
Abstract
Hypothalamic detection of elevated circulating glucose triggers suppression of endogenous glucose production (EGP) to maintain glucose homeostasis. Antipsychotics alleviate symptoms associated with schizophrenia but also increase the risk for impaired glucose metabolism. In the current study, we examined whether two acutely administered antipsychotics from different drug classes, haloperidol (first generation antipsychotic) and olanzapine (second generation antipsychotic), affect the ability of intracerebroventricular (ICV) glucose infusion approximating postprandial levels to suppress EGP. The experimental protocol consisted of a pancreatic euglycemic clamp, followed by kinomic and RNA-seq analyses of hypothalamic samples to determine changes in serine/threonine kinase activity and gene expression, respectively. Both antipsychotics inhibited ICV glucose-mediated increases in glucose infusion rate during the clamp, a measure of whole-body glucose metabolism. Similarly, olanzapine and haloperidol blocked central glucose-induced suppression of EGP. ICV glucose stimulated the vascular endothelial growth factor (VEGF) pathway, phosphatidylinositol 3-kinase (PI3K) pathway, and kinases capable of activating KATP channels in the hypothalamus. These effects were inhibited by both antipsychotics. In conclusion, olanzapine and haloperidol impair central glucose sensing. Although results of hypothalamic analyses in our study do not prove causality, they are novel and provide the basis for a multitude of future studies.
Similar content being viewed by others

Introduction
Antipsychotics remain the cornerstone treatment of schizophrenia and are widely prescribed on- and off-indication across other mental disorders. However, while effective in alleviating symptoms of schizophrenia, antipsychotics adversely impact glucose metabolism. In turn, the leading cause of mortality in schizophrenia remains cardiovascular disease, resulting in premature mortality by 20 to 25 years. While there is a differential metabolic liability which exists between individual agents, recent data supports that all antipsychotics independently of class or individual agent increase the risk of diabetes 3–4 fold above the risk conferred by the illness of schizophrenia itself [1]. While antipsychotics induce significant weight gain which contributes to risk of diabetes, it is also well established that within minutes to hours of administration, olanzapine (Ola) and haloperidol (Hal) increase circulating glucose and insulin concentrations in rodents [2], and furthermore, these acute antipsychotic-induced glucose dysregulatory effects also occur in humans [3, 4]. The mechanisms through which antipsychotics disrupt glucose homeostasis have not been clearly defined, but they can occur independently of increased body weight [4, 5].
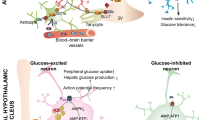
Glucose, which is metabolized via glycolysis to lactate in astrocytes, is the preferred fuel of the central nervous system (CNS). Glucose sensing refers to the ability of cells to detect extracellular glucose and it occurs within as well as outside the CNS (periphery) [6, 7]. Acute (minutes to hours) intracerebroventricular (ICV) administration of glucose decreases plasma glucose concentration [8], inhibits hepatic glucose production (also referred to as endogenous glucose production (EGP)) [8], and improves glucose tolerance [9]. Acute ICV glucose administration also decreases circulating insulin levels [8]. To determine the effect of central glucose on glucose metabolism in vivo without the confounding effect of changes in circulating insulin, a pancreatic euglycemic clamp can be utilized. During a pancreatic euglycemic clamp, insulin secretion is inhibited but basal circulating insulin concentrations are kept constant by exogenous insulin infusion. In addition, plasma glucose concentration is maintained constant at euglycemia. The hypothalamic pathway through which hypothalamic glucose suppresses EGP under clamp conditions was described by Lam et al. [8] almost 20 years ago. Briefly, ICV glucose is converted to lactate, ultimately leading to the activation (opening) of ATP-sensitive K+ channels (KATP channels) located at the plasma membrane of neurons [8]. Suppression of EGP by hyperglycemia is also mediated by central KATP channel activation in humans [10]. Lam et al. [8] and a subsequent study [11] also support the astrocyte-neuron lactate shuttle hypothesis, which states that glucose is converted to lactate in astrocytes before lactate is exported to the extracellular space, taken up by neurons, where it is converted to pyruvate. Independent studies strongly suggest that the hypothalamic pathway downstream of pyruvate and upstream of KATP channel activation is as follows [12]: pyruvate is converted to acetyl-CoA and subsequently malonyl-CoA, which inhibits carnitine palmitoyl transferase-1 (CPT1), resulting in long-chain fatty acyl CoA (LCFA-CoA) accumulation in the cytosol [8, 13, 14]. LCFA-CoA accumulation in the cytosol activates KATP channels [15,16,17], although to the best of our knowledge, it has not been proven that LCFA-CoAs directly activate hypothalamic KATP channels in the context of central glucose-induced EGP suppression. This pathway is summarized in Fig. 1A.

A Overview of current understanding of hypothalamic glucose sensing pathway in the context of endogenous glucose production (EGP) suppression. The dashed arrow indicates that a direct effect has not been reported in the context of ICV glucose. ACC acetyl-CoA carboxylase, CPT1 carnitine palmitoyl transferase-1, GLUT1 glucose transporter-1, GLUT3 glucose transporter-3, KATP channel ATP-sensitive K+ channel, LCFA-CoA long-chain fatty acyl CoA, LDH-A lactate dehydrogenase-A, LDH-B lactate dehydrogenase-B, MCT monocarboxylate transporter, PDH pyruvate dehydrogenase, TCA tricarboxylic acid. Created with BioRender.com. B Schematic representation of the pancreatic euglycemic clamp with ICV and subcutaneous infusions. ICV intracerebroventricular, IV intravenous. Created with BioRender.com. C Glucose infusion rate during clamp period. D Percent suppression in EGP. E Percent change in glucose utilization. n = 5–9/group. *p < 0.05 vs. Veh-Veh.
Various serine/threonine kinases can activate KATP channels in cells, for example cGMP-dependent protein kinase (PKG) indirectly activates KATP channels [18] and extracellular signal-regulated kinase (ERK) [19], cAMP-dependent protein kinase (PKA) [20], and PKC [21] directly phosphorylate, thereby activating, KATP channels. PKC-δ has been implicated in hypothalamic lipid-induced EGP suppression [22]. However, to the best of our knowledge, it is unknown if such serine/threonine kinases mediate hypothalamic glucose-induced EGP suppression. Similarly, alterations in hypothalamic gene expression in the context of hypothalamic glucose-induced EGP suppression has not been reported.
Suppression of EGP by ICV insulin also requires KATP channel activation and is mediated by hypothalamic phosphatidylinositol 3-kinase (PI3K) [23]. We previously reported that Ola blocks the ability of ICV insulin, but not of ICV diazoxide (a KATP channel activator), to suppress EGP [24, 25]. These findings suggest that impairment by Ola of hypothalamus-mediated decrease in EGP may occur upstream of KATP channels or at the level of KATP channels through a mechanism different from diazoxide. It is presently unknown, however, if antipsychotics disrupt nutrient sensing by the CNS to suppress EGP. Thus, our objectives for this study were: (1) to determine in vivo whether two antipsychotics (i.e., Hal and Ola) acutely disrupt hypothalamic glucose sensing resulting in dysregulated glucose homeostasis, and (2) explore ex vivo the effects of central glucose administration on hypothalamic physiological processes at the level of transcription and kinase activity, and hypothesized disruptions by antipsychotics on these glucose-modulated pathways.
