
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder with devastating symptoms, including memory impairments and cognitive deficits. Hallmarks of AD pathology are amyloid-beta (Aβ) deposition forming neuritic plaques and neurofibrillary tangles (NFTs). For many years, AD drug development has mainly focused on directly targeting the Aβ aggregation or the formation of tau tangles, but this disease has no cure so far. Other common characteristics of AD are synaptic abnormalities and dysfunctions such as synaptic damage, synaptic loss, and structural changes in the synapse. Those anomalies happen in the early stages of the disease before behavioural symptoms have occurred. Therefore, better understanding the mechanisms underlying the synaptic dysfunction found in AD and targeting the synapse, especially using early treatment windows, can lead to finding novel and more effective treatments that could improve the lives of AD patients. Researchers have recently started developing different disease-modifying treatments targeting the synapse to rescue and prevent synaptic dysfunction in AD. The main objectives of these new strategies are to halt synaptic loss, strengthen synaptic connections, and improve synaptic density, potentially leading to the rescue or prevention of cognitive impairments. This article aims to address the mechanisms of synaptic degeneration in AD and discuss current strategies that focus on the synapse for AD therapy. Alzheimer’s disease (AD) is a neurodegenerative disorder that significantly impairs memory and causes cognitive and behavioural deficits. Scientists worldwide have tried to find a treatment that can reverse or rescue AD symptoms, but there is no cure so far. One prominent characteristic of AD is the brain atrophy caused by significant synaptic loss and overall neuronal damage, which starts at the early stages of the disease before other AD hallmarks such as neuritic plaques and NFTs. The present review addresses the underlying mechanisms behind synaptic loss and dysfunction in AD and discusses potential strategies that target the synapse.
Similar content being viewed by others

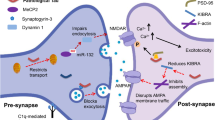

Synapse structure, synapse loss and synaptic dysfunction in AD
Synapses are critical for brain function since they are required for proper communication between neurons. The synapse is the space formed by interneuronal connections that enables neurons to pass electrical and chemical signals to one another. Neuronal communication is initiated at the presynaptic terminal, where vesicles containing different neurotransmitters are released. The neurotransmitter-synthesizing enzymes, type of neurotransmitters released, and transporters are different between inhibitory and excitatory presynaptic terminals. For instance, the most common excitatory neurotransmitter of the central nervous system (CNS) is glutamate [1], whereas GABA is the predominant inhibitory neurotransmitter of the adult brain. On the other hand, the postsynaptic terminals at dendritic spines use specific neurotransmitter receptors such as N-methyl-D-aspartic acid receptors (NMDA receptors) and α-amino-3-hydroxy-5- methyl-4-isoxazole propionic acid receptors (AMPA receptors) to receive and transduce the incoming signals [2].
Synapses are dynamic and change their number, structure, and function, leading to strengthening or weakening their synaptic contacts. This process is called synaptic plasticity and is crucial for cognitive functions such as learning and memory [3]. Synaptic degeneration has been associated with Alzheimer’s disease (AD) [4], and it is clearly found in patients with mild cognitive impairment (MCI) [5]. Other studies have also indicated that synaptic density significantly decreases at stages preceding amyloid plaque deposition in several mice models of AD [6, 7]. Synaptic loss and dysfunction can drastically damage signal transduction and neuronal communication, leading to network failures and abnormalities in the CNS. Thus, synaptic degeneration may be one of the first underlying mechanisms behind memory impairment and cognitive deficits, rather than the accumulation of amyloid-beta (Aβ) plaques and neurofibrillary tangles (NFTs) [8]. Better understanding the causes of synaptic loss in AD might be of critical importance to finding new therapeutic targets for a novel disease-modifying treatment. Although the cause of synaptic loss in AD has not yet been fully elucidated, Aβ, tau, apolipoprotein E (ApoE), and microglia seem to be major players contributing to synaptic dysfunction and neurodegeneration (Figs. 1 and 2).

In healthy conditions, there is a balance between inhibitory and excitatory connections mainly mediated by GABAergic and glutamatergic neurons. In the pathological condition, abnormal APP processing by β-secretase at the Asp1 site increases Aβ monomer generation and Aβ oligomer formation. Aβ oligomers lead to increased neuronal activity by disrupting glutamatergic/GABAergic balance and resulting in LTP impairments. Aβ oligomers can also cause tau missorting from the axon to the somatodendritic compartment and increase tau phosphorylation, which causes further neural toxicity and synaptic dysfunction.

Microglia play a critical role in synaptic refining and elimination. In the pathological condition, there is abnormal microglia activation that leads to synaptic loss. ApoE4 can exacerbate this effect by increasing the C1q accumulation resulting in overactive microglia and increased synaptic loss. ApoE4 can also decrease the phagocytic capacity of astrocytes, leading to abnormal synaptic pruning and increased synaptic debris accumulation, overall causing synaptic dysfunction.
Aβ toxicity mediated synapse loss and synaptic dysfunction
APP processing and Aβ generation
Aβ is a 38–43 amino acid residue peptide produced through the sequential cleavage of the amyloid-beta precursor protein (APP) by β- and γ-secretases. APP is a type I integral membrane glycoprotein ubiquitously expressed, with isoform APP695 predominantly expressed in the CNS [9,10,11,12]. APP can undergo different competitive series of sequential cleavage through either an amyloidogenic or non-amyloid pathogenic pathway. Most of the APP undergoes non-amyloidogenic cleavage processes [13], typically processed by α-secretase at the Leu17 site, producing a large soluble fragment (sAPPα) and a C-terminal fragment (CTF) of 83 amino acids (C83). C83 is further cleaved by γ–secretase, generating a P3a fragment and CTFγ. Beta-site APP cleaving enzyme 1 (BACE1) can also process APP at the Glu11 site, generating C89, which is cleaved by γ-secretase to create a truncated Aβ11-40 as part of the non-amyloidogenic pathways [14, 15]. On the other hand, APP is cleaved first by BACE1 at the Asp1 site for the amyloidogenic pathway to create a secreted form of APP (sAPPβ) and C99. C99 is then further cleaved by γ-secretase, producing Aβ and intracellular CTFγ [16, 17]. Under AD pathological conditions, overproduced Aβ can aggregate and form soluble oligomers, which can then change their conformation into cross-β-sheet fibrils, creating amyloid plaques. Increasing evidence suggests the critical role of soluble Aβ oligomers in the pathogenesis of AD [18, 19]. Unlike many thought, large insoluble Aβ fibrils are not the main responsible form behind neuronal damage.
Aβ oligomers connection to synapse loss and dysfunction in AD
The original amyloid cascade hypothesis mainly focused on large insoluble Aβ fibrils, while very few studies investigated the role of soluble Aβ oligomers in AD-related neuronal toxicity. However, research has found that Aβ oligomers can exist independently of fibrils [20]. Different studies have detected a correlation between the concentration of soluble Aβ oligomers in the brain and synaptic loss and dysfunction [18, 19].
