkers in peripheral blood, including SiMoA, IP-MS, ECLIA, Microfluidic ELISA, Single Molecule Counting®, and Chemiluminescence assays are summarized in Table 3. Some of the listed kits are already available on the diagnostics market as laboratory developed tests (LDTs). Of note, all were introduced before the US FDA’s final rule on LDTs (which significantly increases the agency’s oversight and quality system requirements for these tests) was published. In the following sections, we review the recent advancements that have been achieved in fluid biomarker detection with these and other technologies.
CSF biomarkers – Aβ
Aβ was first shown to be secreted into the CSF in 1992 [47]. Around the same time, the aggregation-prone, 42 amino acid form of Aβ (Aβ42) was determined to be the species of Aβ earliest deposited in plaques [48]. The CSF level of Aβ42 is thus reduced upon plaque formation and is a biomarker of a pathologic state associated with amyloid deposition. It has been suggested that the CSF concentration of Aβ42 begins to decline up to 25 years prior to the onset of AD [49]. Moreover, around 90% of patients with MCI and CSF Aβ42 positivity develop AD within 9–10 years, indicating CSF Aβ42 can predict disease progression [50]. A large meta-analysis by Olsson and colleagues—comprising almost 30,000 AD patients and controls—found a strong association between CSF Aβ42 and AD, reporting a CSF Aβ42 AD to control ratio of 0.56 [51].
The ratio of Aβ42 to Aβ40 (a less amyloidogenic, soluble isoform) is often used. CSF Aβ42/Aβ40 levels measured by immunoassays and IP-MS show concordance with amyloid deposition as determined by amyloid PET [52, 53]. In the Swedish BioFINDER cohort, the Aβ42/Aβ40 ratio better correlated with amyloid-PET and differentiated AD from other neurodegenerative disorders compared to Aβ42 alone [54]. Conversely, CSF Aβ42 alone may have superior prognostic ability than the Aβ42/Aβ40 ratio. In the China Cognition and Aging Study, a difference in CSF Aβ42 levels between AD patients and healthy controls appeared 18 years prior to diagnosis, compared to 14 years for CSF Aβ42/Aβ40 [55]. CSF p-tau/Aβ42, measured by the Elecsys® or Lumipulse® immunoassays, also shows high concordance with amyloid PET [56, 57].
Finally, while post-mortem neuropathology confirmation is rare in fluid biomarkers studies, it has been established for CSF Aβ42. In one such study, Strozyk and colleagues showed an association between lower CSF Aβ42 and increased plaque deposition in the neocortex and hippocampus, examining 155 autopsy samples [58].
CSF biomarkers – tau
Total tau levels in the CSF can be used to estimate the extent of neurodegeneration in AD. The first ELISA protocol for quantifying CSF t-tau was published in 1993 and reported significantly higher CSF t-tau levels in AD patients [59]. In fact, the ratio of CSF t-tau between AD and controls was 2.54 in the meta-analysis by Olsson and colleagues [51]. However, increases in CSF t-tau also occur in acute conditions such as stroke, brain injury, and in disorders without amyloid or tau pathology, such as Creutzfeldt-Jakob disease [60,61,62]. As more of a ‘state marker’ of neuronal injury, t-tau is less specific for AD than Aβ42 or p-tau, and thus is often evaluated within a ratio to other biomarkers. For instance, t-tau/Aβ42 in the CSF strongly correlates with amyloid PET [63, 64].
Since NFTs primarily consist of p-tau, CSF p-tau is consistently increased in AD [51]. Unlike t-tau, p-tau is specific to AD and other tauopathies and remains low in most non-AD neurodegenerative disorders [51, 62]. Many studies have shown CSF p-tau correlates strongly with the severity of NFTs, hippocampal volume, and cortical amyloid deposition [65,66,67].
The three main isoforms of p-tau measured in the context of AD are p-tau181, p-tau217, and p-tau231. Although p-tau181 is the most thoroughly examined and routinely used isoform, recent evidence suggests that CSF p-tau217 can differentiate AD from other neurodegenerative diseases and controls with higher sensitivity and specificity [68]. Further, CSF p-tau217 correlates more strongly with tau PET and longitudinal changes in p-tau217 are greater than p-tau181 [68]. While CSF p-tau217 has the largest fold-change between AD and non-AD, it has been suggested that p-tau231 attains abnormal levels earliest in the AD continuum [69]. Nonetheless, the performance of any analyte heavily depends on the quality of the antibody pair used in the immunoassay. In at least some cases, differences in diagnostic performance seen among analytes may be due to antibody quality rather than reflecting pathophysiological processes.
Assessing these CSF biomarkers in combination would further aid preclinical and differential diagnosis. For instance, the CSF p-tau/Aβ42 demonstrated a sensitivity of 88% and specificity of 100% in the differentiation of AD from other dementias, confirmed in post-mortem studies [70]. In the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort, p-tau/Aβ and t-tau/Aβ most accurately predicted clinical decline in MCI patients over 24 months [64].
CSF biomarkers – novel/atypical targets and limitations
CSF tau assays that measure p-tau205 and the microtubule binding region (MTBR) of tau have shown promising results in tracking tau pathology. The ratio of p-tau205/tau205 in the CSF, determined by IP-MS, more strongly correlates with tau PET than CSF p-tau181 [71]. CSF MTBR containing the residue 243 (MTBR-243), identified by IP-MS, strongly correlates with tau PET and longitudinal increases in insoluble tau [72]. Since these tau species appear later in the disease process (i.e., after p-tau181, 217, and 231), the 2024 AA guidelines categorize these biomarkers as Core 2, which may be useful for biological staging and inform the rate of disease progression [6].
In parallel to the core AD biomarkers, atypical targets and their relation to AD have long been investigated in the CSF. For instance, synaptic proteins are altered in the CSF of preclinical AD patients, preceding symptoms or CSF elevations in t-tau or p-tau [73]. Several groups have described altered levels of neurotransmitters, such as noradrenaline and dopamine, in the CSF of AD patients [74, 75]. Our group has previously demonstrated that the CSF level of irisin—an exercise-induced hormone—positively correlates with CSF Aβ42 and cognitive performance [76, 77]. Markers of glial activation and neuroinflammation in the CSF are also increased in AD and associate with CSF tau, cognitive dysfunction, and cortical thinning [78]. Investigation of biomarkers outside of the established AT(N) framework is crucial for the discovery of novel neuropathological processes and may be relevant in future multi-analyte algorithms to diagnose AD.
An important limitation to the use of CSF biomarkers in clinical practice is that it requires an invasive lumbar puncture, which may cause reluctance in physicians and patients [79, 80]. Common complications include back pain and headache, while rare complications include infections, cerebral hematoma, and cerebral venous thrombosis [81]. Furthermore, CSF acquisition is not a standard clinical procedure; it is restricted to specialized clinics and personnel, and obtaining ethical approval to collect CSF for research or clinical trials can be difficult. It is also important to note that CSF biomarkers reflect the rate of biomarker production and clearance at one-time point [5]. An altered CSF biomarker suggests a pathological state associated with amyloid deposition or NFT formation, unlike neuroimaging biomarkers which can directly measure amyloid or tau load [6].