Materials and methods
Animals
The study protocol was approved by the Centre for Addiction and Mental Health (CAMH) Animal Care Committee and followed the Canadian Council on Animal Care guidelines. Healthy male Sprague Dawley rats (300–400 g, Charles River, Saint-Constant, QC, Canada) were maintained on a 12 h light/dark with ad libitum access to food and water. Animals were pair-housed and acclimatized to the facility for at least 1 week prior to surgeries.
Surgical procedures
Rats underwent ICV cannulation surgery as previously described [24]. Briefly, a cannula was inserted into the third cerebral ventricle under isoflurane anesthesia at the following coordinates: anterior–posterior −2.5 mm, medial–lateral 0.0 mm, and dorsal–ventral −8.0 mm. The cannula was secured in place using stainless steel screws (McCray Optical Supply Inc., Scarborough, ON, Canada) and dental cement, and kept patent with a steel stylet. Following ICV cannulation surgery, rats were housed individually. Seven to eight days post-ICV surgery rats underwent right jugular vein and left carotid artery cannulation surgery. Rats were anesthetized using isoflurane and polyethylene catheters (PE-50, Clay Adams, BD Diagnostic, Mississauga, ON, Canada) capped with silastic tubing (Dow Corning Corp., Midland, MI, USA) were inserted and advanced to the right atrium (jugular vein) and the aortic arch (carotid artery). Catheter lines were externalized dorsally and plugged with a pin. Animals were allowed 3–5 days to recover before they underwent pancreatic euglycemic clamps. Body weight and animal behavior were recorded daily to ensure successful recovery from surgery.
Pancreatic euglycemic clamp
Rats underwent a pancreatic euglycemic clamp to assess changes in glucose metabolism (Fig. 1B). Experiments began at approximately 9–10 a.m. and the clamp began after 4–6 h of fasting. Starting at 0 min and for the duration of the 210 min study, clamp animals received a continuous ICV infusion of vehicle or glucose (2 mM, 5 μl h−1) into the third ventricle of the brain. The dose of ICV glucose infusion was based on postprandial glucose levels in the CNS [8, 26]. A primed (20 μCi) continuous (0.5 μCi min−1) intravenous infusion of [3-3H] glucose (PerkinElmer, Woodbridge, ON, Canada) also began at 0 min and continued for the duration of the clamp to assess glucose kinetics. At the 90 min time point, the clamp phase was initiated via a primed continuous intravenous infusion of somatostatin (3 µg kg−1 min−1) to inhibit endogenous insulin secretion, and exogenous insulin (1.1 mU kg−1 min−1) to replace insulin at basal levels. After the 90 min time point, intravenous infusion of a 25% glucose solution was initiated and its rate adjusted as required to maintain euglycemia (i.e., basal blood glucose concentrations). A single subcutaneous (s.c.) injection of Ola (3 mg per kg of body weight; Toronto Research Chemicals, Toronto, ON, Canada), Hal (0.25 mg per kg of body weight; Toronto Research Chemicals) or vehicle was administered immediately prior to initiation of the clamp phase, i.e., prior to 90 min. Dose of both antipsychotics was chosen based on clinically relevant dopamine D2 brain receptor occupancies [27,28,29]. These doses also correspond to previous work done by our group using acute antipsychotic administration and hyperinsulinemic euglycemic clamps [29]. Blood samples were taken from the carotid artery at 10 min intervals to measure plasma glucose, insulin, and [3–3H] glucose specific activity. Plasma and tissue samples were collected and stored at −80 °C for subsequent analyses. During the pancreatic euglycemic clamp, there are two physiological and isotopic steady states: a basal steady state (60–90 min) and a clamp steady state (180–210 min). The basal steady state is also referred to as the basal period while the clamp steady state is also known as the clamp period. Treatments are written in the order ICV-s.c. and the experimental groups in the study were: vehicle-vehicle (Veh-Veh), glucose-vehicle (Glu-Veh), glucose-olanzapine (Glu-Ola), vehicle-olanzapine (Veh-Ola), glucose-haloperidol (Glu-Hal), and vehicle-haloperidol (Veh-Hal). Rats were randomly assigned to an experimental group; the investigator was not blinded. At the end of the clamp, hypothalamus samples were collected [30] and stored at −80 °C for future analyses.
Plasma analyses
Plasma glucose was measured using an Analox GM9 glucose analyzer (Analox Instruments Ltd., Amblecote, UK). [3–3H] glucose radioactivity in plasma was determined by deproteination with barium hydroxide and zinc sulfate and subsequent evaporation to remove tritiated water. Total glucose turnover (rate of appearance of endogenous + exogenous glucose), which in steady state corresponds to glucose utilization (Rd), was quantified based on plasma specific activity (i.e., radioactivity/glucose concentration) during the basal period and the last 30 min of the clamp. Glucose infusion rate refers to the rate of exogenous glucose infusion during the last 30 min of the clamp. EGP was calculated by subtracting the glucose infusion rate from total glucose turnover. Percent suppression of EGP was calculated as follows: 100*(basal EGP-clamp EGP)/basal EGP. Percent change in glucose utilization was calculated using the formula: 100*(clamp Rd-basal Rd)/basal Rd. Plasma insulin was measured by ELISA (Mercodia, Winston Salem, NC, USA).
Kinome array analysis
Kinome array analyses were carried out as previously described [31]. Briefly, hypothalamic samples were processed and loaded onto chips containing immobilized peptide substrates containing consensus phosphorylation sequences that are targeted by serine/threonine kinases. This technique permits detection of activity changes in tissue according to treatment group in a large array of kinases, captured in real time. The signal intensity detected by the PamStation12 instrument (PamGene International) is proportional to the extent to which phosphoantibodies bind phosphorylated peptides. For each peptide, group comparisons were as follows: Glu-Veh vs. Veh-Veh, Glu-Ola vs. Glu-Veh, and Glu-Hal vs. Glu-Veh. To determine which kinases were responsible for changes in the phosphorylation of the peptides, random sampling analysis of kinases identified with GPS 3.0, Kinexus Phosphonet (Kinexus Bioinformatics), PhosphoELM, and PhosphoSite Plus was performed. Kinases with Z score ≥ │1.5│, which corresponds to a frequency approximately less than or equal to 6.7 %, were included in the study. Z score, also known as standardized deviate, is a way of standardizing a variable, for example kinase activity, such that probabilities in a normal distribution corresponding to a Z score can be easily obtained from commonly available tables [32]. Hence, the greater the absolute value of Z score, the lower the probability of the change in kinase activity being by random chance. The acronyms for kinases refer to families (i.e., kinome random sampling analyzer ID) and members of kinase families are in the Supplementary Material; additional detail for the identification of a given kinase family is provided within the main text when the kinase is of high relevance. Kinase network modeling was also done as in Bentea et al. [31]; in essence, this involved using databases to determine how protein kinases whose activity was altered in a given comparison interact with each other.