Soluble Aβ oligomers can bind at the synapse and cause synaptic loss and synaptic changes in shape, size, and composition [21]. Aβ oligomers have been found to colocalize with PSD-95 in AD transgenic mice, indicating their presence in the postsynaptic terminal and modifying the synaptic structure, composition, and function [22]. Aβ oligomers exposure can lead to fewer functional spines and abnormally shaped spines similar to those found in mental retardation [21]. Likewise, soluble Aβ oligomers from the cerebral cortex of AD subjects or released by human APP-transfected cells were found to change the number, composition, and shape of synapses, leading to synaptic plasticity deficits and cognitive function impairments [23, 24].
Several studies using in vitro and in vivo samples have shown that soluble Aβ oligomers can induce inhibition of long-term potentiation (LTP) [24, 25]. Recordings of the field excitatory postsynaptic potentials (fEPSP) in wild-type mouse samples treated with Aβ oligomers indicated that hippocampal LTP impairments were caused by disruption of the glutamatergic/GABAergic balance [26]. Furthermore, synthetic Aβ aggregates can inhibit NMDA receptor-dependent LTP and diminish AMPA receptor mEPSC (miniature excitatory postsynaptic current) amplitude and frequency [27, 28]. All these studies support the hypothesis that Aβ oligomers can have a toxic effect on neural connections, resulting in synaptic loss and dysfunction.
Drug development research has also focused on the oligomer theory and placed substantial efforts to create a novel therapy for AD treatment. However, most attempts to date have failed to provide an effective disease-modifying cure for AD.
Tau toxicity leads to synaptic loss and dysfunction
Tau mediates Aβ-induced synaptic loss and dysfunction
Tau is a microtubule-associated protein (MAP) that is mostly enriched in the axons of neurons. Aggregation of Aβ and formation of tau tangles into NFTs are hallmarks of Alzheimer’s disease (AD). Synaptic density loss has also been correlated with increased phosphorylated tau (p-Tau), and Aβ levels [29], indicating that p-Tau also plays a role in synaptic toxicity in AD. For instance, endogenous tau was found to move to the somatodendritic area after treatment with Aβ oligomers, causing spinal density loss [30].
Decreased levels of endogenous tau can also reduce Aβ-induced synaptic loss and dysfunction as well as improve cognitive function in AD transgenic mice expressing human APP [31]. These findings suggest that tau can mediate synaptic dysfunction in the presence of Aβ oligomers and Aβ aggregates.
Tau directly induces synaptic loss and dysfunction
Tau can have toxic effects on the synapse resulting in density loss and synaptic dysfunction independent from Aβ [32]. Tau is predominantly found in axons, but misfolded tau oligomers can be located in the pre and postsynaptic terminals in AD [33]. Interestingly, high tau levels in the postsynaptic terminal have been linked with synaptic loss in tau-transgenic mice [34, 35], and were shown to cause synaptic dysfunction [36, 37]. Of relevance, tau-induced synaptic loss was found to occur before neurodegeneration in transgenic mice models [38]. Transgenic mice with overexpression of human tau have also been associated with fewer synaptic proteins, loss of spinal density and LTP impairment leading to cognitive and memory deficits [39, 40]. Missorting tau from the presynaptic to the postsynaptic compartment can happen during the early onset of AD in mouse models, suggesting that this abnormal tau localization could be partially responsible for the early synaptic dysfunction seen in AD [41]. More research needs to be conducted to understand further the early role of tau on AD-related synaptic dysfunction.
As mentioned, amyloid plaques and NFTs are both pathological hallmarks that have been associated with the progression of AD and ongoing synaptic loss. However, some research suggests that synaptic dysfunction may occur earlier than these common AD symptoms [6, 7]. Therefore, other mechanisms and pathways that trigger synaptic dysfunction in AD need to be investigated, such as genetic factors, inflammation and phagocytosis. Modelling all these interlinked pathological pathways using novel in vitro and in vivo models that best represent the synaptic degeneration occurring in AD, instead of using the traditional Aβ toxicity cells or animals, might lead to more promising treatment avenues.
Genetic risk factor ApoE4 exacerbates synapse loss and dysfunction
Apolipoprotein E (ApoE), specifically ApoE4, is the strongest genetic risk factor associated with sporadic AD [42]. In humans, ApoE has three allelic genetic variants leading to three protein isoforms, ApoE2, ApoE3 and ApoE4, with ApoE3 being the most common isoform (77.9%), while ApoE2 (8.4%) and ApoE4 (13.7%) are much less common and thus can be considered variants [43]. ApoE2 can play a protective role and reduce the risk of AD [44, 45]. On the other hand, inheriting even one copy of ApoE4 is associated with a 3-fold increased risk of AD, whereas two copies of ApoE4 is associated with a 15-fold increased risk [43]. It has been reported that ApoE4 can promote synaptic loss and degeneration through abnormal synaptic pruning by astrocytes, microglia, protein changes or through Aβ, and tau-dependent pathways. However, the role of ApoE in AD is very complex and needs to be further studied.
Proteomic comparative analysis of AD synapses showed that ApoE4 was associated with increased synaptic protein changes in AD [46]. In AD patient induced pluripotent stem cells (iPSCs)-derived cerebral organoids, ApoE4 can aggravate synaptic loss and neurodegeneration [47]. Moreover, synaptic pruning by astrocytes is mediated and dependent on the ApoE allele, with ApoE2 increasing the phagocytic capacity of astrocytes and ApoE4 decreasing the rate of synaptic pruning by astrocytes, leading to the accumulation of synaptic debris and synaptic dysfunction [48]. Similarly, other studies also showed that ApoE4 could decrease synaptic density in ApoE4 transgenic mice independently of Aβ presence [49].
On the other hand, by injecting AAV4-human-ApoE4 into the lateral ventricles of traditionally used APP/PS1 mice, ApoE4 enhanced Aβ oligomer concentration in tissue fluid, increased abnormal neurites and caused synaptic loss around plaques [50]. ApoE4 can also aggravate tau pathology in AD patient-derived cerebral organoids [47] and lead to synaptic degeneration. For instance, in MAPT mice infected with ApoE4, there is significant hippocampal synaptic loss [51]. In a recent study using a new mice model with selective deletion of ApoE4 from astrocytes, removal of ApoE4 reduced tau-induced synaptic degeneration and microglial phagocytosis at the synapse [52]. These new findings suggest that ApoE4 may contribute to synaptic degeneration via both Aβ/tau-dependent and independent mechanisms. While targeting ApoE, especially ApoE4, may be a promising treatment that rescues synaptic damage, the role of ApoE in synaptic loss in AD needs to be further examined.
Microglia mediates synaptic loss and dysfunction by playing a protective role
Microglia are the main macrophages located in the CNS. They are especially important during brain development since they play a crucial role in synaptic remodelling, pruning and plasticity [53, 54]. However, there is still much to investigate regarding the precise mechanisms behind the communication between synapses and microglia and how microglia conduct synaptic refining and pruning. One potential mechanism is the classical complement cascade [55, 56]. In this mechanism, microglia are characterized as the primary source of complement component 1q (C1q) and complement component 3 (C3) protein [57, 58], which are part of the innate immune response. C1q can activate C3, which then modulates synaptic refining by eliminating dysfunctional synapses. C1q and C3 are developmentally regulated, presenting upregulated levels during early developmental stages and downregulated expression in the mature CNS [55, 59, 60]. C1q or C3 Knockout (KO) mice show synaptic pruning and refining deficits, causing neural circuit issues [55, 59].