Blood-based biomarkers – Aβ
Plasma Aβ42/Aβ40 levels decrease with advancing clinical stage and can be used to identify Aβ pathology early in the AD continuum [82]. As mentioned previously, IP-MS methods have so far performed better than immunoassays in predicting amyloid PET status [45, 83]. IP-MS measures of Aβ40/Aβ42 correlate with CSF Aβ42 and clinical staging and successfully predict amyloid PET status [84, 85]. Further, the risk of converting to amyloid PET positivity was 15-fold greater for individuals with abnormal Aβ42/Aβ40 levels [86]. The IP-MS-based PrecivityAD (C2N) test for Aβ42/Aβ40 was the first LDT for plasma AD biomarkers to reach the market in 2020. In 2022, the current version of this test, which is aided by age and ApoE proteotype determination (Table 3), predicted amyloid-PET status in cohort samples totaling 686 participants, with an AUC of 0.88 [87]. The inclusion of age and ApoE proteotype—two established risk factors for brain amyloidosis—improved the accuracy of plasma Aβ42/Aβ40 for identifying amyloid-PET status [88].
One major issue encountered with plasma Aβ42/Aβ40 is the small difference seen between Aβ-positive and Aβ-negative individuals. The Aβ42/Aβ40 ratio is reduced by only 8–15% in plasma compared to 40–60% in CSF due to peripheral expression of Aβ [89]. Although less reliant on antibody performance, IP-MS is also subject to the pre-analytical variables that affect immunoassays in general and, as highlighted by data from the Alzheimer’s Association quality control program, the inter-laboratory variability for AD biomarker assays can be quite high (20–30%, published CSF data), especially for Aβ [90]. The small effect size that must be measured in plasma Aβ assays, together with pre-analytical factors known to disproportionally impact Aβ (such as analyte stability and surface adsorption) make the performance of plasma Aβ42/Aβ40 more so impacted by intra- and inter-assay variation than other plasma biomarkers [91]. Plasma Aβ42/Aβ40 may thus have additional hurdles to overcome before widespread adoption. The search for CNS-specific forms of Aβ in the plasma, such as by isolating neuronal-derived vesicles (discussed below), may help overcome the biological factors that currently limit the robustness of Aβ as a BBM for AD.
Blood-based biomarkers – tau
Plasma p-tau has emerged as a promising biomarker for AD that may reflect both Aβ and tau pathology [92]. Due to limitations with detecting Aβ mentioned above, many non-MS-based tests have focused on p-tau, particularly p-tau181, p-tau217, and p-tau231. Available p-tau immunoassays use antibodies against phosphorylated sites on the N-terminal or mid-domain of tau, as these fragments are more soluble and more frequently secreted compared to aggregation-prone C-terminal tau [93].
Plasma p-tau181 repeatedly correlates with tau and amyloid PET and CSF p-tau181 [94,95,96,97]. This biomarker is also increased in preclinical AD stages and further increases with advancing clinical stage [94,95,96,97]. Plasma p-tau181 can differentiate AD from cognitively unimpaired older adults, MCI, and non-AD neurodegenerative diseases [94,95,96, 98, 99]. Higher baseline concentrations of plasma p-tau181 are associated with future development of AD pathology in individuals with normal cognition and MCI [94, 96, 98, 100]. Notably, plasma p-tau181 can predict conversion to AD up to eight years prior to death and, when combined with Aβ42 and NfL, can predict AD eight years prior to clinical onset [98, 101]. In a large clinical-based cohort, higher baseline levels of blood p-tau181, quantified using SiMoA, were associated with an accelerated time to AD onset [100]. Blood p-tau181 was also a better predictor of 5-year AD risk than blood Aβ42/Aβ40, t-tau, and NfL [100]. Furthermore, longitudinal measurements have shown low intra-individual variability in plasma p-tau181, indicating that it may be useful in measuring treatment responses [102].
Plasma p-tau217 can also track CNS changes across the AD continuum and may outperform plasma p-tau181 in several analyses. In a head-to-head comparison of 10 plasma p-tau assays, IP-MS for p-tau217 performed better than all other plasma p-tau assays for predicting Aβ status and progression from MCI to AD [103]. The immunoassays for p-tau217 (Janssen and Eli Lilly) and p-tau181 (ADx Neuroscience, Washington University) also performed well for both outcomes [103], and a novel ECL-based plasma p-tau217 assay developed by Meso Scale Discovery (MSD) outperformed p-tau181 for differentiating AD and controls [104]. Notably, the fold change in plasma p-tau217 between AD and controls is consistently higher than in plasma p-tau181 [104,105,106]. Plasma p-tau217 also correlates with amyloid and tau PET, CSF and plasma p-tau181, CSF p-tau 217, and with amyloid plaques and tau-containing NFTs in studies of autopsy-confirmed AD [105, 107,108,109]. Amyloid PET positive, tau PET negative individuals can be differentiated from controls with plasma p-tau217 levels, suggesting that plasma p-tau217 may change before tau aggregation is detectable by PET [110]. In fact, plasma %p-tau217 (p-tau217/non-phosphorylated tau) analyzed by MS was equivalent to FDA-approved CSF tests in determining amyloid PET and superior in determining tau PET [111]. Moreover, individuals with MCI who progressed to AD dementia up to six years later had higher baseline levels of plasma p-tau217 than non-converters [112]. Plasma p-tau217 not only performs well in clinical staging, but it may outperform plasma p-tau 181 in differentiating AD from normal cognition and other neurodegenerative disorders [106, 107, 112].
Plasma p-tau231 has gained traction as a biomarker for AD pathology, showing a correlation with CSF p-tau231, tau PET, and amyloid PET [113]. Plasma p-tau231 measured by Quanterix’s SiMoA can distinguish AD from non-AD neurodegenerative disorders, amyloid PET negative controls, and amyloid PET negative MCI [113]. However, plasma p-tau231 detected by ADx Neuroscience’s SiMoA kit could not identify AD or asymptomatic amyloid PET positivity [114]. Furthermore, plasma p-tau231 did not outperform p-tau181 in differentiating AD from non-AD neurodegenerative disorders and performs inferiorly to CSF p-tau 231 in detecting amyloid PET positivity [113, 115]. While plasma p-tau231 and p-tau217 are similarly associated with longitudinal changes in amyloid PET, concentrations of plasma p-tau231 may increase earlier than amyloid PET thresholds are reached or increases in plasma p-tau181 are detected [113, 116]. Despite these promising results, longitudinal measures of plasma p-tau231 failed to predict cognitive decline in preclinical AD [117]. While plasma p-tau231 may reach a significant threshold early in AD pathology, succeeding cognitive decline may be better indicated by other p-tau isoforms. Plasma p-tau231 may detect AD pathology before amyloid or tau PET, but larger-scale longitudinal studies and assay standardization are warranted.
Since the majority of plasma total-tau originates from peripheral sources, higher CNS t-tau in AD is unlikely to cause significant increases in plasma t-tau [105]. Recently, an antibody that selectively binds brain-derived tau (TauJ.5H3) was developed [118]. Initial studies report that blood-based brain-derived tau has similar diagnostic performance to CSF t-tau in distinguishing AD from controls and other neurodegenerative disorders, and shows weaker correlations with age, comorbidities, and race/ethnicity than other BBMs [118, 119]. Another promising avenue for detecting CNS-specific t-tau in the periphery is through the isolation of neuronal-derived vesicles from the blood (discussed below).