RNA sequencing (RNA-seq) analysis
RNA sequencing analysis was performed as previously described [33]. Briefly, total RNA was extracted from hypothalamic samples using RNeasy Mini kit (Qiagen, Hilden, Germany) with in-column DNase treatment using RNase-Free DNase (Qiagen). RNA samples were then sent for commercial sequencing service including sequencing library preparation and next-generation sequencing. On average ~40 million paired end 150 bp reads were generated per sample, which were aligned to rat reference genome Rnor_6.0 provided by Ensembl using HISAT2 aligner. Count data were then generated for the reads aligned to exons and transcripts using the GenomicFeature and GenomicAlignments packages in R and gene model (GTF file) provided by Ensembl. On average, 32 million unique reads aligned to 32,754 genes considered for all downstream analysis, were obtained per sample. After removing genes with low expression (fpm ≤1 in less than a half of samples), 16,056 genes were included in differential expression analysis. The experimental design allowed the following contrasts to be examined using the DESeq2 R package: Glu-Veh vs. Veh-Veh, Glu-Ola vs. Glu-Veh, and Glu-Hal vs. Glu-Veh. Biological pathways affected in different contrasts were determined using the following three gene set enrichment analysis (GSEA) approaches: (1) full transcriptome pathway analysis, including leading edge gene analysis, using fgsea R package, (2) targeted pathway analysis consisting of top 10% of upregulated and downregulated regulated genes using enrichR R package, (3) comparison of (1) and (2) (i.e., cross-pod comparison), which identified shared Gene Ontology (GO) pathways between full transcriptome pathway analysis and targeted pathway analysis. GO pathways are obtained from the GO Consortium, which contains data of biological pathways derived from experiments. Perturbagens that have concordant and discordant effects of the gene signatures consisting of L1000 genes were retrieved for each contrast using integrative LINCS (iLINCS). Mechanism of action and the gene targets were annotated for each perturbagen using L1000 fireworks database and DrugBank database. In other words, a perturbagen with a concordant mechanism of action is a compound that, based on information found in databases, results in a similar gene expression profile for a given comparison in our study, while a perturbagen with a discordant mechanism of action is a compound that, also based on information found in databases, causes the opposite gene expression profile for a given comparison in our study.
Statistical analyses
For pancreatic clamp data, statistical analyses were done with GraphPad Prism 9 and significance was set at p < 0.05. Sample size estimate was based on previously performed experiments. Clamp results are shown as mean ± SEM. One-way ANOVA with Dunnett’s post hoc test was performed to assess differences between groups (comparison to Veh-Veh).
Results
Plasma glucose and insulin concentrations were similar across experimental groups (Supplementary Table). Glucose infusion rate is a measure of whole-body glucose metabolism. Glucose infusion rate was increased by ICV glucose (p < 0.05, Glu-Veh vs. Veh-Veh, Fig. 1C) and it was similar between the other groups and Veh-Veh (Fig. 1C). Hence, both Ola and Hal impaired whole-body glucose metabolism stimulated by ICV glucose. ICV glucose suppressed EGP (p < 0.05, Glu-Veh vs. Veh-Veh, Fig. 1D), but no other group was significantly different from Veh-Veh, indicating that Ola and Hal blocked the ability of ICV glucose to reduce EGP. EGP suppression for Glu-Hal was similar compared to Veh-Veh (p = 0.134). Glucose utilization was not affected in any of the experimental groups (Fig. 1E). As in our previous work [24, 25], during pancreatic (basal insulin) euglycemic clamps there was no effect of either Hal or Ola in the absence of a homeostatic (i.e., glucose or insulin) challenge on any clamp derived parameters. This is in keeping with other studies which employed a basal pancreatic euglycemic clamp, or measured glucose kinetics following Ola injection under basal conditions (i.e., no insulin infusion) reporting no effect of Ola on glucose homeostasis [34, 35]. This suggests that a nutrient or hormonal stimulus may be necessary to observe acute antipsychotic-induced glucose dysregulation.
To understand the underlying mechanisms of alterations in glucose homeostasis caused by antipsychotics, high-throughput protein kinase activity profiling of hypothalamus samples was conducted using serine/threonine kinase (STK) chips. A heat map displaying the extent of phosphorylation of the top 50 variable peptides across experimental groups is shown in Supplementary Fig. 1. For each comparison, the Z score for each kinase is in Supplementary Fig. 2, while the log2 fold change for each peptide is in Supplementary Fig. 3. The kinome array results represent family of kinases. Interpretation of kinome array results focused on kinases that can modulate key steps in the established hypothalamic pathway required for ICV glucose to inhibit EGP (Fig. 1A). Given lack of Ola or Hal effects on clamp parameters in absence of a nutrient stimulus, we focused our analyses on kinases activated or deactivated by ICV glucose, and respective effects on glucose-stimulated kinase activity by each antipsychotic. The kinases with differential activity between ICV glucose-infused groups are shown in the Venn diagram in Fig. 2A. It is important to note that the Venn diagram does not provide information regarding the direction (increase vs. decrease) in kinase activity. Kinases located in regions that overlap indicate that their activity changed in more than one group comparison, while kinases in regions that do not overlap indicate that their activity only changed in one group comparison. The Venn diagram shows that, in the context of ICV glucose, while Hal and Ola mostly affect the activity of the same kinases, Ola has unique effects on kinase activity that Hal does not. Directionality of changes in kinase activity for each comparison is also shown, starting with the Glu-Veh vs. Veh-Veh comparison in Fig. 2B. The black dots represent relative signal intensity of peptides for a given kinase in the expected range, i.e., not affected by treatment, namely log2 fold change −0.3 to 0.3, which is equivalent to a fold change of 0.8 to 1.2. If, for a given kinase, relative signal intensity of peptides was above 1.2-fold change or below 0.8-fold change, they would be shown as red rots, i.e., different among treatments. When Glu-Veh was compared to Veh-Veh, various kinases were found to be activated, since the red rots are located in the log2 fold change >0.3 region (Fig. 2B). Specifically, ICV glucose-treated hypothalamic tissue demonstrated activation of the following kinases: TAO, STE7, SGK (SGK1-3), RSK, PLK, PKG, PKA, p21-activated kinase (PAKB; PAK4-6), JNK, ERK, DYRK, DMPK, cyclin dependent kinase family (CDK), AMP-activated protein kinase (AMPK), and Akt (Fig. 2B). ICV glucose did not inhibit serine/threonine kinases in the hypothalamus. The corresponding kinase network is shown in Fig. 2C, with Akt and ERK showing prominence (diameter size) for kinases with a Z score >2. In other words, Akt and ERK have the highest number of interactions with other protein kinases.