In AD mice models and human patients, microglia quickly surrounds amyloid plaques and are found in high numbers around newly created neuritic plaques [61], with different studies suggesting its involvement in Aβ clearance [62]. C1q is significantly increased in AD mice models and even in wild-type mice injected with Aβ oligomers [63, 64]. C1q depletion or exposure to C1q activator blockers in AD mouse models decreases synaptic loss and dysfunction [63, 65]. Similarly, Aβ oligomer treatment did not cause synaptic dysfunction in C1q-KO mice [63]. Interestingly, accumulation of C1q protein in the hippocampus could be dependent on the ApoE allele present, with a recent study suggesting that in ApoE4 KI mice, there is significantly higher C1q accumulation in hippocampus [48], which could lead to synaptic dysfunction. These results support the crucial role of C1q on synaptic elimination and suggest that overactive microglia and overexpression of C1q might be partially responsible for synaptic dysfunction in AD.
Microglia can also express NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3). Interestingly, NLRP3, the adaptor protein ASC and pro-caspase 1, form the NLRP3 inflammasome, a crucial part of the innate immune response [66]. NLRP3 inflammasome can colocalize with amyloid plaques leading to the reduction of Aβ clearance and more Aβ aggregates [67]. In a recent study, the deficiency of functional NLRP3 inflammasome caused significant reduction in tau phosphorylation and aggregation [68]. An inhibitor of NLRP3 inflammasome called dapansutrile or OLT117 improved synaptic plasticity and caused less microglia activation in AD mouse models [69]. These studies suggest the validity of researching neuroinflammation as a potential target for AD treatment that could rescue synaptic density and plasticity.
As explained in this initial section, there is plenty of evidence to show a significant increase in synaptic loss and dysfunction during the early stages of AD. This synaptic dysfunction can be linked to Aβ oligomers, Aβ toxicity, tau phosphorylation, ApoE isoform, increased microglia activity and inflammation. Research should continue investigating the causes of synaptic loss in neurodegenerative diseases since it is one of the early markers of these pathologies, particularly in AD. In the next section, current treatments that specifically target the AD-related synaptic changes will be reviewed.
Targeting the synapse for Alzheimer’s Disease treatments
Potential AD disease-modifying therapies have also focused on the synapse and preventing or rescuing synaptic density loss. This section will examine these AD treatments, what specific part of the synapse they target, and whether the US Food and Drug Administration (FDA) has approved them to treat AD (Table 1).
Targeting kinase activity to improve synaptic density
One of the drug candidates to restore synaptic dysfunction is AZD0530 or saracatinib. AZD0530 inhibits a family of protein kinases known as Src [70]. Several preclinical trials have tested this medication for AD using transgenic AD mouse models taking advantage of the inhibition caused by this compound on Fyn kinase. Of relevance, the formation of Aβ oligomers can lead to activation of Fyn, which results in tau phosphorylation, and is linked to AD [71, 72]. Oral administration of AZD0530 can block the activation of Fyn kinase in neuronal synapses, potentially leading to synaptic density improvements under pathological conditions. Preclinical trials have shown significant improvement of synaptic density without affecting Aβ plaques [73], indicating that this compound truly focuses on the synapse rather than targeting Aβ. Other preclinical studies in AD transgenic mice indicated that AZD030 could rescue synaptic loss and memory impairments [74] and increase synaptic density in the hippocampus [75]. A phase Ib clinical trial found that AZD0530 is safe and relatively well tolerated in AD patients and can pass the blood-brain barrier [76]. However, a recent phase 2a randomized clinical trial using this compound on AD patients found no statistical significance compared to placebo-controlled groups in cerebral metabolic decline and other biomarkers of the pathology [77]. More research should be conducted to understand the mechanism of this compound better and if it has other off-target effects on other members of the Src family besides Fyn kinase inhibition. Furthermore, the effect of different dosages and long-term administration should be further investigated in clinical trials for AD treatment.
Similarly, masitinib is another inhibitor of protein kinases. Interestingly, masitinib has also been implicated in blocking Fyn kinases and studied in neurodegenerative diseases [78]. This drug can block the survival and function of mast cells, which are crucial for inflammatory responses linked to synaptic loss in different pathologies. In a recent study using a transgenic AD mice model, masitinib was found to improve spatial learning and cognition by potentially protecting the synapse [79]. A phase 2 placebo-controlled study of this drug was conducted by administering it in adjunction to other compounds on AD patients [80]. As an add-on therapy, masitinib was shown to decrease cognitive decline and was well tolerated [80]. A very recent phase 3 trial for this drug in AD patients is undergoing analysis (ClinicalTrials.gov Identifier: NCT01872598). Remarkably, a study using human induced pluripotent stem cells (hiPSCs) and human neural stem cells found that Fyn kinase inhibitors, including AZD0530 and masitinib, can decrease Fyn kinase activity and Aβ42 release [81]. However, these effects were only found for a subset of neurons derived from AD patients with high APP Tyr phosphorylation [81]. These results suggest that certain AD patients with high APP phosphorylation might benefit more from Fyn kinase inhibitor treatment than other patients, indicating the potential importance of using early biomarkers to identify APP phosphorylation levels before treatment administration.
The wingless pathway (Wnt) also relies on kinase activity and is crucial in synaptic formation and maintenance. Inhibitors of specific targets within this signalling pathway have been studied as a potential treatment for reversing synaptic loss observed in AD. Fasudil inhibits ROCK, which is part of the Wnt pathway. Fasudil and its active metabolite hydroxyfasudil can cross the blood-brain barrier and enter the CNS [82]. Fasudil, along with bone marrow stromal cells, was found to rescue synaptic loss and enhance learning and memory in AD transgenic mouse model [83]. Furthermore, in an AD-induced rat model, fasudil was found to improve behavioural symptoms of AD [84]. However, this compound has not been approved for clinical AD trials, potentially due to possible severe side effects and toxicity. Interestingly, preclinical studies using a derivative of fasudil called FSD-C10 suggest more promising results and fewer safety concerns than fasudil [85] while also benefiting memory and synaptic density [86].