Use of BBMs in clinical practice and interventional trials
BBMs have potential utility for early and accurate AD diagnosis, monitoring of disease progression and treatment effects, and screening for clinical trial eligibility. Compared to neuroimaging and CSF biomarkers, BBMs are minimally invasive, cost-effective, highly scalable, and available outside specialized centers and in low-resource settings [120, 121]. In clinical practice, BBMs have the potential to address the high rates of misdiagnosis and underdiagnosis in AD caused by current testing limitations, which in turn will allow for the optimization of therapeutic intervention [120] In remote settings without access to blood-collection facilities, AD biomarkers may even be quantified by SiMoA in dried blood spots [122].
BBMs can also improve the design of clinical trials investigating AD drugs. PET and CSF biomarkers are commonly used to screen and recruit large numbers of individuals for trials, in a costly, time-consuming process, where patients with or without AD-specific pathology are selected. Karikari et al. estimate cost-savings of 58% in the recruitment phase of AD clinical trials by pre-screening participants with a blood p-tau test prior to PET scans compared to screening all participants with a PET scan alone [93].
In the past 5 years, the use of BBMs as endpoints in AD interventional trials has increased substantially (Fig. 1). BBMs are included in 26.5% of trials that list biomarkers as primary or secondary endpoints (Tables 1 and 2; Supplementary Table 1) and most frequently in trials of small molecule therapeutics (e.g., Simufilam, Suvorexant, AR1001) (Fig. 2).
Detecting amyloidosis with BBMs will be critical for the success of anti-Aβ immunotherapies (Table 2), as amyloid-positive individuals, who stand to benefit from anti-Aβ treatment, must be selected and monitored [6]. Furthermore, it is important to recruit pre-amyloid individuals (e.g., with plasma p-tau231) so disease-modifying therapies can be initiated before irreversible downstream pathology occurs [113]. Given the importance of tracking Aβ in response to Aβ immunotherapy and the usefulness of BBMs for this purpose, it was notable to find that, among trials for anti-Aβ immunotherapies, only those for Solanezumab included BBMs as primary or secondary endpoints (Table 2). While virtually all large-scale AD immunotherapy trials in recent years have measured and published data on BBMs, few have listed them as endpoints. PET and CSF biomarkers are often prioritized, despite their higher cost and risks, with BBMs being used only for exploratory analyses. This is an interesting reflection of each sponsor’s confidence in BBMs at the time of study design and is likely to shift as the biomarker field progresses.
Interpretation of BBMs
Numerous factors, such as age, genotype, and sex are known to have an influence on AD pathology and biomarker levels and should be considered when interpreting BBM data [88, 123]. As BBMs are incorporated in clinical settings, diagnostic workups and cutoff values can be expected to be refined based on demographic factors and genotype (e.g., C2N’s plasma Aβ42/Aβ40 tests incorporate age and ApoE proteotype). Additionally, certain medical conditions such as previous stroke, diabetes, high body mass index (BMI), dyslipidemia, and chronic kidney disease (CKD) have already reported to influence BBM levels [124,125,126,127]. In community-based samples, CKD was associated with higher levels of plasma p-tau181, p-tau217, Aβ40, Aβ42, NfL, and total tau [124,125,126,127]. While this particular effect may be caused by altered clearance of plasma proteins, others may be a result of direct influence on AD pathology, which is not yet completely understood. The exact extent to which comorbidities can impact the reliability of reference values and the interpretation of BBM data still needs to be determined. In a recent report, Mielke and colleagues found that excluding individuals with either CKD, myocardial infarction or stroke significantly impacted the determination of a normal range for plasma p-tau in community-based samples [124]. In contrast, using cohort samples, other authors found that creatinine (as a proxy for kidney function) and BMI were associated with BBM levels, but did not alter their interpretation in a meaningful way [128].
The impact of population diversity, socioeconomic disparities, and lifestyle on biomarker levels and their interpretation have also only begun to be investigated. Multi-ethnic American studies show conflicting results on the impacts of race/ethnicity on BBM levels [108, 125, 126, 129]. While Mohs et al. found different BBM levels among non-Hispanic Blacks, non-Hispanic Whites, and Hispanics, race/ethnicity did not impact the ability of BBMs to predict amyloid PET positivity [129]. In Chinese cohorts, large studies have confirmed the excellent diagnostic performance of BBMs seen in Western populations, despite differences in APOE4 prevalence, diet, and lifestyle [101, 130]. Contrarily, little data is available on AD biomarkers in developing countries where large longitudinal cohorts are less common. In one notable study including 746 participants of Caribbean-Hispanic ethnicity, 91% of them from the Dominican Republic, authors distinguished AD from cognitively normal controls using plasma p-tau181 with a performance similar to that shown for US- and Europe-based cohorts [131]. While work is still ongoing to locally validate BBMs and account for possible confounding factors, the conservative use of cutoff values advised by AA guidelines, with the inclusion of an indeterminate zone, may be crucial [6].
Neuronal derived EVs as a source of BBMs
Extracellular vesicles (EVs) are cell-derived membranous structures that vary in their biogenesis, release, composition, and interaction with cells. Numerous physiological roles have been proposed for EVs, particularly as a mode of intercellular communication. Regardless of their physiological relevance, EVs hold promise as a source of biomarkers. EVs carry a sample of the proteins, lipids, and nucleic acids found in their cell of origin [132]. Neuronal-derived EVs (NEVs) may carry membrane proteins specific to neurons, allowing for their identification and immunoprecipitation, as well as AD-specific biomarkers.
Evidence suggests that NEVs can cross the BBB and accumulate in the blood and that their concentration and cargo reflect the pathophysiological changes in AD [133, 134]. Varied levels of Aβ, tau, synaptic proteins, insulin resistance-associated proteins, and RNAs have been detected in NEVs isolated from the plasma of AD patients compared to MCI or healthy controls [135,136,137,138]. Furthermore, there are myriad targets that show potential as AD biomarkers in the CSF (e.g. irisin) but are not neuron-specific and thus not relevant when measured in whole plasma. However, the isolation of NEVs from the blood may allow for the detection of CNS-specific changes of non-CNS specific biomarkers.
Isolating NEVs
Several methods for isolating EVs exist, differing in specificity, yield, and ease of use. Ultracentrifugation (UC) was the first to be described and has been considered the gold standard method of EV isolation, but several alternative methods have attracted interest and investment in recent years [134]. UC results in high purity, especially when combined with a density gradient (dgUC), but is time-consuming, requires expensive equipment and large volumes of starting material, and has low recovery [134, 139]. Ultrafiltration (UF) and size-exclusion chromatography (SEC) separate samples according to their size with high ease of use but also low recovery [134, 139]. Polymer-based precipitation can isolate EVs using water-excluding polymers, such as polyethylene glycol (PEG), which precipitate the less soluble EVs [134, 139]. Precipitation is an easy, high-yield method with commercially available kits such as ExoQuick ULTRA or miRCURY [134, 139]. The main disadvantages are the co-precipitation of blood proteins, which reduces purity, and the interference from polymers in downstream assays [139]. Generally, a combination of isolation methods is used to improve EV purity and yield.