A Venn diagram for ICV glucose-infused groups that indicates overlap in kinase activity alterations with Z ≥ │1.5│per comparison. B Log2 fold change in relative signal intensity of peptides for kinases with Z score > │1.5│for comparison of Glu-Veh vs. Veh-Veh. C Kinase network model for comparison of Glu-Veh vs. Veh-Veh, where the circle diameter increases with interaction number. D Log2 fold change in relative signal intensity of peptides for kinases with Z score > │1.5│for comparison of Glu-Hal vs. Glu-Veh. E Log2 fold change in relative signal intensity of peptides for kinases with Z score > │1.5│for comparison of Glu-Ola vs. Glu-Veh. n = 4/group.
In the context of ICV glucose, both Hal and Ola inhibited kinase activity (Fig. 2D, E). The kinases inhibited by Hal were also inhibited by Ola, but Ola inhibited more kinases than Hal (Fig. 2D, E). For example, Ola, but not Hal, inhibited PKCA. Hal decreased the activity of kinases activated by ICV glucose, except STE7, SGK, and PAKB, while Ola decreased the activity of all kinases activated by ICV glucose, except STE7 (Fig. 2D, E). Hence, among the kinases stimulated by ICV glucose, SGK and PAKB are differentially affected by Hal and Ola (Fig. 2A). SGK is one of the key downstream effectors of the PI3K pathway; the other key downstream effector of PI3K is Akt [36]. Since ICV glucose-induced Akt activity is inhibited by Hal and Ola (Fig. 2B, D, E), both Hal and Ola inhibit PI3K signaling, with Hal being more selective towards the PI3K-Akt branch. As previously mentioned, hypothalamic PI3K mediates suppression of EGP by ICV insulin [23]. To the best of our knowledge, it has not been reported that PI3K in the hypothalamus mediates suppression of EGP by ICV glucose.
Pathway analysis of kinome array results indicated which pathways were generally stimulated by ICV glucose (Fig. 3A) and which pathways were generally inhibited when Hal or Ola was co-administered (Fig. 3B, C). Notably, two pathways that were stimulated by ICV glucose, namely the hypoxia-inducible factor 1 (HIF-1) and vascular endothelial growth factor (VEGF) pathways, were inhibited by Hal and Ola. It has been reported that glucose upregulates expression of VEGF through various mechanisms, including ROS-induced activation of the transcription factor HIF-1 [37,38,39,40]. VEGF stimulates various mediators, including PI3K, PKC, nitric oxide synthase (NOS), and ERK via VEGF receptor 2 [41, 42].

A Pathway analysis (KEGG 2021 Human) based on kinome array results for comparison of Glu-Veh vs. Veh-Veh. B Pathway analysis (KEGG 2021 Human) based on kinome array results for comparison of Glu-Hal vs. Glu-Veh. C Pathway analysis (KEGG 2021 Human) based on kinome array results for comparison of Glu-Ola vs. Glu-Veh. n = 4/group.
RNA-seq analysis was also performed to examine transcriptional changes caused by ICV glucose with and without antipsychotic treatment (Supplementary Fig. 4). Interpretation of results focused on mediators that are implicated in regulation of glucose metabolism. Leading edge (LE) gene analysis shows the gene expression driving most of the pathways for each comparison. For the Glu-Veh vs. Veh-Veh comparison, top upregulated LE genes included Vegfa and insulin-like growth factor 1 (Igf1), while top downregulated genes included inflammatory cytokines (tumor necrosis factor (Tnf), interleukin 1β (Il1b), and interleukin 6 (Il6)) (Fig. 4A). For the Glu-Hal vs. Glu-Veh comparison, the pathways affected by Hal were due to upregulation of inflammatory cytokines and downregulation of ion channels (type 2 ryanodine receptor (Ryr2), voltage-gated calcium channel 1.3 (Cacna1d), and voltage-gated potassium channel 7.1 (Kcnq1)) (Fig. 4B). Similar to Hal, Ola causes alterations in pathways due to upregulation of inflammatory cytokine expression and interestingly, Ola has an additive effect on Igf1 in the context of ICV glucose (Fig. 4C).

Leading edge gene analysis for (A) Glu-Veh vs. Veh-Veh, (B) Glu-Hal vs. Glu-Veh, and (C) Glu-Ola vs. Glu-Veh. n = 4/group except for Veh-Veh, for which n = 3.
Top and bottom 10 cross-pod common pathways are in Fig. 5. Cross-pod common pathways are GO pathways present in both full transcriptome pathway analysis and targeted pathway analysis, which uses the top 10% upregulated and downregulated genes. As expected, ICV glucose upregulated hypothalamic oxidoreductase activity, which is associated with increased electron transport chain activity, and long-chain fatty acid biosynthetic process, the first step of which is the conversion of acetyl-CoA to malonyl-CoA (Fig. 5A). ICV glucose also downregulated chemokine activity and chemokine receptor binding (Fig. 5A). When co-administered with ICV glucose, Hal or Ola generally stimulated chemokine and proinflammatory pathways and Ola also downregulated oxidoreductase activity (Fig. 5B, C).

Cross-pod common pathway analysis for (A) Glu-Veh vs. Veh-Veh, (B) Glu-Hal vs. Glu-Veh, and (C) Glu-Ola vs. Glu-Veh. n = 4/group except for Veh-Veh, for which n = 3.
Lastly, perturbagen analysis was performed to identify compounds that imitate or inhibit the transcriptional effects of each treatment comparison. A glucocorticoid receptor agonist, whose effects include dampening inflammation and promoting fatty acid synthesis [43, 44], was the top concordant mechanism of action (MoA) for ICV glucose, while VEGFR inhibitor was among the top discordant MoAs for ICV glucose (Fig. 6A), indicating that glucose may act as VEGFR stimulator. For Glu-Hal vs. Glu-Veh, the top concordant MoAs included Akt inhibitor and sodium channel blocker (Fig. 6B). The latter is consistent with reports that Hal is an inhibitor of voltage-gated sodium channels [45, 46]. For Glu-Ola vs. Glu-Veh, PI3K inhibitor and VEGFR inhibitor were among the top concordant MoAs (Fig. 6C).

A Perturbagens from MoA analysis for Glu-Veh vs. Veh-Veh. B Perturbagens MoA analysis for Glu-Hal vs. Glu-Veh. C Perturbagens MoA analysis for Glu-Ola vs. Glu-Veh. n = 4/group except for Veh-Veh, for which n = 3. D Proposed hypothalamic mechanisms for ICV glucose-induced suppression of EGP that were also inhibited by antipsychotics based on the findings in this study are in blue. Each dashed line indicates that a relationship has been found in the literature, but it was not tested in our study. eNOS endothelial nitric oxide synthase, ERK extracellular signal-regulated kinase, GLUT3 glucose transporter-3, HCAR1 hydrocarboxylic acid receptor 1, HIF-1 hypoxia-inducible factor 1, KATP channel ATP-sensitive K+ channel, MCT monocarboxylate transporter, NO nitric oxide, PI3K phosphatidylinositol 3-kinase, PKA cAMP-dependent protein kinase, PKG cGMP-dependent protein kinase, ROS reactive oxygen species, TCA tricarboxylic acid, VEGF vascular endothelial growth factor. Created with BioRender.com.