Targeting the Glutamatergic pathway to protect synapses
Memantine is an NMDA receptor antagonist that inhibits Ca2+ influx happening extrasynaptically and targets the receptor under pathological conditions. One reported advantage of this drug is that it is a low-affinity, non-competitive antagonist of NMDA receptors [87], avoiding the adverse side effects of prolonged receptor blockage on learning and memory. Memantine was synthesized by Eli Lilly laboratories and was used intravenously in AD patients in 1986 [88]. This project covered only a small sample size of 20 patients with two different doses and showed no benefit on neuropsychiatric functioning, but it detected damaging side effects. In the late 1990s, memantine was used again in a larger, placebo-controlled clinical study of moderate to severe AD patients [89]. This project found positive effects of the use of memantine for behavioural performance and acceptable drug tolerance. Another clinical study in the early 2000s confirmed these findings [90], leading to the approval of this drug by the European Agency for the Evaluation of Medical Products (EMA) and later by the FDA. Since then, it has been associated with decreased neuronal toxicity and linked to improved AD symptoms. In more recent mouse model studies, memantine was shown to rescue symptoms of AD pathology, corroborating previous findings [91, 92]. Although memantine has been approved for AD treatment, other studies have shown limited efficacy of this drug even when administered in adjunction with cholinesterase inhibitors [93]. There is still controversy on how effective memantine is in AD, with researchers suggesting it might depend on the disease stage and symptoms of AD patients [94]. A more selective blocker of abnormal NMDA receptor activity is NitroSynapsin, formerly known as nitromemantine, which protects synapses and has been shown to work in vivo and in vitro [95]. A recent study in rodent models and hiPSC-derived neurons found that using NitroSynapsin can protect against synaptic loss caused by αSyn oligomer damage, which has been previously associated with neurodegenerative diseases such as Parkinson’s and AD [96]. Furthermore, NitroSynapsin was able to rescue hyperexcitability of neurons caused by abnormal electrical events in an AD hiPSC neuronal organoid model, showing more promising results than memantine [97].
Riluzole is a glutamate modulator that has been FDA approved to treat amyotrophic lateral sclerosis. Preclinical trials using rodent models of AD and ageing have found that riluzole can prevent age-related cognitive decline [98], and protect against Aβ-induced learning and memory deficits [99]. Even though its mechanisms to improve AD symptoms are not entirely understood, it is suggested that riluzole has neuroprotective effects, decreases glutamatergic toxicity and induces clustering of dendritic spines, which increases synaptic plasticity under pathological conditions. Phase 2 clinical trials for AD using riluzole have been recently conducted and showed less decline in cerebral glucose metabolism than placebo groups, which is correlated with slower cognitive decline [100]. Other clinical trials are ongoing for this drug and its prodrug troriluzole (ClinicalTrials.gov Identifier: NCT03605667).
Targeting the cholinergic pathway to increase synaptic strength
Anti-cholinesterases have been very well studied to treat AD. Within this drug category, galantamine, rivastigmine, and donepezil are FDA-approved for AD treatment. The role of anti-cholinesterases in the treatment of AD is to increase the availability of acetylcholine at the synapse by inhibiting the hydrolysis of acetylcholine, leading to stronger cholinergic synapses. Several preclinical and clinical studies have indicated the efficacy of these drugs as AD treatment. Of relevance, clinical studies have found that donepezil can delay the progression of AD and improve cognitive function, especially in early AD stages [101,102,103]. Similarly, large AD clinical studies that indicate the treatment efficacy of galantamine [104, 105] and rivastigmine [106, 107] have also been reported.
Furthermore, anti-cholinesterases have been administered together with NMDA receptor antagonists, such as memantine, to treat neurodegenerative diseases. For example, namzaric is a combination of memantine and donepezil, and it is FDA approved for AD treatment. Several clinical studies in AD patients have found that using memantine together with anti-cholinesterases benefits cognition, memory, and behaviour [108,109,110]. However, a recent meta-analysis shows that namzaric and other anti-cholinesterases, such as galantamine and rivastigmine, have limited efficacy as an AD therapy [93]. Similarly, another meta-analysis in 2013 indicated that cognitive enhancers such as memantine and anti-cholinesterases did not improve cognition [111]. One reason for this limitation might be that these compounds cannot rescue severe synaptic loss caused by AD, showing once more the importance of early drug administration in AD. A particular characteristic of the cholinergic system is the circadian rhythm in its activity, meaning that it is more active during waking hours [112]. Enhanced acetylcholine levels during sleeping hours can then lead to side effects such as insomnia and other sleep-related disorders. Therefore, analyzing the timing of administration and duration (half-life) of anti-cholinesterases based on their circadian rhythm is necessary to improve current AD treatment.
Targeting the serotonin pathway to strengthen the synapse
Several studies have associated enhancements in cognitive function with the administration of serotonin reuptake inhibitors (SSRI) in dementia patients with depression during AD and MCI progression or in addition to other drugs [113,114,115]. SSRIs such as fluoxetine and sertraline work by inhibiting serotonin’s reuptake, increasing serotonin’s availability in the synapse and strengthening communication between neurons. However, there is controversy on the effects of SSRI as an effective treatment for cognitive impairment and dementia associated with AD. Different studies have indicated the benefits of SSRI for cognition in dementia patients [114], while many others have found no significant effects or detrimental effects [116,117,118,119].
One SSRI that is commonly studied is fluoxetine. This drug was shown to prevent cognitive decline in AD animal models [120,121,122], increase the synaptic protein expression as well as spine density in the hippocampus of rodents [123, 124] and protect synapses while preventing neuronal apoptosis in primary cultures and AD animal models. In a more recent study, fluoxetine improved cognitive behaviours in AD transgenic mice while rescuing synaptic density in the early stages of the pathology [122]. These studies show that this compound’s positive effects on cognition might be related to rescuing synaptic loss in the early stages of AD. Fluoxetine has also been used to treat depression and anxiety in clinical studies, with reports of improved cognition and reduced depression in AD patients [125]. In contrast, another study and a metanalysis paper indicated no significant effect in depression scores for depressive AD patients after fluoxetine treatment compared to placebo [126, 127]. Furthermore, as previously mentioned, several other studies find no effect or even detrimental effects of antidepressants such as fluoxetine on dementia and cognition. Overall, SSRI antidepressants have been used as adjuncts with other treatments or alone to treat depression in AD patients. There is still controversy regarding their efficacy in enhancing cognition in dementia patients. Different dosages of the compounds, the timing of the SSRI administration, and the duration of administration of the drugs could be the cause behind these controversial results.
Neurotrophic factors to rescue synaptic loss
Early stages of AD in patients have shown compensatory mechanisms that potentially increase neurogenesis and synaptic plasticity [128,129,130]. These attempts to reverse the progressive loss of neurons fail in the long term, and the disease progresses. Researchers consider that one potential reason for this failure is insufficient neurotrophic factors in the CNS, specifically at the synapse, to sustain prolonged compensatory mechanisms that improve synaptic plasticity and neurogenesis [130]. For instance, several studies have found abnormal levels of different neurotrophic factors, such as brain-derived neurotrophic factor (BDNF), VGF and nerve growth factor (NGF), in AD patients [131,132,133,134].
Treatments using neurotrophic factors have limitations due to the delivery method. Neurotrophic factors do not cross the blood-brain barrier and therefore need different delivery methods to be administered successfully for AD treatment. A phase 1 clinical trial using NGF gene therapy and delivery has been conducted in AD patients showing that it slowed cognitive decline without severe adverse side effects [135]. Similarly, clinical studies using intracerebral injections of adeno-associated viral vectors expressing NGF have shown no adverse side effects in AD patients [136, 137]. However, more studies need to investigate the efficacy of these treatments and their long-term effects. Several studies have focused on orally administering neurotrophic tetra-peptide compound P021 to transgenic AD mouse models during early AD stages. P021 was shown to increase neurogenesis and synaptic plasticity while enhancing cognitive function and rescuing synaptic loss in AD rodent models [138,139,140,141]. More studies using neurotrophic factors as AD treatments need to be conducted to investigate dosage and critical administration periods. Furthermore, different delivery methods for neurotrophic factors are currently being examined, such as encapsulated delivery [142, 143] and NGF mimetic peptides or small molecules such as LM11A-31 [144] (ClinicalTrials.gov Identifier: NCT03069014), which can help to overcome the side effects linked to the route of administration in humans.