Importantly, EV isolation based on physical properties alone cannot differentiate NEVs from peripherally derived EVs. Combining these methods with immunoaffinity isolation targeting EV-specific surface proteins is the only technique devised thus far to obtain NEVs. A common protocol to isolate NEVS involves an antibody targeting the L1 cell adhesion molecule (L1-CAM), a neuronal protein that is sorted to EVs [140]. This method has been used to isolate NEVs containing Aβ42, t-tau, p-tau181, NfL, and synaptic proteins [141]. Although L1-CAM is expressed by neurons, it is also expressed by peripheral tissues, raising important concerns about the specificity of NEVs obtained through L1-CAM IP. Notably, in a 2021 report, authors were unable to find L1-CAM immunoreactivity in EV-enriched SEC fractions isolated from human plasma, despite using an ultrasensitive SiMoA assay as readout [142].
In a continued effort to find targets for isolating NEVs, the ATPase Na + /K+ transporting subunit alpha 3 (ATP1A3) was recently tested, showing promising results [143]. ATP1A3 is a neuron-enriched protein found in EVs isolated from human brain tissue and plasma. Although ATP1A3 expression patterns (as reported by proteinatlas.org) suggest that IP using ATP1A3 might show much higher neuronal specificity than L1-CAM, ATP1A3 expression is also not exclusive to neurons, allowing for the possibility of contamination from peripheral EVs.
Further, low recovery is a common problem with EV IP techniques. In addition to partial or inconsistent elution of EVs from beads, the low concentration of NEVs in human plasma—evidenced by the very low starting concentration of neuron-specific proteins in whole blood—makes isolating useful amounts of pure NEVs challenging [139]. Likewise, detecting protein targets in pure NEV samples can be challenging even for current ultrasensitive methods. Many authors—such as You et al. when describing the novel ATP1A3 method—have resorted to single-event techniques to detect biomarkers in plasma NEVs (e.g., fluorescent nanoparticle tracking analysis, nanoparticle flow cytometry, single-EV visualization by dSTORM) [143]. These, however, are often semi-quantitative in nature and are much further from diagnostic implementation than traditional immunoassays or IP-MS.
More recently, affinity-based proteomic techniques (e.g. Olink, NULISA), which are based on oligonucleotide-conjugated antibodies quantified via polymerase chain reaction or next-generation sequencing, have allowed for a combination of attomolar sensitivity and very high multiplexing (Table 3) [144, 145]. Although the field has so far focused primarily on high-sensitivity, single-target assays, these novel technologies may open to door to multi-analyte biomarker discovery, particularly in samples such as NEVs.
AD-related biomarkers in NEVs
Several studies have used ExoQuick followed by L1-CAM IP to enrich NEVs from plasma and subsequently quantify their AD-related biomarker content [146,147,148,149]. Levels of Aβ42, t-tau, and p-tau181 in plasma NEVs were shown to increase with advancing clinical stage and to correlate with CSF biomarkers [146,147,148,149]. Fiandaca and colleagues found that NEV Aβ42, t-tau, and p-tau181 could predict the development of AD up to 10 years prior to clinical diagnosis [146]. Similarly, Winston and colleagues report that NEV Aβ42 and p-tau181 could predict patient conversion from MCI to AD in the following three years [147].
As mentioned above, the recently described ATP1A3 NEVs also show potential as sources of AD biomarkers. By detecting pan-Aβ immunoreactivity in immobilized NEVs, authors could discern AD patients from MCI and controls in a total of 30 cohort samples [143]. These results indicate that core AD biomarkers found in blood-derived NEVs may be of diagnostic and prognostic value. Nonetheless, these studies are limited in size and standardized methods to isolate NEVs have not been established.
NEVs may also carry non-core AD biomarkers. Markers of insulin signaling (e.g., p-Ser IRS-1, p-Tyr IRS-1) are altered in NEVs of AD patients compared to controls [150]. These differences are identifiable up to 10 years before the clinical onset of AD [150]. Additionally, NEV levels of synaptic proteins, such as synaptotagmin, synaptophysin, and neurogranin, may be reduced years before AD onset [136]. Differential expression of micro-RNAs in NEVs has distinguished patients with AD from those with stable MCI, MCI that progresses to AD, and other neurodegenerative disorders [138, 151]. Furthermore, a transcriptomic analysis of EVs from post-mortem brain tissue identified an enrichment of inflammation-associated mRNAs and depletion of synaptic signaling mRNAs in AD compared to healthy controls [152]. It is to be expected that novel NEV biomarkers will be discovered in the coming years as the field continues to develop.
While NEVs are an exciting avenue in the field of BBMs of AD, they are in their infancy compared to the clinically validated core AD biomarkers. The current lack of consensus and standardized protocols regarding NEV isolation methods prevents NEVs from living up to their potential as a source of AD-related biomarkers. Establishing reliable and reproducible NEV isolation and detection methods will encourage the discovery of novel targets and improve our ability to measure CNS-specific changes in the blood.
Conclusion
The field of AD biomarkers has developed rapidly in the last decade, closely following technological advancements. The emergence and optimization of ultrasensitive immunoassays and MS techniques are now very close to establishing an equivalence between CSF and blood biomarkers. In turn, core AD biomarkers quantified in these biofluids have reached diagnostic and prognostic accuracies similar to those of PET. Widespread adoption of blood as a source of core AD biomarkers will benefit both clinical practice and interventional trials, due to the technical and financial ease of sampling blood over CSF. Most recently, NEVs isolated from the blood have shown promising results for investigating core and atypical AD biomarkers. Not unlike what happened for BBMs over the past decade, an effort for standardizing NEV protocols is warranted before NEVs can be seen as a robust and reliable source of AD biomarkers.
References
Kumar A, Singh A, Ekavali. A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep. 2015;67:195–203.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc. 2011;7:280–92.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–9.
Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA‐AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–62.
Jack Jr. CR, Andrews JS, Beach TG, Buracchio T, Dunn B, et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimers Dement. 2024;20:5143–69.
McDonald CR, McEvoy LK, Gharapetian L, Fennema-Notestine C, Hagler DJ, Holland D, et al. Regional rates of neocortical atrophy from normal aging to early Alzheimer disease. Neurology. 2009;73:457–65.
Scahill RI, Schott JM, Stevens JM, Rossor MN, Fox NC. Mapping the evolution of regional atrophy in Alzheimer’s disease: unbiased analysis of fluid-registered serial MRI. Proc Natl Acad Sci USA. 2002;99:4703–7.
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services task force on Alzheimer’s disease. Neurology. 1984;34:939–44.
Bobinski M, de Leon MJ, Wegiel J, DeSanti S, Convit A, Saint Louis LA, et al. The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer’s disease. Neuroscience. 1999;95:721–5.
Dickerson BC, Fenstermacher E, Salat DH, Wolk DA, Maguire RP, Desikan R, et al. Detection of cortical thickness correlates of cognitive performance: reliability across MRI scan sessions, scanners, and field strengths. NeuroImage. 2008;39:10–18.
Killiany RJ, Gomez-Isla T, Moss M, Kikinis R, Sandor T, Jolesz F, et al. Use of structural magnetic resonance imaging to predict who will get Alzheimer’s disease. Ann Neurol. 2000;47:430–9.
Lee S, Lee H, Kim KW. Magnetic resonance imaging texture predicts progression to dementia due to Alzheimer disease earlier than hippocampal volume. J Psychiatry Neurosci. 2020;45:7–14.
Costafreda SG, Dinov ID, Tu Z, Shi Y, Liu C-Y, Kloszewska I, et al. Automated hippocampal shape analysis predicts the onset of dementia in mild cognitive impairment. NeuroImage. 2011;56:212–9.