Discussion
Hypothalamic glucose sensing is established to play a key role in regulation of energy balance. Elevated glucose levels are sensed by the hypothalamus resulting in inhibition of food intake and reduction of glucose production. Conversely, glucopenia (or inhibition of hypothalamic glucose sensing) promotes energy intake and is associated with counterregulatory hormone release to counter low glucose levels and increase fatty acid release from adipose tissue [47,48,49,50]. While previous work by our group has demonstrated that Ola can inhibit central insulin action resulting in dysregulated hepatic glucose production and feeding [24, 51], the effects of antipsychotics on brain glucose sensing have not been examined. Importantly, glucose can act independently from insulin (i.e., known as “glucose effectiveness”) to regulate hepatic glucose production [52] and we have shown that antipsychotics inhibit glucose effectiveness evaluated using an intravenous glucose tolerance test (IVGTT) in humans [3]. In the current set of data, we demonstrate that Ola and Hal reduce the rate of exogenous glucose infusion required to maintain euglycemia during ICV glucose administration, indicating impaired whole-body glucose metabolism due to the ability of hypothalamic glucose to suppress EGP. To further investigate mechanisms by which Hal and Ola disrupt glucose effectiveness in the hypothalamus, we employed kinome array and RNA-seq analyses of hypothalamic samples. We report that ICV glucose stimulates the VEGF pathway, PI3K pathway, and various kinases that can directly activate KATP channels in the hypothalamus. Ola and Hal inhibit these effects of ICV glucose. Although the ex vivo analyses described herein do not establish causality, they are unbiased approaches to narrow down the hypothalamic pathway(s) likely to mediate glucose-induced EGP suppression and whole-body glucose metabolism. Our summary of proposed hypothalamic pathways involving alterations in serine/threonine kinase activity and gene expression through which ICV glucose suppresses EGP and which are inhibited by antipsychotics are shown in Fig. 6D.
PI3K in the hypothalamus is necessary for ICV insulin to suppress EGP [23]. While ICV glucose stimulated Akt and SGK, 2 branches that are downstream of PI3K, Ola inhibited both kinases and Hal inhibited Akt. Moreover, PI3K inhibitor is a concordant MoA only for Ola. These findings suggest that Hal is a less potent inhibitor of PI3K and may explain why there is a trend towards Hal leaving ICV glucose-induced EGP suppression intact in our study. PI3K can also be activated by factors other than insulin, including VEGF [42]. Indeed, kinome array and RNA-seq results indicate that the VEGF pathway is stimulated by ICV glucose and blocked by antipsychotics. Interestingly, lactate, a metabolite of glucose that suppresses EGP when administered ICV [8], increases VEGF expression by engaging with its cell surface receptor, hydrocarboxylic acid receptor 1 (HCAR1) [53]. Another factor that has been reported to be upstream of PI3K is HIF-1 and we found that the HIF-1 pathway is activated by ICV glucose, but inhibited by antipsychotics. Accordingly, it has been reported that hypothalamic HIF-1 inhibition reduces glucose tolerance [54]. A burst in reactive oxygen species (ROS), for example due to increased glucose availability and metabolism, stabilizes and increases the activity of HIF-1 [38, 40]. HIF-1 augments the production of VEGF [39], which may act in an autocrine or paracrine manner because both astrocytes and neurons express VEGF and VEGR2 [55, 56]. Pyruvate dehydrogenase (PDH) converts pyruvate to acetyl-CoA in the mitochondria. Since Ola and Hal can inhibit PDH through various mechanisms, including direct interaction (in vitro) and decreased gene expression [57, 58], the adverse effects of antipsychotics on glucose sensing could be due to blunted ROS production. Taken together, our results suggest that ICV glucose may stimulate a hypothalamic glucose → HIF-1 → VEGF → PI3K pathway that is inhibited by antipsychotics. However, an alternative explanation is that if Ola and Hal inhibit PDH, then the generation of acetyl-CoA and malonyl-CoA would be diminished, thereby reducing CPT-1 inhibition and decreasing cytosolic levels of LCFA-CoA, which can activate KATP channels, resulting in suppression of EGP. Indeed, activation of PDH in the hypothalamus is sufficient to suppress EGP [8]. Although our RNA-seq results suggest that neither fatty acid synthesis nor oxidation are affected by antipsychotics (Fig. 4), it may be possible that there are multiple parallel pathways through which hypothalamic glucose suppresses EGP.
The primary event that needs to occur for hypothalamic glucose sensing is entry of glucose into parenchyma and based on previous reports, glucose entry into astrocytes via GLUT1 is likely to be pivotal [8]. We cannot exclude the possibility that, in our study, antipsychotics impaired glucose entry into cells, thereby impairing glucose sensing, because it has been reported that: (1) Ola and Hal can reduce glucose uptake [59, 60], (2) reduction in hypothalamic GLUT1 blunts suppression of EGP by hypothalamic glucose [61], and (3) VEGF can increase GLUT1 expression [62].
Ultimately, the ability of ICV glucose to suppress EGP depends on activation of hypothalamic KATP channels [8]. We found that ICV glucose stimulates the activity of kinases capable of directly activating KATP channels, namely ERK and PKA [19, 20], while Hal and Ola inhibit them. Interestingly, ERK can be activated by VEGF and HCAR1 [42, 63], while PKA can be activated by VEGF [64]. Moreover, VEGF activates nitric oxide synthase (NOS) via PKA and Akt [41, 42, 64]. NOS generates nitric oxide (NO) and the NO-PKG pathway can indirectly activate KATP channels [18]. Interestingly, we found that while ICV glucose increased PKG activity, Hal and Ola inhibited it. It has been reported that Hal can directly inhibit KATP channels, but it is unclear if the high concentrations required for this inhibition are reached in vivo, especially in our study, where we used a single subcutaneous injection [65]. Thus, the inhibition of the kinases may be indirect, by inhibition of glucose uptake and thereby their activation by glucose.
Another way that kinases can affect KATP channel function is by controlling the rate at which KATP channels are trafficked to the cell membrane. Two kinases activated by ICV glucose and inhibited by antipsychotics in our study, namely AMPK and PKA, have been found to promote trafficking of KATP channels to the cell membrane [66]. It has also been reported that Hal and Ola inhibit AMPK [57, 67]. The possibility that KATP channel trafficking is impaired by antipsychotics is further substantiated by our RNA-seq results, which indicate that Ola and Hal generally diminish expression of genes involved in remodeling of the cytoskeleton (Fig. 5), a process that is required for KATP channel trafficking [66].