Summary
In this section, we have mentioned five different targets for AD treatment that focus on improving synaptic function and health. Overall, targeting kinase activity could be a potentially viable avenue for AD treatment. Specifically, Fyn kinase and ROCK inhibitors have the advantage of reducing synaptic dysfunction in different AD models. However, their limitations associated with off-target effects need to be considered more carefully since severe side effects have been associated with these inhibitors. Additionally, further studies to more conclusively determine if subsets of AD patients are more likely to benefit from kinase inhibitors are crucial and could explain the results of using Fyn inhibitors with different degrees of success in humans.
As an alternative approach, targeting the glutamatergic pathway has been studied as a potential target for AD treatment for many years. Memantine, the only glutamatergic drug FDA-approved for AD treatment, has been found to improve synaptic function. However, there is controversy associated with the efficacy of these drugs in AD, with several studies reporting minimal positive effects. Presumably, the reasons behind this controversy could be associated with drug administration at a late AD stage when it is difficult for glutamatergic-mediated drugs to rescue synaptic damage. This shows once more the importance of intervening early and identifying pathogenic windows in AD, wherein drug administration will be safe and therapeutically effective. The other class of FDA-approved AD drugs is anti-cholinesterases. They have been shown to benefit synaptic function and can be administered together with memantine. However, similarly to treatments targeting the glutamatergic pathway, anti-cholinesterases have been shown to have controversial results, including limited efficacy and adverse side effects.
Regarding the serotonin pathway, SSRIs might offer more benefits to AD patients with depression. These drugs alone or administered in adjunction to other AD-approved treatments have had controversial results regarding their efficacy in improving cognition in dementia. Interestingly, SSRI administration appears to have mixed results, with some studies indicating its benefits to synaptic function in AD, while others report no improvements in cognition and even detrimental effects. Alternatively, treatments using neurotrophic factors might help rescue ongoing synaptic loss in AD, but their main limitation lies in difficulties surrounding their route of administration. Further studies using novel methods such as encapsulated delivery of neurotrophic factors might prove to be very beneficial in AD treatment.
Conclusions
The study of synaptic changes in AD might be one crucial path to identifying viable targets for AD treatment. Determining these targets that can reverse or prevent synaptic density loss can be an effective avenue for new disease-modifying therapies for AD patients. In this review, several compounds that help prevent or rescue synaptic degeneration were examined regarding their targets within the synapse and their efficacy. Future studies should focus on the pharmacokinetics of the compounds and the critical windows for the treatment administration, while investigating efficient dosage depending on the AD stage. One reason behind the reported failure of AD treatments might be that the medications are administered too late since the first cognitive symptoms usually show after drastic brain changes and severe synaptic loss have already occurred [5]. Therefore, more studies should focus on detecting AD at earlier stages by developing new diagnostic tools such as effective biomarkers targeting the early synaptic changes. Furthermore, almost all studies mentioned in this review did not consider the sex-difference effects of the treatments. Given that AD has been shown in several studies to be more prevalent in females, more studies should examine if the compounds cause different effects depending on the sex of the patients or animal models. Finally, the development of new diagnostic tools to detect synaptic loss at early events of AD pathology could be crucial for the efficacy of timed-treatment administration, allowing the identification of early AD windows and helping prevent significant synaptic damage.
References
Jan LY, Jan YN. L-glutamate as an excitatory transmitter at the Drosophila larval neuromuscular junction. J Physiol. 1976;262:215–36.
Takumi Y, Ramirez-Leon V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci. 1999;2:618–24.
Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, et al. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–9.
Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurological Sci. 1987;78:151–64.
Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–84.
Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–8.
Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, et al. Transsynaptic progression of amyloid-beta-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron. 2010;68:428–41.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80.
Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci USA. 1987;84:4190–4.
Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, et al. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–4.
Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–6.
Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science. 1987;235:877–80.
Li Y, Zhou W, Tong Y, He G, Song W. Control of APP processing and Abeta generation level by BACE1 enzymatic activity and transcription. FASEB J. 2006;20:285–92.
Zhang S, Wang Z, Cai F, Zhang M, Wu Y, Zhang J, et al. BACE1 cleavage site selection critical for amyloidogenesis and Alzheimer’s Pathogenesis. J Neurosci. 2017;37:6915–25.
Deng Y, Wang Z, Wang R, Zhang X, Zhang S, Wu Y, et al. Amyloid-beta protein (Abeta) Glu11 is the major beta-secretase site of beta-site amyloid-beta precursor protein-cleaving enzyme 1(BACE1), and shifting the cleavage site to Abeta Asp1 contributes to Alzheimer pathogenesis. Eur J Neurosci. 2013;37:1962–9.
Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci USA. 1999;96:6959–63.
Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, et al. Presenilins are required for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1. Nat Cell Biol. 2000;2:463–5.
Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–62.
McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–6.
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–53.
Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807.
Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA. 2009;106:4012–7.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9.
Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556-61.
Lei M, Xu H, Li Z, Wang Z, O’Malley TT, Zhang D, et al. Soluble Abeta oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol Dis. 2016;85:111–21.
Zhao D, Watson JB, Xie CW. Amyloid beta prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J Neurophysiol. 2004;92:2853–8.
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–38.
Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK. Co-occurrence of Alzheimer’s disease ss-amyloid and tau pathologies at synapses. Neurobiol Aging. 2010;31:1145–52.
Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–50.
Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011;31:700–11.
Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–54.
Tai HC, Wang BY, Serrano-Pozo A, Frosch MP, Spires-Jones TL, Hyman BT. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s disease. Acta Neuropathologica Commun. 2014;2:146.
Hoffmann NA, Dorostkar MM, Blumenstock S, Goedert M, Herms J. Impaired plasticity of cortical dendritic spines in P301S tau transgenic mice. Acta Neuropathologica Commun. 2013;1:82.
Jaworski T, Lechat B, Demedts D, Gielis L, Devijver H, Borghgraef P, et al. Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am J Pathol. 2011;179:2001–15.
Kopeikina KJ, Polydoro M, Tai HC, Yaeger E, Carlson GA, Pitstick R, et al. Synaptic alterations in the rTg4510 mouse model of tauopathy. J Comp Neurol. 2013;521:1334–53.
Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, et al. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol. 2010;223:385–93.
Dejanovic B, Huntley MA, De Maziere A, Meilandt WJ, Wu T, Srinivasan K, et al. Changes in the synaptic proteome in tauopathy and rescue of Tau-Induced synapse loss by C1q antibodies. Neuron. 2018;100:1322–36 e1327.
Dickstein DL, Brautigam H, Stockton SD Jr, Schmeidler J, Hof PR. Changes in dendritic complexity and spine morphology in transgenic mice expressing human wild-type tau. Brain Struct Funct. 2010;214:161–79.
Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci. 2009;29:10741–9.
Pickett EK, Henstridge CM, Allison E, Pitstick R, Pooler A, Wegmann S, et al. Spread of tau down neural circuits precedes synapse and neuronal loss in the rTgTauEC mouse model of early Alzheimer’s disease. Synapse. 2017;71:e21965.
Zheng L, Duan J, Duan X, Zhou W, Chen C, Li Y, et al. Association of apolipoprotein E (ApoE) Polymorphism with Alzheimer’s Disease in Chinese Population. Curr Alzheimer Res. 2016;13:912–7.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–56.
Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11:667.
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–81.
Hesse R, Hurtado ML, Jackson RJ, Eaton SL, Herrmann AG, Colom-Cadena M, et al. Comparative profiling of the synaptic proteome from Alzheimer’s disease patients with focus on the APOE genotype. Acta Neuropathologica Commun. 2019;7:214.
Zhao J, Fu Y, Yamazaki Y, Ren Y, Davis MD, Liu CC, et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat Commun. 2020;11:5540.
Chung WS, Verghese PB, Chakraborty C, Joung J, Hyman BT, Ulrich JD, et al. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci USA. 2016;113:10186–91.
Jain S, Yoon SY, Leung L, Knoferle J, Huang Y. Cellular source-specific effects of apolipoprotein (apo) E4 on dendrite arborization and dendritic spine development. PLoS ONE. 2013;8:e59478.
Hudry E, Dashkoff J, Roe AD, Takeda S, Koffie RM, Hashimoto T, et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. 2013;5:212ra161.
Kang SS, Ahn EH, Liu X, Bryson M, Miller GW, Weinshenker D, et al. ApoE4 inhibition of VMAT2 in the locus coeruleus exacerbates Tau pathology in Alzheimer’s disease. Acta Neuropathol. 2021;142:139–58.
Wang C, Xiong M, Gratuze M, Bao X, Shi Y, Andhey PS, et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron. 2021;109:1657–74 e1657.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–8.
Paolicelli RC, Gross CT. Microglia in development: linking brain wiring to brain environment. Neuron Glia Biol. 2011;7:77–83.
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–78.
Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89.
Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, et al. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation. 2017;14:48.
Haga S, Ikeda K, Sato M, Ishii T. Synthetic Alzheimer amyloid beta/A4 peptides enhance production of complement C3 component by cultured microglial cells. Brain Res. 1993;601:88–94.
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705.
Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013;33:13460–74.
Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–4.
Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun. 2015;6:6176.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–6.
Reichwald J, Danner S, Wiederhold KH, Staufenbiel M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J Neuroinflammation. 2009;6:35.
Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J Neurosci. 2004;24:6457–65.
Zhang Y, Dong Z, Song W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct Target Ther. 2020;5:37.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8.
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669–73.
Lonnemann N, Hosseini S, Marchetti C, Skouras DB, Stefanoni D, D’Alessandro A, et al. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2020;117:32145–54.
Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP, et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy] -5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–88.
Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, et al. The complex PrPc-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J Neurosci. 2012;32:16857–71.
Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, et al. Alzheimer amyloid-Î 2 oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012;15:1227–35.
Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol. 2015;77:953–71.
Smith LM, Zhu R, Strittmatter SM. Disease-modifying benefit of Fyn blockade persists after washout in mouse Alzheimer’s model. Neuropharmacology. 2018;130:54–61.
Toyonaga T, Smith LM, Finnema SJ, Gallezot JD, Naganawa M, Bini J, et al. In vivo synaptic density imaging with 11C-UCB-J detects treatment effects of saracatinib in a mouse model of Alzheimer disease. J Nucl Med. 2019;60:1780–6.
Nygaard HB, Wagner AF, Bowen GS, Good SP, MacAvoy MG, Strittmatter KA, et al. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimers Res Ther. 2015;7:35–35.
Van Dyck CH, Nygaard HB, Chen K, Donohue MC, Raman R, Rissman RA, et al. Effect of AZD0530 on cerebral metabolic decline in Alzheimer Disease: a randomized clinical trial. JAMA Neurol. 2019;76:1219–29.
Trias E, Ibarburu S, Barreto-Núñez R, Varela V, Moura IC, Dubreuil P, et al. Evidence for mast cells contributing to neuromuscular pathology in an inherited model of ALS. JCI Insight. 2017;2:e95934.
Li T, Martin E, Abada Y-S, Boucher C, Cès A, Youssef I, et al. Effects of chronic masitinib treatment in APPPS1dE9 transgenic mice modeling Alzheimer’s Disease. J Alzheimer’s Dis. 2020;76:1339–45.
Piette F, Belmin J, Vincent H, Schmidt N, Pariel S, Verny M, et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trail. Alzheimer’s Res Ther. 2011;3:1–11.
Iannuzzi F, Sirabella R, Canu N, Maier TJ, Annunziato L, Matrone C. Fyn tyrosine kinase elicits amyloid precursor protein Tyr682 phosphorylation in neurons from Alzheimer’s Disease Patients. Cells. 2020;9:1807.
Sellers KJ, Elliott C, Jackson J, Ghosh A, Ribe E, Rojo AI, et al. Amyloid β synaptotoxicity is Wnt-PCP dependent and blocked by fasudil. Alzheimers Dement. 2018;14:306–17.
Yu J, Yan Y, Gu Q, Kumar G, Yu H, Zhao Y, et al. Fasudil in combination with Bone Marrow Stromal Cells (BMSCs) Attenuates Alzheimer’s Disease-Related changes through the regulation of the peripheral immune system. Front Aging Neurosci. 2018;10:216–16.
Kumar M, Bansal N. Fasudil hydrochloride ameliorates memory deficits in rat model of streptozotocin-induced Alzheimer’s disease: Involvement of PI3-kinase, eNOS and NFκB. Behavioural Brain Res. 2018;351:4–16.
Xin YL, Yu JZ, Yang XW, Liu CY, Li YH, Feng L, et al. FSD-C10: a more promising novel ROCK inhibitor than Fasudil for treatment of CNS autoimmunity. Biosci Rep. 2015;35:247.
Gu QF, Yu JZ, Wu H, Li YH, Liu CY, Feng L, et al. Therapeutic effect of Rho kinase inhibitor FSD-C10 in a mouse model of alzheimer’s disease. Exp Therapeutic Med. 2018;16:3929–38.
Bormann J. Memantine is a potent blocker of N-methyl-D-aspartate (NMDA) receptor channels. Eur J Pharm. 1989;166:591–2.
Fleischhacker WW, Buchgeher A, Schubert H. Memantine in the treatment of senile dementia of the Alzheimer type. Prog Neuropsychopharmacol Biol Psychiatry. 1986;10:87–93.
Winblad B, Poritis N. Memantine in severe dementia: results of the 9M-Best Study (Benefit and efficacy in severely demented patients during treatment with memantine). Int J Geriatr Psychiatry. 1999;14:135–46.
Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in moderate-to-severe Alzheimer’s disease. N. Engl J Med. 2003;348:1333–41.
Devi L, Ohno M. Cognitive benefits of memantine in Alzheimer’s 5XFAD model mice decline during advanced disease stages. Pharmacol Biochem Behav. 2016;144:60–66.