Sabuncu MR, Desikan RS, Sepulcre J, Yeo BTT, Liu H, Schmansky NJ, et al. The dynamics of cortical and hippocampal atrophy in Alzheimer disease. Arch Neurol. 2011;68:1040–8.
Jack CR, Shiung MM, Gunter JL, O’Brien PC, Weigand SD, Knopman DS, et al. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology. 2004;62:591–600.
O’Brien JT, Paling S, Barber R, Williams ED, Ballard C, McKeith IG, et al. Progressive brain atrophy on serial MRI in dementia with Lewy bodies, AD, and vascular dementia. Neurology. 2001;56:1386–8.
Sperling RA, Jack CR, Black SE, Frosch MP, Greenberg SM, Hyman BT, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367–85.
Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol. 2004;55:306–19.
Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–45.
Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2008;29:1456–65.
Okello A, Koivunen J, Edison P, Archer HA, Turkheimer FE, Någren K, et al. Conversion of amyloid positive and negative MCI to AD over 3 years: An 11C-PIB PET study. Neurology. 2009;73:754–60.
Jack CR Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132:1355–65.
Laforce R Jr, Soucy J-P, Sellami L, Dallaire-Théroux C, Brunet F, Bergeron D, et al. Molecular imaging in dementia: past, present, and future. Alzheimers Dement. 2018;14:1522–52.
Doraiswamy PM, Sperling RA, Johnson K, Reiman EM, Wong TZ, Sabbagh MN, et al. Florbetapir F 18 amyloid PET and 36-month cognitive decline: a prospective multicenter study. Mol Psychiatry. 2014;19:1044–51.
Ong KT, Villemagne VL, Bahar-Fuchs A, Lamb F, Langdon N, Catafau AM, et al. Aβ imaging with 18F-florbetaben in prodromal Alzheimer’s disease: a prospective outcome study. J Neurol Neurosurg Psychiatry. 2015;86:431–6.
Vandenberghe R, Van Laere K, Ivanoiu A, Salmon E, Bastin C, Triau E, et al. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann Neurol. 2010;68:319–29.
Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al. Use of florbetapir-PET for imaging β-amyloid pathology. JAMA. 2011;305:275–83.
Sabri O, Sabbagh MN, Seibyl J, Barthel H, Akatsu H, Ouchi Y, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: phase 3 study. Alzheimers Dement. 2015;11:964–74.
Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kövari E, Perl DP, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60:1495–1500.
Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med. 2016;8:338ra66.
Ossenkoppele R, Smith R, Mattsson-Carlgren N, Groot C, Leuzy A, Strandberg O, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a head-to-head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78:961–71.
Jie CVML, Treyer V, Schibli R, Mu L. TauvidTM: the first FDA-approved PET tracer for imaging tau pathology in Alzheimer’s disease. Pharmaceuticals. 2021;14:110.
Bischof GN, Dodich A, Boccardi M, van Eimeren T, Festari C, Barthel H, et al. Clinical validity of second-generation tau PET tracers as biomarkers for Alzheimer’s disease in the context of a structured 5-phase development framework. Eur J Nucl Med Mol Imaging. 2021;48:2110–20.
Malarte M-L, Gillberg P-G, Kumar A, Bogdanovic N, Lemoine L, Nordberg A. Discriminative binding of tau PET tracers PI2620, MK6240 and RO948 in Alzheimer’s disease, corticobasal degeneration and progressive supranuclear palsy brains. Mol Psychiatry. 2023;28:1272–83.
Edison P, Rowe CC, Rinne JO, Ng S, Ahmed I, Kemppainen N, et al. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry. 2008;79:1331–8.
Abdelhak A, Foschi M, Abu-Rumeileh S, Yue JK, D’Anna L, Huss A, et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat Rev Neurol. 2022;18:158–72.
Bridel C, van Wieringen WN, Zetterberg H, Tijms BM, Teunissen CE, the NFL Group Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta-analysis. JAMA Neurol. 2019;76:1035–48.
Dakterzada F, López-Ortega R, Arias A, Riba-Llena I, Ruiz-Julián M, Huerto R, et al. Assessment of the concordance and diagnostic accuracy between elecsys and lumipulse fully automated platforms and innotest. Front Aging Neurosci. 2021;13:604119.
Willemse EAJ, Tijms BM, van Berckel BNM, Le Bastard N, van der Flier WM, Scheltens P, et al. Comparing CSF amyloid-beta biomarker ratios for two automated immunoassays, Elecsys and Lumipulse, with amyloid PET status. Alzheimers Dement Diagn Assess Dis Monit. 2021;13:e12182.
Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol. 2010;28:595–9.
Li D, Mielke MM. An update on blood-based markers of Alzheimer’s disease using the SiMoA platform. Neurol Ther. 2019;8:73–82.
Therriault J, Ashton NJ, Pola I, Triana-Baltzer G, Brum WS, Di Molfetta G, et al. Comparison of two plasma p-tau217 assays to detect and monitor Alzheimer’s pathology. eBioMedicine. 2024;102:105046.
Ashton NJ, Puig-Pijoan A, Milà-Alomà M, Fernández-Lebrero A, García-Escobar G, González-Ortiz F, et al. Plasma and CSF biomarkers in a memory clinic: head-to-head comparison of phosphorylated tau immunoassays. Alzheimers Dement. 2023;19:1913–24.
Janelidze S, Teunissen CE, Zetterberg H, Allué JA, Sarasa L, Eichenlaub U, et al. Head-to-head comparison of 8 plasma amyloid-β 42/40 assays in Alzheimer disease. JAMA Neurol. 2021;78:1375–82.
Verberk IMW, Misdorp EO, Koelewijn J, Ball AJ, Blennow K, Dage JL, et al. Characterization of pre-analytical sample handling effects on a panel of Alzheimer’s disease–related blood-based biomarkers: results from the standardization of Alzheimer’s Blood Biomarkers (SABB) working group. Alzheimers Dement. 2022;18:1484–97.
Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, et al. Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature. 1992;359:325–7.
Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron. 1994;13:45–53.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608.
Buchhave P, Minthon L, Zetterberg H, Wallin ÅK, Blennow K, Hansson O. Cerebrospinal fluid levels ofβ-Amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. 2012;69:98–106.
Olsson B, Lautner R, Andreasson U, Öhrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15:673–84.
Doecke JD, Ward L, Burnham SC, Villemagne VL, Li Q-X, Collins S, et al. Elecsys CSF biomarker immunoassays demonstrate concordance with amyloid-PET imaging. Alzheimers Res Ther. 2020;12:36.
Wisch JK, Gordon BA, Boerwinkle AH, Luckett PH, Bollinger JG, Ovod V, et al. Predicting continuous amyloid PET values with CSF and plasma Aβ42/Aβ40. Alzheimers Dement Diagn Assess Dis Monit. 2023;15:e12405.
Janelidze S, Zetterberg H, Mattsson N, Palmqvist S, Vanderstichele H, Lindberg O, et al. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: better diagnostic markers of Alzheimer disease. Ann Clin Transl Neurol. 2016;3:154–65.
Jia J, Ning Y, Chen M, Wang S, Yang H, Li F, et al. Biomarker changes during 20 years preceding Alzheimer’s disease. N Engl J Med. 2024;390:712–22.
Schindler SE, Gray JD, Gordon BA, Xiong C, Batrla-Utermann R, Quan M, et al. Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimers Dement. 2018;14:1460–9.