However, we have recently reported that Ola does not block the ability of diazoxide, which stimulates KATP channels, to suppress EGP in experimental conditions similar to the current study [25]. Hence, either the dose of diazoxide overpowered the inhibitory effects of Ola on KATP channels or Ola’s impairment of glucose sensing occurs predominantly through other (upstream) mechanisms. Regarding the former possibility, it is relevant to note that diazoxide and ERK and PKA act on different subunits of the KATP channel. The KATP channel consists of channel-forming and regulatory subunits, namely Kir6.2 and SUR1 respectively in neurons [68, 69]. Diaxozide interacts with the SUR1 subunit [70] while ERK and PKA phosphorylate the Kir6.2 subunit [19, 20]. It is unclear if stimulation of one subunit can override reduced activation of another subunit.
It has been reported that in the mediobasal hypothalamus, activation of AMPK ablates EGP suppression by glucose infused into the mediobasal hypothalamus [71]. In contrast, we found that ICV glucose activates hypothalamic AMPK. We assessed AMPK activity in the entire hypothalamus, not just parenchyma in a specific location such as the mediobasal hypothalamus, so it is feasible that glucose has different effects on AMPK activity in different regions and cell types of the hypothalamus. In agreement with our studies, AMPK activation has been reported to be necessary for glucose sensing by two populations of hypothalamic neurons important for the regulation of whole-body glucose metabolism and energy balance, namely proopiomelanocortin (POMC) and agouti-related protein (AgRP) neurons [72].
Our study has various limitations. First, our findings are hypothesis-generating and do not establish causality. The links between potential mediators in our proposed mechanism are based on published literature. We also did not assess the effects of ICV glucose with or without antipsychotic administration on LCFA-CoA and phosphatidylinositol 3,4,5-triphosphate (PIP3, which is generated by PI3K), both of which can interact with and activate KATP channels [15,16,17, 73]. Although a hypothalamic LCFA-CoA → PKC-δ pathway mediates lipid-induced EGP suppression [22], we only found that ICV glucose activated the PKC family of kinases at a Z score < │1.5│(Fig. 2C), which does not robustly support a role for hypothalamic PKC in mediating ICV glucose-induced suppression of EGP. However, this may not completely rule out the possibility of a causal role for hypothalamic PKC-δ pathway in glucose sensing because the kinome array does not differentiate between PKC isoforms. It has been reported that Ola depletes Krebs cycle intermediates and malonyl-CoA in skeletal muscle [74]. If this occurs in neurons, a decrease in cytosolic LCFA-CoA and KATP channel inactivation would be expected. Second, we used entire hypothalamus samples for ex vivo analyses and therefore, we cannot pinpoint to which cell types our findings apply.
In conclusion, we report that ICV glucose activates various kinases as well as the VEGF pathway in the hypothalamus and that Hal and Ola disrupt central glucose effectiveness. Our findings have clinical implications; glucose suppresses EGP via the CNS in humans [10] and the loss of central glucose sensing in the presence of antipsychotics may at least partly explain the high rates of impaired glucose metabolism in patients treated with these agents. Blockade of central glucose sensing would also be expected to result in impairments of appetite regulation [47, 48], and hepatic secretion of triglyceride rich lipoproteins [49], potentially explaining dysregulation of feeding and lipid homeostasis by these agents. As a final point, we are reminded that glycolysis in glial cells generates lactate, which beyond a key role in regulation of metabolic homeostasis, represents the primary fuel source of neuronal cells and is critical to homeostatic functions such as cognition [75]. Thus, examining central glucose sensing in the illness of schizophrenia and its interactions with antipsychotic treatments has the potential to provide clues also as to mechanisms of drug action, and possibly failure of these drugs to improve key domains of psychopathology such as cognitive function. Overall, our results provide the groundwork for intervention studies to attenuate antipsychotic-induced impairments of glucose metabolism at the level of the liver and other tissues.
References
Rajkumar AP, Horsdal HT, Wimberley T, Cohen D, Mors O, Borglum AD, et al. Endogenous and antipsychotic-related risks for diabetes mellitus in young people with schizophrenia: a Danish population-based cohort study. Am J Psychiatry. 2017;174:686–94.
Boyda HN, Tse L, Procyshyn RM, Wong D, Wu TK, Pang CC, et al. A parametric study of the acute effects of antipsychotic drugs on glucose sensitivity in an animal model. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:945–54.
Hahn MK, Wolever TM, Arenovich T, Teo C, Giacca A, Powell V, et al. Acute effects of single-dose olanzapine on metabolic, endocrine, and inflammatory markers in healthy controls. J Clin Psychopharmacol. 2013;33:740–6.
Teff KL, Rickels MR, Grudziak J, Fuller C, Nguyen HL, Rickels K. Antipsychotic-induced insulin resistance and postprandial hormonal dysregulation independent of weight gain or psychiatric disease. Diabetes. 2013;62:3232–40.
Kowalchuk C, Castellani LN, Chintoh A, Remington G, Giacca A, Hahn MK. Antipsychotics and glucose metabolism: how brain and body collide. Am J Physiol Endocrinol Metab. 2019;316:E1–5.
López-Gambero AJ, Martínez F, Salazar K, Cifuentes M, Nualart F. Brain glucose-sensing mechanism and energy homeostasis. Mol Neurobiol. 2019;56:769–96.
Donovan CM, Watts AG. Peripheral and central glucose sensing in hypoglycemic detection. Physiology. 2014;29:314–24.
Lam TK, Gutierrez-Juarez R, Pocai A, Rossetti L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science. 2005;309:943–7.
Osundiji MA, Lam DD, Shaw J, Yueh CY, Markkula SP, Hurst P, et al. Brain glucose sensors play a significant role in the regulation of pancreatic glucose-stimulated insulin secretion. Diabetes. 2012;61:321–8.
Carey M, Lontchi-Yimagou E, Mitchell W, Reda S, Zhang K, Kehlenbrink S, et al. Central K(ATP) channels modulate glucose effectiveness in humans and rodents. Diabetes. 2020;69:1140–8.
Abraham MA, Rasti M, Bauer PV, Lam TKT. Leptin enhances hypothalamic lactate dehydrogenase A (LDHA)-dependent glucose sensing to lower glucose production in high-fat-fed rats. J Biol Chem. 2018;293:4159–66.
Li RJW, Zhang SY, Lam TKT. Interaction of glucose sensing and leptin action in the brain. Mol Metab. 2020;39:101011.
Guay C, Madiraju SR, Aumais A, Joly E, Prentki M. A role for ATP-citrate lyase, malic enzyme, and pyruvate/citrate cycling in glucose-induced insulin secretion. J Biol Chem. 2007;282:35657–65.
Wang Y, Yu W, Li S, Guo D, He J, Wang Y. Acetyl-CoA carboxylases and diseases. Front Oncol. 2022;12:836058.