Folch J, Busquets O, Ettcheto M, Sánchez-López E, Castro-Torres RD, Verdaguer E, et al. Memantine for the treatment of Dementia: a review on its current and future applications. J Alzheimer’s Dis. 2018;62:1223–40.
Knight R, Khondoker M, Magill N, Stewart R, Landau S. A systematic review and meta-analysis of the effectiveness of acetylcholinesterase inhibitors and memantine in treating the cognitive symptoms of dementia. Dement Geriatr Cogn Disord. 2018;45:131–51.
Tampi RR, van Dyck CH. Memantine: Efficacy and safety in mild-to-severe Alzheimer’s disease. Neuropsychiatr Dis Treat. 2007;3:245–58.
Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci USA. 2013;110:E2518–2527.
Trudler D, Sanz-Blasco S, Eisele YS, Ghatak S, Bodhinathan K, Akhtar MW, et al. alpha-Synuclein oligomers induce glutamate release from astrocytes and excessive extrasynaptic NMDAR activity in neurons, thus contributing to synapse loss. J Neurosci. 2021;41:2264–73.
Ghatak S, Dolatabadi N, Gao R, Wu Y, Scott H, Trudler D, et al. NitroSynapsin ameliorates hypersynchronous neural network activity in Alzheimer hiPSC models. Mol Psychiatry. 2021;26:5751–65.
Pereira AC, Lambert HK, Grossman YS, Dumitriu D, Waldman R, Jannetty SK, et al. Glutamatergic regulation prevents hippocampal-dependent age-related cognitive decline through dendritic spine clustering. Proc Natl Acad Sci USA. 2014;111:18733–8.
Mokhtari Z, Baluchnejadmojarad T, Nikbakht F, Mansouri M, Roghani M. Riluzole ameliorates learning and memory deficits in Abeta25-35-induced rat model of Alzheimer’s disease and is independent of cholinoceptor activation. Biomed Pharmacother. 2017;87:135–44.
Matthews DC, Mao X, Dowd K, Tsakanikas D, Jiang CS, Meuser C, et al. Riluzole, a glutamate modulator, slows cerebral glucose metabolism decline in patients with Alzheimer’s disease. Brain. 2021;144:3742-55.
Burns A, Rossor M, Hecker J, Gauthier S, Petit H, Möller HJ, et al. The effects of donepezil in Alzheimer’s disease - Results from a multinational trial. Dement Geriatr Cogn Disord. 1999;10:237–44.
Johannsen P, Salmon E, Hampel H, Xu Y, Richardson S, Qvitzau S, et al. Assessing therapeutic efficacy in a progressive disease: a study of donepezil in Alzheimer’s disease. CNS Drugs. 2006;20:311–25.
Seltzer B, Zolnouni P, Nunez M, Goldman R, Kumar D, Ieni J, et al. Efficacy of donepezil in early-stage Alzheimer disease: a randomized placebo-controlled trial. Arch Neurol. 2004;61:1852–6.
Wilcock GK, Lilienfeld S, Gaens E. Efficacy and safety of galantamine in patients with mild to moderate Alzheimer’s disease: multicentre randomised controlled trial. Br Med J. 2000;321:1445–9.
Tariot PN, Solomon PR, Morris JC, Kershaw P, Lilienfeld S, Ding C. A 5-month, randomized, placebo-controlled trial of galantamine in AD. Neurology. 2000;54:2269–76.
Rösler M, Anand R, Cicin-Sain A, Gauthier S, Agid Y, Dal-Bianco P, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: International randomised controlled trial. Br Med J. 1999;318:633–40.
Small GW, Kaufer D, Mendiondo MS, Quarg P, Spiegel R. Cognitive performance in Alzheimer’s disease patients receiving rivastigmine for up to 5 years. Int J Clin Pract. 2005;59:473–7.
Lopez OL, Becker JT, Wahed AS, Saxton J, Sweet RA, Wolk DA, et al. Long-term effects of the concomitant use of memantine with cholinesterase inhibition in Alzheimer disease. J Neurol Neurosurg Psychiatry. 2009;80:600–7.
Riepe MW, Adler G, Ibach B, Weinkauf B, Tracik F, Gunay I. Domain-specific improvement of cognition on memantine in patients with Alzheimer’s disease treated with rivastigmine. Dement Geriatr Cogn Disord. 2007;23:301–6.
Rountree SD, Chan W, Pavlik VN, Darby EJ, Siddiqui S, Doody RS. Persistent treatment with cholinesterase inhibitors and/or memantine slows clinical progression of Alzheimer disease. Alzheimer’s Res Ther. 2009;1:7.
Tricco AC, Soobiah C, Berliner S, Ho JM, Ng CH, Ashoor HM, et al. Efficacy and safety of cognitive enhancers for patients with mild cognitive impairment: a systematic review and meta-analysis. CMAJ. 2013;185:1393–401.
Davis B, Sadik K. Circadian cholinergic rhythms: Implications for cholinesterase inhibitor therapy. Dement Geriatr Cogn Disord. 2006;21:120–9.
Finkel SI, Mintzer JE, Dysken M, Krishnan KRR, Burt T, McRae T. A randomized, placebo-controlled study of the efficacy and safety of sertraline in the treatment of the behavioral manifestations of Alzheimer’s disease in outpatients treated with donepezil. Int J Geriat Psychiat. 2004;19:9–18.
Mowla A, Mosavinasab M, Pani A. Does fluoxetine have any effect on the cognition of patients with mild cognitive impairment? J Clin Psychopharm. 2007;27:67–70.
Bartels C, Wagner M, Wolfsgruber S, Ehrenreich H, Schneider A. Impact of SSRI therapy on risk of conversion from mild cognitive impairment to Alzheimer’s dementia in individuals with previous depression. Am J Psychiatry. 2018;175:232–41.
Banerjee S, Hellier J, Dewey M, Romeo R, Ballard C, Baldwin R, et al. Sertraline or mirtazapine for depression in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-controlled trial. Lancet. 2011;378:403–11.
Munro CA. Cognitive response to pharmacological treatment for depression in Alzheimer Disease: secondary outcomes from the Depression in Alzheimer’s Disease Study (DIADS). Am J Geriatr Psychiatry. 2004;12:491–8.
Olafsson K, Jørgensen S, Jensen HV, Bille A, Arup P, Andersen J. Fluvoxamine in the treatment of demented elderly patients: a double‐blind, placebo‐controlled study. Acta Psychiatr Scandinavica. 1992;85:453–6.
Porsteinsson AP, Drye LT, Pollock BG, Devanand DP, Frangakis C, Ismail Z, et al. Effect of citalopram on agitation in Alzheimer disease: The CitAD randomized clinical trial. JAMA - J Am Med Assoc. 2014;311:682–91.
Ivković M, Damjanović A, Jasović-Gasić M, Paunović VR. The effects of fluoxetine on cognitive functions in animal model of Alzheimer’s disease. Psychiatr Danubina. 2004;16:15–20.