Campbell MR, Ashrafzadeh-Kian S, Petersen RC, Mielke MM, Syrjanen JA, van Harten AC, et al. P-tau/Aβ42 and Aβ42/40 ratios in CSF are equally predictive of amyloid PET status. Alzheimers Dement Diagn Assess Dis Monit. 2021;13:e12190.
Strozyk D, Blennow K, White LR, Launer LJ. CSF Aβ 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology. 2003;60:652–6.
Vandermeeren M, Mercken M, Vanmechelen E, Six J, Van de Voorde A, Martin J-J, et al. Detection of proteins in normal and Alzheimer’s disease cerebrospinal fluid with a sensitive sandwich enzyme-linked immunosorbent assay. J Neurochem. 1993;61:1828–34.
Hesse C, Rosengren L, Vanmechelen E, Vanderstichele H, Jensen C, Davidsson P, et al. Cerebrospinal fluid markers for Alzheimer’s disease evaluated after acute ischemic stroke. J Alzheimers Dis. 2000;2:199–206.
Ost M, Nylén K, Csajbok L, Ohrfelt AO, Tullberg M, Wikkelsö C, et al. Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology. 2006;67:1600–4.
Skillbäck T, Rosén C, Asztely F, Mattsson N, Blennow K, Zetterberg H. Diagnostic performance of cerebrospinal fluid total tau and phosphorylated tau in Creutzfeldt-Jakob disease: results from the Swedish Mortality Registry. JAMA Neurol. 2014;71:476–83.
Janelidze S, Pannee J, Mikulskis A, Chiao P, Zetterberg H, Blennow K, et al. Concordance between different amyloid immunoassays and visual amyloid positron emission tomographic assessment. JAMA Neurol. 2017;74:1492–501.
Hansson O, Seibyl J, Stomrud E, Zetterberg H, Trojanowski JQ, Bittner T, et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14:1470–81.
Tapiola T, Alafuzoff I, Herukka S-K, Parkkinen L, Hartikainen P, Soininen H, et al. Cerebrospinal fluid β-Amyloid 42 and tau proteins as biomarkers of alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66:382–9.
Buerger K, Ewers M, Pirttilä T, Zinkowski R, Alafuzoff I, Teipel SJ, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain. 2006;129:3035–41.
de Souza LC, Chupin M, Lamari F, Jardel C, Leclercq D, Colliot O, et al. CSF tau markers are correlated with hippocampal volume in Alzheimer’s disease. Neurobiol Aging. 2012;33:1253–7.
Janelidze S, Stomrud E, Smith R, Palmqvist S, Mattsson N, Airey DC, et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat Commun. 2020;11:1683.
Ashton NJ, Benedet AL, Pascoal TA, Karikari TK, Lantero-Rodriguez J, Brum WS, et al. Cerebrospinal fluid p-tau231 as an early indicator of emerging pathology in Alzheimer’s disease. EBioMedicine. 2022;76:103836.
Seeburger JL, Holder DJ, Combrinck M, Joachim C, Laterza O, Tanen M, et al. Cerebrospinal fluid biomarkers distinguish postmortem-confirmed Alzheimer’s disease from other dementias and healthy controls in the OPTIMA cohort. J Alzheimers Dis. 2015;44:525–39.
Barthélemy NR, Saef B, Li Y, Gordon BA, He Y, Horie K, et al. CSF tau phosphorylation occupancies at T217 and T205 represent improved biomarkers of amyloid and tau pathology in Alzheimer’s disease. Nat Aging. 2023;3:391–401.
Horie K, Salvadó G, Barthélemy NR, Janelidze S, Li Y, He Y, et al. CSF MTBR-tau243 is a specific biomarker of tau tangle pathology in Alzheimer’s disease. Nat Med. 2023;29:1954–63.
Lleó A, Núñez-Llaves R, Alcolea D, Chiva C, Balateu-Paños D, Colom-Cadena M, et al. Changes in synaptic proteins precede neurodegeneration markers in preclinical Alzheimer’s disease cerebrospinal fluid. Mol Cell Proteomics. 2019;18:546–60.
Lourenco MV, Ribeiro FC, Santos LE, Beckman D, Melo HM, Sudo FK, et al. Cerebrospinal fluid neurotransmitters, cytokines, and chemokines in Alzheimer’s and Lewy body diseases. J Alzheimers Dis. 2021;82:1067–74.
Henjum K, Watne LO, Godang K, Halaas NB, Eldholm RS, Blennow K, et al. Cerebrospinal fluid catecholamines in Alzheimer’s disease patients with and without biological disease. Transl Psychiatry. 2022;12:151.
Lourenco MV, Ribeiro FC, Sudo FK, Drummond C, Assunção N, Vanderborght B, et al. Cerebrospinal fluid irisin correlates with amyloid‐β, BDNF, and cognition in Alzheimer’s disease. Alzheimers Dement Diagn Assess Dis Monit. 2020;12:e12034.
Lourenco MV, Frozza RL, de Freitas GB, Zhang H, Kincheski GC, Ribeiro FC, et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat Med. 2019;25:165–75.
Janelidze S, Mattsson N, Stomrud E, Lindberg O, Palmqvist S, Zetterberg H, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91:e867–77.
Blazel MM, Lazar KK, Van Hulle CA, Ma Y, Cole A, Spalitta A, et al. Factors associated with lumbar puncture participation in Alzheimer’s disease research. J Alzheimers Dis. 2020;77:1559–67.
Judge D, Roberts J, Khandker RK, Ambegaonkar B, Black CM. Physician practice patterns associated with diagnostic evaluation of patients with suspected mild cognitive impairment and Alzheimer’s disease. Int J Alzheimer’s Dis. 2019;2019:e4942562.
Engelborghs S, Niemantsverdriet E, Struyfs H, Blennow K, Brouns R, Comabella M, et al. Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimers Dement Diagn Assess Dis Monit. 2017;8:111–26.
Palmqvist S, Janelidze S, Stomrud E, Zetterberg H, Karl J, Zink K, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease-related β-amyloid status. JAMA Neurol. 2019;76:1060–9.
Keshavan A, Pannee J, Karikari TK, Rodriguez JL, Ashton NJ, Nicholas JM, et al. Population-based blood screening for preclinical Alzheimer’s disease in a British birth cohort at age 70. Brain. 2021;144:434–49.
Tosun D, Veitch D, Aisen P, Jack CR Jr, Jagust WJ, et al. Detection of β-amyloid positivity in Alzheimer’s disease neuroimaging initiative participants with demographics, cognition, MRI and plasma biomarkers. Brain Commun. 2021;3:fcab008.
Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature. 2018;554:249–54.
Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA, et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93:e1647–59.
Hu Y, Kirmess KM, Meyer MR, Rabinovici GD, Gatsonis C, Siegel BA, et al. Assessment of a plasma amyloid probability score to estimate amyloid positron emission tomography findings among adults with cognitive impairment. JAMA Netw Open. 2022;5:e228392.
Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–31.
Hansson O, Edelmayer RM, Boxer AL, Carrillo MC, Mielke MM, Rabinovici GD, et al. The Alzheimer’s association appropriate use recommendations for blood biomarkers in Alzheimer’s disease. Alzheimers Dement. 2022;18:2669–86.