Larsson O, Deeney JT, Bränström R, Berggren PO, Corkey BE. Activation of the ATP-sensitive K+ channel by long chain acyl-CoA. A role in modulation of pancreatic beta-cell glucose sensitivity. J Biol Chem. 1996;271:10623–6.
Gribble FM, Proks P, Corkey BE, Ashcroft FM. Mechanism of cloned ATP-sensitive potassium channel activation by oleoyl-CoA. J Biol Chem. 1998;273:26383–7.
Yang HQ, Martinez-Ortiz W, Hwang J, Fan X, Cardozo TJ, Coetzee WA. Palmitoylation of the K(ATP) channel Kir6.2 subunit promotes channel opening by regulating PIP(2) sensitivity. Proc Natl Acad Sci USA. 2020;117:10593–602.
Chai Y, Lin YF. Stimulation of neuronal KATP channels by cGMP-dependent protein kinase: involvement of ROS and 5-hydroxydecanoate-sensitive factors in signal transduction. Am J Physiol Cell Physiol. 2010;298:C875–92.
Lin YF, Chai Y. Functional modulation of the ATP-sensitive potassium channel by extracellular signal-regulated kinase-mediated phosphorylation. Neuroscience. 2008;152:371–80.
Lin YF, Jan YN, Jan LY. Regulation of ATP-sensitive potassium channel function by protein kinase A-mediated phosphorylation in transfected HEK293 cells. Embo J. 2000;19:942–55.
Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ. Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc Natl Acad Sci USA. 2000;97:9058–63.
Ross R, Wang PY, Chari M, Lam CK, Caspi L, Ono H, et al. Hypothalamic protein kinase C regulates glucose production. Diabetes. 2008;57:2061–5.
Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8:1376–82.
Kowalchuk C, Teo C, Wilson V, Chintoh A, Lam L, Agarwal SM, et al. In male rats, the ability of central insulin to suppress glucose production is impaired by olanzapine, whereas glucose uptake is left intact. J Psychiatry Neurosci. 2017;42:424–31.
Kowalchuk C, Castellani L, Kanagsundaram P, McIntyre WB, Asgariroozbehani R, Giacca A, et al. Olanzapine-induced insulin resistance may occur via attenuation of central K(ATP) channel-activation. Schizophr Res. 2021;228:112–7.
Routh VH, Hao L, Santiago AM, Sheng Z, Zhou C. Hypothalamic glucose sensing: making ends meet. Front Syst Neurosci. 2014;8:236.
Kapur S, Wadenberg ML, Remington G. Are animal studies of antipsychotics appropriately dosed? Lessons from the bedside to the bench. Can J Psychiatry. 2000;45:241–6.
Kapur S, VanderSpek SC, Brownlee BA, Nobrega JN. Antipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: a suggested solution based on in vivo occupancy. J Pharm Exp Ther. 2003;305:625–31.
Chintoh AF, Mann SW, Lam L, Giacca A, Fletcher P, Nobrega J, et al. Insulin resistance and secretion in vivo: effects of different antipsychotics in an animal model. Schizophr Res. 2009;108:127–33.
Ziotopoulou M, Mantzoros CS, Hileman SM, Flier JS. Differential expression of hypothalamic neuropeptides in the early phase of diet-induced obesity in mice. Am J Physiol Endocrinol Metab. 2000;279:E838–45.
Bentea E, Depasquale EAK, O’Donovan SM, Sullivan CR, Simmons M, Meador-Woodruff JH, et al. Kinase network dysregulation in a human induced pluripotent stem cell model of DISC1 schizophrenia. Mol Omics. 2019;15:173–88.
Armitage P, Berry G, Matthews JNS. Statistical methods in medical research. 4th ed. Malden, Massachusetts, USA: Blackwell Science; 2002.
Shukla R, Newton DF, Sumitomo A, Zare H, McCullumsmith R, Lewis DA, et al. Molecular characterization of depression trait and state. Mol Psychiatry. 2022;27:1083–94.
Albaugh VL, Judson JG, She P, Lang CH, Maresca KP, Joyal JL, et al. Olanzapine promotes fat accumulation in male rats by decreasing physical activity, repartitioning energy and increasing adipose tissue lipogenesis while impairing lipolysis. Mol Psychiatry. 2011;16:569–81.
Girault EM, Alkemade A, Foppen E, Ackermans MT, Fliers E, Kalsbeek A. Acute peripheral but not central administration of olanzapine induces hyperglycemia associated with hepatic and extra-hepatic insulin resistance. PLOS ONE. 2012;7:e43244.
Faes S, Dormond O. PI3K and AKT: unfaithful partners in cancer. Int J Mol Sci. 2015;16:21138–52.
Natarajan R, Bai W, Lanting L, Gonzales N, Nadler J. Effects of high glucose on vascular endothelial growth factor expression in vascular smooth muscle cells. Am J Physiol. 1997;273:H2224–31.
Zhao T, Zhu Y, Morinibu A, Kobayashi M, Shinomiya K, Itasaka S, et al. HIF-1-mediated metabolic reprogramming reduces ROS levels and facilitates the metastatic colonization of cancers in lungs. Sci Rep. 2014;4:3793.
Harms KM, Li L, Cunningham LA. Murine neural stem/progenitor cells protect neurons against ischemia by HIF-1alpha-regulated VEGF signaling. PLoS ONE. 2010;5:e9767.
Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–8.
Wang X, Bove AM, Simone G, Ma B. Molecular bases of VEGFR-2-mediated physiological function and pathological role. Front Cell Dev Biol. 2020;8:599281.
Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. 2016;17:611–25.
Gerber AN, Newton R, Sasse SK. Repression of transcription by the glucocorticoid receptor: a parsimonious model for the genomics era. J Biol Chem. 2021;296:100687.
Wang JC, Gray NE, Kuo T, Harris CA. Regulation of triglyceride metabolism by glucocorticoid receptor. Cell Biosci. 2012;2:19.
Wakamori M, Kaneda M, Oyama Y, Akaike N. Effects of chlordiazepoxide, chlorpromazine, diazepam, diphenylhydantoin, flunitrazepam and haloperidol on the voltage-dependent sodium current of isolated mammalian brain neurons. Brain Res. 1989;494:374–8.
Lenkey N, Karoly R, Lukacs P, Vizi ES, Sunesen M, Fodor L, et al. Classification of drugs based on properties of sodium channel inhibition: a comparative automated patch-clamp study. PLoS ONE. 2010;5:e15568.
Davis JD, Wirtshafter D, Asin KE, Brief D. Sustained intracerebroventricular infusion of brain fuels reduces body weight and food intake in rats. Science. 1981;212:81–3.
Miselis RR, Epstein AN. Feeding induced by intracerebroventricular 2-deoxy-D-glucose in the rat. Am J Physiol. 1975;229:1438–47.
Lam TK, Gutierrez-Juarez R, Pocai A, Bhanot S, Tso P, Schwartz GJ, et al. Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nat Med. 2007;13:171–80.
Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI. Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes. 1995;44:180–4.
Castellani LN, Wilkin J, Abela AR, Benarroch L, Ahasan Z, Teo C, et al. Effects of acute olanzapine exposure on central insulin-mediated regulation of whole body fuel selection and feeding. Psychoneuroendocrinology. 2018;98:127–30.
Mevorach M, Giacca A, Aharon Y, Hawkins M, Shamoon H, Rossetti L. Regulation of endogenous glucose production by glucose per se is impaired in type 2 diabetes mellitus. J Clin Investig. 1998;102:744–53.
Morland C, Andersson KA, Haugen ØP, Hadzic A, Kleppa L, Gille A, et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor HCAR1. Nat Commun. 2017;8:15557.
Gaspar JM, Mendes NF, Corrêa-da-Silva F, Lima-Junior JC, Gaspar RC, Ropelle ER, et al. Downregulation of HIF complex in the hypothalamus exacerbates diet-induced obesity. Brain Behav Immun. 2018;73:550–61.
Freitas-Andrade M, Carmeliet P, Stanimirovic DB, Moreno M. VEGFR-2-mediated increased proliferation and survival in response to oxygen and glucose deprivation in PlGF knockout astrocytes. J Neurochem. 2008;107:756–67.
Luck R, Urban S, Karakatsani A, Harde E, Sambandan S, Nicholson L, et al. VEGF/VEGFR2 signaling regulates hippocampal axon branching during development. Elife. 2019;8:e49818.
Stapel B, Kotsiari A, Scherr M, Hilfiker-Kleiner D, Bleich S, Frieling H, et al. Olanzapine and aripiprazole differentially affect glucose uptake and energy metabolism in human mononuclear blood cells. J Psychiatr Res. 2017;88:18–27.
Sacks W, Esser AH, Sacks S. Inhibition of pyruvate dehydrogenase complex (PDHC) by antipsychotic drugs. Biol Psychiatry. 1991;29:176–82.
Vestri HS, Maianu L, Moellering DR, Garvey WT. Atypical antipsychotic drugs directly impair insulin action in adipocytes: effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacology. 2007;32:765–72.
Dwyer DS, Pinkofsky HB, Liu Y, Bradley RJ. Antipsychotic drugs affect glucose uptake and the expression of glucose transporters in PC12 cells. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:69–80.
Chari M, Yang CS, Lam CK, Lee K, Mighiu P, Kokorovic A, et al. Glucose transporter-1 in the hypothalamic glial cells mediates glucose sensing to regulate glucose production in vivo. Diabetes. 2011;60:1901–6.
Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S, et al. Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell. 2016;165:882–95.
Li G, Wang HQ, Wang LH, Chen RP, Liu JP. Distinct pathways of ERK1/2 activation by hydroxy-carboxylic acid receptor-1. PLoS ONE. 2014;9:e93041.
Lu Y, Xiong Y, Huo Y, Han J, Yang X, Zhang R, et al. Grb-2-associated binder 1 (Gab1) regulates postnatal ischemic and VEGF-induced angiogenesis through the protein kinase A-endothelial NOS pathway. Proc Natl Acad Sci USA. 2011;108:2957–62.
Yang SB, Proks P, Ashcroft FM, Rupnik M. Inhibition of ATP-sensitive potassium channels by haloperidol. Br J Pharm. 2004;143:960–7.
Chen PC, Kryukova YN, Shyng SL. Leptin regulates KATP channel trafficking in pancreatic β-cells by a signaling mechanism involving AMP-activated protein kinase (AMPK) and cAMP-dependent protein kinase (PKA). J Biol Chem. 2013;288:34098–109.
Koppel J, Jimenez H, Adrien L, Greenwald BS, Marambaud P, Cinamon E, et al. Haloperidol inactivates AMPK and reduces tau phosphorylation in a tau mouse model of Alzheimer’s disease. Alzheimers Dement. 2016;2:121–30.
Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366.
Szeto V, Chen NH, Sun HS, Feng ZP. The role of K(ATP) channels in cerebral ischemic stroke and diabetes. Acta Pharm Sin. 2018;39:683–94.
Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. Embo J. 1997;16:1145–52.
Yang CS, Lam CK, Chari M, Cheung GW, Kokorovic A, Gao S, et al. Hypothalamic AMP-activated protein kinase regulates glucose production. Diabetes. 2010;59:2435–43.
Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Investig. 2007;117:2325–36.
Plum L, Ma X, Hampel B, Balthasar N, Coppari R, Münzberg H, et al. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Investig. 2006;116:1886–901.
Albaugh VL, Vary TC, Ilkayeva O, Wenner BR, Maresca KP, Joyal JL, et al. Atypical antipsychotics rapidly and inappropriately switch peripheral fuel utilization to lipids, impairing metabolic flexibility in rodents. Schizophr Bull. 2012;38:153–66.
Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, et al. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–23.
Acknowledgements
MKH is supported in part by an Academic Scholars Award from the Department of Psychiatry, University of Toronto, and has grant support from the Banting and Best Diabetes Centre (BBDC), the Canadian Institutes of Health Research (CIHR), PSI Foundation, Ontario, holds the Kelly and Michael Meighen Chair in Psychosis Prevention, and the Cardy Schizophrenia Research Chair. She is also supported by the Danish Diabetes Academy, and a Steno Diabetes Centre Fellowship Award. This work was also supported by The National Institute of Mental Health [grant numbers MH107487, MH121102] and National Institute of Health [grant number AG057598], both awarded to REM. SMA is supported in part by an Academic Scholars Award from the Department of Psychiatry, University of Toronto, and has grant support from CIHR, PSI Foundation, Ontario, and the CAMH Discovery Fund. SP was supported by a Discovery Fund Postdoctoral Fellowship from CAMH. RS is supported by Bebensee Schizophrenia Research Fellowship, by the Ian Douglas Bebensee Foundation, Toronto, Canada. SW was supported by the Ontario Graduate Scholarship and the Banting & Best Diabetes Centre—Novo Nordisk Studentship from the University of Toronto.
Author information
Authors and Affiliations
Contributions
LNC, CK, and MKH conceived the study. LNC, SP, CK, LH, KA, WGR, XZ, AC, GR, SMA, AG, REM, MKH designed experiments. LNC, SP, CK, RA, RS, SW, KA, WGR, XZ, and EA performed experiments. LNC, SP, KA, WGR, and XZ analyzed the data. SP wrote the paper and KA, WGR, XZ, AG, REM, and MKH assisted with interpretation of the data. The paper was reviewed and approved by all authors.
Corresponding author
Ethics declarations
Competing interests
MKH received consultant fees from Alkermes.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Castellani, L.N., Pereira, S., Kowalchuk, C. et al. Antipsychotics impair regulation of glucose metabolism by central glucose. Mol Psychiatry 27, 4741–4753 (2022). https://doi.org/10.1038/s41380-022-01798-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-022-01798-y
- Springer Nature Limited