Jin L, Gao LF, Sun DS, Wu H, Wang Q, Ke D, et al. Long-term ameliorative effects of the antidepressant fluoxetine exposure on cognitive deficits in 3 × TgAD Mice. Mol Neurobiol. 2017;54:4160–71.
Zhou CN, Chao FL, Zhang Y, Jiang L, Zhang L, Fan JH, et al. Fluoxetine delays the cognitive function decline and synaptic changes in a transgenic mouse model of early Alzheimer’s disease. J Comp Neurol. 2019;527:1378–87.
Hajszan T, MacLusky NJ, Leranth C. Short-term treatment with the antidepressant fluoxetine triggers pyramidal dendritic spine synapse formation in rat hippocampus. Eur J Neurosci. 2005;21:1299–303.
Zheng J, Xu DF, Li K, Wang HT, Shen PC, Lin M, et al. Neonatal exposure to fluoxetine and fluvoxamine alteres spine density in mouse hippocampal CA1 pyramidal neurons. Int J Clin Exp Pathol. 2011;4:162–8.
Rozzini L, Vicini Chilovi B, Conti M, Bertoletti E, Zanetti M, Trabucchi M, et al. Efficacy of SSRIs on cognition of Alzheimer’s disease patients treated with cholinesterase inhibitors. Int Psychogeriatr. 2010;22:114–9.
Orgeta V, Tabet N, Nilforooshan R, Howard R. Efficacy of antidepressants for depression in Alzheimer’s Disease: systematic review and meta-analysis. J Alzheimer’s Dis. 2017;58:725–33.
Petracca GM, Chemerinski E, Starkstein SE. A double-blind, placebo-controlled study of fluoxetine in depressed patients with Alzheimer’s disease. Int Psychogeriatr. 2001;13:233–40.
Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, et al. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101:343–7.
Li B, Yamamori H, Tatebayashi Y, Shafit-Zagardo B, Tanimukai H, Chen S, et al. Failure of neuronal maturation in Alzheimer disease dentate gyrus. J Neuropathol Exp Neurol. 2008;67:78–84.
Mukaetova-Ladinska EB, Garcia-Siera F, Hurt J, Gertz HJ, Xuereb JH, Hills R, et al. Staging of cytoskeletal and β-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s disease. Am J Pathol. 2000;157:623–36.
Ferrer I, Marín C, Rey MJ, Ribalta T, Goutan E, Blanco R, et al. BDNF and full-length and Truncated TrkB expression in Alzheimer disease. Implications in Therapeutic strategies. J Neuropathol Exp Neurol. 1999;58:729–39.
Cocco C, D’Amato F, Noli B, Ledda A, Brancia C, Bongioanni P, et al. Distribution of VGF peptides in the human cortex and their selective changes in Parkinson’s and Alzheimer’s diseases. J Anat. 2010;217:683–93.
Bruno MA, Leon WC, Fragoso G, Mushynski WE, Almazan G, Cuello AC. Amyloid beta-induced nerve growth factor dysmetabolism in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:857–69.
Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol Cell Neurosci. 2001;18:210–20.
Tuszynski MH, Thal L, Pay M, Salmon DP, Sang U, H., Bakay R, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11:551–5.
Rafii MS, Baumann TL, Bakay RAE, Ostrove JM, Siffert J, Fleisher AS, et al. A phase1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimer’s Dement: J Alzheimer’s Assoc. 2014;10:571–81.
Rafii MS, Tuszynski MH, Thomas RG, Barba D, Brewer JB, Rissman RA, et al. Adeno-associated viral vector (serotype 2)-nerve growth factor for patients with Alzheimer disease a randomized clinical trial. JAMA Neurol. 2018;75:834–41.
Baazaoui N, Iqbal K. Prevention of Amyloid-β and Tau pathologies, associated neurodegeneration, and cognitive deficit by early treatment with a neurotrophic compound. J Alzheimer’s Dis. 2017;58:215–30.
Baazaoui N, Iqbal K. Prevention of dendritic and synaptic deficits and cognitive impairment with a neurotrophic compound. Alzheimer’s Res Ther. 2017;9:45–45.
Kazim SF, Blanchard J, Dai CL, Tung YC, LaFerla FM, Iqbal IG, et al. Disease modifying effect of chronic oral treatment with a neurotrophic peptidergic compound in a triple transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2014;71:110–30.
Li B, Wanka L, Blanchard J, Liu F, Chohan MO, Iqbal K, et al. Neurotrophic peptides incorporating adamantane improve learning and memory, promote neurogenesis and synaptic plasticity in mice. FEBS Lett. 2010;584:3359–65.
Eyjolfsdottir H, Eriksdotter M, Linderoth B, Lind G, Juliusson B, Kusk P, et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: Application of a second-generation encapsulated cell biodelivery device. Alzheimers Res Ther. 2016;8:30–30.
Eriksdotter M, Navarro-Oviedo M, Mitra S, Wahlberg L, Linderoth B, Tjernberg LO, et al. Cerebrospinal fluid from Alzheimer patients affects cell-mediated nerve growth factor production and cell survival in vitro. Exp Cell Res. 2018;371:175–84.
James ML, Belichenko NP, Shuhendler AJ, Hoehne A, Andrews LE, Condon C, et al. [(18)F]GE-180 PET detects reduced microglia activation after LM11A-31 Therapy in a Mouse Model of Alzheimer’s Disease. Theranostics. 2017;7:1422–36.
Funding
This work was supported by the Key Laboratory of Alzheimer’s Disease Of Zhejiang Province, the grants from the Canadian Institutes of Health Research (CIHR) Project Grant PJT-166127 to WS, the National Natural Science Foundation of China (No. 81801091) to LP, and the Natural Science Foundation of Shandong Province of China (No. ZR2019BH076) to LP. WS was the holder of the Tier 1 Canada Research Chair in Alzheime’s Disease, and IBL is supported by the UBC Four Year Fellowship and the Jock & Irene Graham Brain Research Endowment.
Author information
Authors and Affiliations
Contributions
All authors were involved in literature search and writing of the article. WS supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Peng, L., Bestard-Lorigados, I. & Song, W. The synapse as a treatment avenue for Alzheimer’s Disease. Mol Psychiatry 27, 2940–2949 (2022). https://doi.org/10.1038/s41380-022-01565-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-022-01565-z
- Springer Nature Limited
This article is cited by
-
Secoisolariciresinol diglucoside attenuates neuroinflammation and cognitive impairment in female Alzheimer’s disease mice via modulating gut microbiota metabolism and GPER/CREB/BDNF pathway
Journal of Neuroinflammation (2024)
-
Microbiota–gut–brain axis and its therapeutic applications in neurodegenerative diseases
Signal Transduction and Targeted Therapy (2024)
-
miR-143-3p modulates depressive-like behaviors via Lasp1 in the mouse ventral hippocampus
Communications Biology (2024)
-
Apolipoprotein E in Alzheimer’s Disease: Focus on Synaptic Function and Therapeutic Strategy
Molecular Neurobiology (2024)
-
Olfactory Three-Needle Electroacupuncture Improved Synaptic Plasticity and Gut Microbiota of SAMP8 Mice by Stimulating Olfactory Nerve
Chinese Journal of Integrative Medicine (2024)