Mattsson N, Andreasson U, Persson S, Carrillo MC, Collins S, Chalbot S, et al. CSF biomarker variability in the Alzheimer’s association quality control program. Alzheimers Dement. 2013;9:251–61.
Benedet AL, Brum WS, Hansson O, Karikari TK, Zimmer ER, Zetterberg H, et al. The accuracy and robustness of plasma biomarker models for amyloid PET positivity. Alzheimers Res Ther. 2022;14:26.
Mielke MM, Frank RD, Dage JL, Jeromin A, Ashton NJ, Blennow K, et al. Comparison of plasma phosphorylated tau species with amyloid and tau positron emission tomography, neurodegeneration, vascular pathology, and cognitive outcomes. JAMA Neurol. 2021;78:1108–17.
Karikari TK, Ashton NJ, Brinkmalm G, Brum WS, Benedet AL, Montoliu-Gaya L, et al. Blood phospho-tau in Alzheimer disease: analysis, interpretation, and clinical utility. Nat Rev Neurol. 2022;18:400–18.
Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26:379–86.
Thijssen EH, La Joie R, Wolf A, Strom A, Wang P, Iaccarino L, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med. 2020;26:387–97.
Shen X-N, Huang Y-Y, Chen S-D, Guo Y, Tan L, Dong Q, et al. Plasma phosphorylated-tau181 as a predictive biomarker for Alzheimer’s amyloid, tau and FDG PET status. Transl Psychiatry. 2021;11:1–10.
Martínez-Dubarbie F, Guerra-Ruiz A, López-García S, Lage C, Fernández-Matarrubia M, Infante J, et al. Accuracy of plasma Aβ40, Aβ42, and p-tau181 to detect CSF Alzheimer’s pathological changes in cognitively unimpaired subjects using the Lumipulse automated platform. Alzheimers Res Ther. 2023;15:163.
Lantero Rodriguez J, Karikari TK, Suárez-Calvet M, Troakes C, King A, Emersic A, et al. Plasma p-tau181 accurately predicts Alzheimer’s disease pathology at least 8 years prior to post-mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol. 2020;140:267–78.
Tropea TF, Waligorska T, Xie SX, Nasrallah IM, Cousins KAQ, Trojanowski JQ, et al. Plasma phosphorylated tau181 predicts cognitive and functional decline. Ann Clin Transl Neurol. 2023;10:18–31.
Planche V, Bouteloup V, Pellegrin I, Mangin J-F, Dubois B, Ousset P-J, et al. Validity and performance of blood biomarkers for Alzheimer disease to predict dementia risk in a large clinic-based cohort. Neurology. 2023;100:e473–84.
Cai H, Pang Y, Fu X, Ren Z, Jia L. Plasma biomarkers predict Alzheimer’s disease before clinical onset in Chinese cohorts. Nat Commun. 2023;14:6747.
Karikari TK, Benedet AL, Ashton NJ, Lantero Rodriguez J, Snellman A, Suárez-Calvet M, et al. Diagnostic performance and prediction of clinical progression of plasma phospho-tau181 in the Alzheimer’s disease neuroimaging initiative. Mol Psychiatry. 2021;26:429–42.
Janelidze S, Bali D, Ashton NJ, Barthélemy NR, Vanbrabant J, Stoops E, et al. Head-to-head comparison of 10 plasma phospho-tau assays in prodromal Alzheimer’s disease. Brain. 2023;146:1592–601.
Kivisäkk P, Fatima HA, Cahoon DS, Otieno B, Chacko L, Minooei F, et al. Clinical evaluation of a novel plasma pTau217 electrochemiluminescence immunoassay in Alzheimer’s disease. Sci Rep. 2024;14:629.
Barthélemy NR, Horie K, Sato C, Bateman RJ. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J Exp Med. 2020;217:e20200861.
Thijssen EH, La Joie R, Strom A, Fonseca C, Iaccarino L, Wolf A, et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol. 2021;20:739–52.
Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, et al. Discriminative accuracy of plasma Phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2020;324:772–81.
Brickman AM, Manly JJ, Honig LS, Sanchez D, Reyes-Dumeyer D, Lantigua RA, et al. Plasma p-tau181, p-tau217, and other blood-based Alzheimer’s disease biomarkers in a multi-ethnic, community study. Alzheimers Dement. 2021;17:1353–64.
Salvadó G, Ossenkoppele R, Ashton NJ, Beach TG, Serrano GE, Reiman EM, et al. Specific associations between plasma biomarkers and postmortem amyloid plaque and tau tangle loads. EMBO Mol Med. 2023;15:e17123.
Janelidze S, Berron D, Smith R, Strandberg O, Proctor NK, Dage JL, et al. Associations of plasma Phospho-Tau217 levels with tau positron emission tomography in early alzheimer disease. JAMA Neurol. 2021;78:149–56.
Barthélemy NR, Salvadó G, Schindler SE, He Y, Janelidze S, Collij LE, et al. Highly accurate blood test for Alzheimer’s disease is similar or superior to clinical cerebrospinal fluid tests. Nat Med. 2024;30:1085–95.
Mattsson-Carlgren N, Janelidze S, Palmqvist S, Cullen N, Svenningsson AL, Strandberg O, et al. Longitudinal plasma p-tau217 is increased in early stages of Alzheimer’s disease. Brain. 2020;143:3234–41.
Ashton NJ, Pascoal TA, Karikari TK, Benedet AL, Lantero-Rodriguez J, Brinkmalm G, et al. Plasma p-tau231: a new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol. 2021;141:709–24.
Meyer SD, Vanbrabant J, Schaeverbeke JM, Reinartz M, Luckett ES, Dupont P, et al. Phospho‐specific plasma p‐tau181 assay detects clinical as well as asymptomatic Alzheimer’s disease. Ann Clin Transl Neurol. 2022;9:734.
Therriault J, Servaes S, Tissot C, Rahmouni N, Ashton NJ, Bened AL, et al. Equivalence of plasma p-tau217 with cerebrospinal fluid in the diagnosis of Alzheimer’s disease. Alzheimers Dement. 2023;19:4967–77.
Milà-Alomà M, Ashton NJ, Shekari M, Salvadó G, Ortiz-Romero P, Montoliu-Gaya L, et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat Med. 2022;28:1797–801.
Mattsson-Carlgren N, Salvadó G, Ashton NJ, Tideman P, Stomrud E, Zetterberg H, et al. Prediction of longitudinal cognitive decline in preclinical Alzheimer disease using plasma biomarkers. JAMA Neurol. 2023;80:360–9.
Gonzalez-Ortiz F, Turton M, Kac PR, Smirnov D, Premi E, Ghidoni R, et al. Brain-derived tau: a novel blood-based biomarker for Alzheimer’s disease-type neurodegeneration. Brain. 2023;146:1152–65.
Gonzalez-Ortiz F, Kirsebom B-E, Contador J, Tanley JE, Selnes P, Gísladóttir B, et al. Plasma brain-derived tau is an amyloid-associated neurodegeneration biomarker in Alzheimer’s disease. Nat Commun. 2024;15:2908.
Hansson O. Biomarkers for neurodegenerative diseases. Nat Med. 2021;27:954–63.
Teunissen CE, Verberk IMW, Thijssen EH, Vermunt L, Hansson O, Zetterberg H, et al. Blood-based biomarkers for Alzheimer’s disease: towards clinical implementation. Lancet Neurol. 2022;21:66–77.
Huber H, Blennow K, Zetterberg H, Boada M, Jeromin A, Weninger H, et al. Biomarkers of Alzheimer’s disease and neurodegeneration in dried blood spots—a new collection method for remote settings. Alzheimers Dement. 2024;20:2340–52.
Koran MEI, Wagener M, Hohman TJ, Alzheimer’s Neuroimaging Initiative Sex differences in the association between AD biomarkers and cognitive decline. Brain Imaging Behav. 2017;11:205–13.
Mielke MM, Dage JL, Frank RD, Algeciras-Schimnich A, Knopman DS, Lowe VJ, et al. Performance of plasma phosphorylated tau 181 and 217 in the community. Nat Med. 2022;28:1398–405.
O’Bryant SE, Petersen M, Hall J, Johnson LA, Team for the H-HS Medical comorbidities and ethnicity impact plasma Alzheimer’s disease biomarkers: Important considerations for clinical trials and practice. Alzheimers Dement. 2023;19:36–43.
Ramanan VK, Graff-Radford J, Syrjanen J, Shir D, Algeciras-Schimnich A, Lucas J, et al. Association of plasma biomarkers of Alzheimer disease with cognition and medical comorbidities in a biracial cohort. Neurology. 2023;101:e1402–11.
Syrjanen JA, Campbell MR, Algeciras-Schimnich A, Vemuri P, Graff-Radford J, Machulda MM, et al. Associations of amyloid and neurodegeneration plasma biomarkers with comorbidities. Alzheimers Dement. 2022;18:1128–40.
Pichet Binette A, Janelidze S, Cullen N, Dage JL, Bateman RJ, Zetterberg H, et al. Confounding factors of Alzheimer’s disease plasma biomarkers and their impact on clinical performance. Alzheimers Dement. 2023;19:1403–14.
Mohs RC, Beauregard D, Dwyer J, Gaudioso J, Bork J, MaGee-Rodgers T, et al. The Bio-Hermes study: biomarker database developed to investigate blood-based and digital biomarkers in community-based, diverse populations clinically screened for Alzheimer’s disease. Alzheimers Dement. 2024;20:2752–65.
Wu X, Xiao Z, Yi J, Ding S, Gu H, Wu W, et al. Development of a plasma biomarker diagnostic model incorporating ultrasensitive digital immunoassay as a screening strategy for Alzheimer disease in a Chinese population. Clin Chem. 2021;67:1628–39.
Honig LS, Kang MS, Lee AJ, Reyes-Dumeyer D, Piriz A, Soriano B, et al. Evaluation of plasma biomarkers for A/T/N classification of Alzheimer disease among adults of caribbean hispanic ethnicity. JAMA Netw Open. 2023;6:e238214.
Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–83.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–28.
Badhwar A, Haqqani AS. Biomarker potential of brain-secreted extracellular vesicles in blood in Alzheimer’s disease. Alzheimers Dement Diagn Assess Dis Monit. 2020;12:e12001.
Manolopoulos A, Delgado-Peraza F, Mustapic M, Pucha KA, Nogueras-Ortiz C, Daskalopoulos A, et al. Comparative assessment of Alzheimer’s disease-related biomarkers in plasma and neuron-derived extracellular vesicles: a nested case-control study. Front Mol Biosci. 2023;10:1254834.
Goetzl EJ, Kapogiannis D, Schwartz JB, Lobach IV, Goetzl L, Abner EL, et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016;30:4141–8.
Mullins RJ, Mustapic M, Goetzl EJ, Kapogiannis D. Exosomal biomarkers of brain insulin resistance associated with regional atrophy in Alzheimer’s disease. Hum Brain Mapp. 2017;38:1933–40.
Kumar A, Su Y, Sharma M, Singh S, Kim S, Peavey JJ, et al. MicroRNA expression in extracellular vesicles as a novel blood-based biomarker for Alzheimer’s disease. Alzheimers Dement. 2023;19:4952–66.
Martins TS, Vaz M, Henriques AG. A review on comparative studies addressing exosome isolation methods from body fluids. Anal Bioanal Chem. 2023;415:1239–63.
Fauré J, Lachenal G, Court M, Hirrlinger J, Chatellard-Causse C, Blot B, et al. Exosomes are released by cultured cortical neurones. Mol Cell Neurosci. 2006;31:642–8.
Vandendriessche C, Kapogiannis D, Vandenbroucke RE. Biomarker and therapeutic potential of peripheral extracellular vesicles in Alzheimer’s disease. Adv Drug Deliv Rev. 2022;190:114486.
Norman M, Ter-Ovanesyan D, Trieu W, Lazarovits R, Kowal EJK, Lee JH, et al. L1CAM is not associated with extracellular vesicles in human cerebrospinal fluid or plasma. Nat Methods. 2021;18:631–4.
You Y, Zhang Z, Sultana N, Ericsson M, Martens YA, Sun M, et al. ATP1A3 as a target for isolating neuron-specific extracellular vesicles from human brain and biofluids. Sci Adv. 2023;9:eadi3647.
Whelan CD, Mattsson N, Nagle MW, Vijayaraghavan S, Hyde C, Janelidze S, et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol Commun. 2019;7:169.
Feng W, Beer JC, Hao Q, Ariyapala IS, Sahajan A, Komarov A, et al. NULISA: a proteomic liquid biopsy platform with attomolar sensitivity and high multiplexing. Nat Commun. 2023;14:7238.
Fiandaca MS, Kapogiannis D, Mapstone M, Boxer A, Eitan E, Schwartz JB, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case‐control study. Alzheimers Dement. 2015;11:600.
Winston CN, Goetzl EJ, Akers JC, Carter BS, Rockenstein EM, Galasko D, et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement Diagn Assess Dis Monit. 2016;3:63–72.
Jia L, Qiu Q, Zhang H, Chu L, Du Y, Zhang J, et al. Concordance between the assessment of Aβ42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimers Dement. 2019;15:1071–80.
Li T-R, Yao Y-X, Jiang X-Y, Dong Q-Y, Yu X-F, Wang T, et al. β-Amyloid in blood neuronal-derived extracellular vesicles is elevated in cognitively normal adults at risk of Alzheimer’s disease and predicts cerebral amyloidosis. Alzheimers Res Ther. 2022;14:66.
Kapogiannis D, Boxer A, Schwartz JB, Abner EL, Biragyn A, Masharani U, et al. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. 2015;29:589–96.
Ting YT, Geng LC, Chao GS, Yi Z, Chang WP. The serum exosome derived MicroRNA-135a, -193b, and-384 were potential Alzheimer’s disease biomarkers. Biomed Environ Sci. 2018;31:87–96.
Ransom LS, Liu CS, Dunsmore E, Palmer CR, Nicodemus J, Ziomek D, et al. Human brain small extracellular vesicles contain selectively packaged, full-length mRNA. Cell Rep. 2024;43:114061.
Author information
Authors and Affiliations
Contributions
FGDF and TRH conceptualized the article. TRH wrote the original draft. TRH and LES created the figures and tables. TRH, LES, FTM, and FGDF reviewed and edited the manuscript. All authors approve the submission of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hunter, T.R., Santos, L.E., Tovar-Moll, F. et al. Alzheimer’s disease biomarkers and their current use in clinical research and practice. Mol Psychiatry (2024). https://doi.org/10.1038/s41380-024-02709-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41380-024-02709-z
- Springer Nature Limited